

Abstract
Acute Myeloid Leukemia is a devastating and heterogeneous, hematological malignancy characterized by the uncontrolled proliferation of undifferentiated myeloid progenitor cells—blasts. Mutations in certain mitochondrial proteins, such as IDH2 have been shown to contribute to leukemogenesis. However, the role of mutations in mitochondrial-encoded Electron Transport Chain (ETC) genes have thus far not been well elucidated in AML. Here, we use TCGA data to characterize mutations in the ETC genes and their association with clinical outcomes in AML. We found that mitochondrial ETC mutations—in Complex I, III, IV and/or V (ATP Synthase)—were present in 8% of patients with AML and were significantly more frequent in older patients. Patients with ETC mutations had worse overall survival than ETC wild type patients (OS: 9.3 vs 20.1 months; p-value: 0.007). Additionally, mutations in either or both Complex I and IV were associated with TP53 mutations (p-value: 0.009), and among TP53 mutated patients, mutations in either or both Complex I and IV were significantly associated with worse overall survival (OS: 0.85 vs 9.4 months; p-value: 0.008). Elucidation of the mechanisms by which ETC mutations contribute to AML pathogenesis and progression would facilitate the development of novel therapeutic targets.
Introduction
Oxidative phosphorylation (OXPHOS) is a key intracellular process that generates ATP to power the cell. It is the last step in cellular respiration whereby cofactors, such as NADH and FADH2, generated in previous steps in respiration donate electrons to the Electron Transport Chain (ETC) to generate a H+ gradient that drives ATP synthase to convert ADP to ATP1. The ETC comprises of four complexes and an ATP synthase and is located on the inner membrane of the mitochondria1. During OXPHOS, however, some electrons may leak from the different electron transport chain complexes and bind with molecular oxygen to form superoxide anions. Superoxide anions bind to iron-sulfur cores of important cellular enzymes and inactivate them, causing shut down of pivotal intracellular processes2,3. Superoxide anions are detoxified by an important mitochondrial antioxidant called SOD2, which converts superoxide anions to H2O2. H2O2 is further detoxified by glutathione peroxidase 1 (GPX1). H2O2 may also interact with Fe2+ to generate OH−, or hydroxyl radicals4,5. Thus, mutations or defects in complexes of the ETC could have significant effects on the metabolic balance of the mitochondria.
OXPHOS downregulation resulting from a decrease in mtDNA content was reported in several malignancies including breast cancer, gastric cancer, hepatocellular carcinoma and non–small cell lung cancer (NSCLC), and was found to be associated with poor clinical outcome and correlated with invasive and metastatic tumors6,7. On the other hand, recent studies suggest that particular cancers such as acute lymphoblastic leukemia (ALL), non-Hodgkin lymphoma, prostate cancer, head and neck cancer, and thyroid cancer among others exhibit increased mtDNA content and these malignant cells rely heavily on OXPHOS for their energy source and survival8. Mitochondrial DNA (mtDNA) encodes 13 subunits of the ETC complexes: I, III, IV and V (ATP Synthase)9. Mutations in mtDNA have been identified in several malignancies10. Mitochondrial mutations were found to be important drivers in prostate cancer aggression11 and may contribute to hepatocellular carcinoma and colorectal cancer progression10,12.
Acute Myeloid Leukemia (AML) is a devastating and heterogeneous hematological malignancy characterized by the uncontrolled proliferation of undifferentiated myeloid progenitor cells—known as blasts13. The altered metabolic state of AML cells and the role of the mitochondria has been shown to contribute to its pathogenesis14–16. Mutations in the mitochondrial enzyme isocitrate dehydrogenase-2 (IDH2) have been identified as oncogenic drivers of AML. They convert alpha-ketoglutarate into the R-enantiomer of 2-hydroxyglutarate, which is associated with DNA hypermethylation, epigenetic changes, reduced ATP synthase activity and overall reduced mitochondrial energy metabolism17–19.
While mitochondrial DNA mutations have been previously reported in hematologic malignancies including AML10,20, mutations in the ETC genes encoded by the mitochondria have not been fully characterized in AML. Thus, we used data of patients with AML from the Cancer Genome Atlas (TCGA) to investigate mitochondrial ETC mutations in AML and assessed their association with clinical and molecular characteristics and with patients’ clinical outcome.
Results
Frequency of ETC complex gene mutations
Of the 200 patients included in this analysis, 16 (8%) patients had a mutation in one or more of the mitochondrial encoded ETC complexes genes. Each mitochondrial encoded ETC gene codes a different subunit of each complex. Four patients had mutations in Complex I. Complex I has seven mitochondrial encoded genes-MT-ND1, MT-ND2, MT-ND3, MT-ND4, MT-ND4L, MT-ND5 and MT-ND6—where one patient has a missense mutation in MT-ND1 and MT-ND5, two patients have missense mutations in MT-ND5 and one patient has a truncating mutation in ND5. Three patients had missense mutations in the mitochondrial encoded MT-CYB gene in Complex III. Complex IV has three mitochondrial encoded genes—MT-CO1, MT-CO2 and MT-CO3. Two patients had missense mutations in MT-CO1, five patients had missense mutations in MT-CO2 and two patients had missense mutations in MT-CO3. One patient had a missense mutation in MT-ATP6 of the ATP Synthase; but no patients had mutations in MT-ATP8. One patient had a mutation in both Complex I and Complex IV. Because Complex II is composed of four nuclear gene encoded subunits, mutations in Complex II were not included in this study. Frequencies of mutations are summarized in Table 1.
Table 1.
Frequency of Mutations in the mtDNA ETC genes in Patients with AML (+denotes one patient with mutations in both ND1 and ND5).
| Complex | Gene | Amino Acid Change | Mutation Type | No. of Patients |
|---|---|---|---|---|
| Complex I | ND1 | Y43H+ | Missense | 1 (0.5%) |
| ND2 | — | — | — | |
| ND3 | — | — | — | |
| ND4 | — | — | — | |
| ND4L | — | — | — | |
| ND5 | S345P, S476P | Missense | 4 (2.0%) | |
| L260P+ | Missense | |||
| N452S | Missense | |||
| P242Qfs*? | Truncating | |||
| ND6 | — | — | — | |
| Complex III | CYB | V73M | Missense | 3 (1.50%) |
| M89I | Missense | |||
| A302T | Missense | |||
| Complex IV | CO1 | G435E | Missense | 2 (1.0%) |
| M237V | Missense | |||
| CO2 | L126S | Missense | 5 (2.5%) | |
| D173N | Missense | |||
| V191M | Missense | |||
| T66A | Missense | |||
| D112N | Missense | |||
| CO3 | L175P | Missense | 2 (1.0%) | |
| V254A | Missense | |||
| ATP Synthase | ATP6 | M60T | Missense | 1 (0.5%) |
| ATP8 | — | — | — |
Association of ETC complex gene mutations with clinical characteristics
We assessed the association between ETC gene mutations and primary patient characteristics. The results are summarized in Table 2. No significant difference in median white blood cell count, peripheral blood blasts and bone marrow blasts were detected between patients with mutations and patients without mutations. Mutations in mitochondrial DNA of any of the ETC genes were significantly associated with age (median: 68.5 vs 57; p-value: 0.001), the M0 FAB classification of AML (%: 26.7 vs 8.2; p-value: 0.04) and poor molecular risk (%: 50 vs 23.4; p-value: 0.035).
Table 2.
Clinical characteristics of patients with AML according to mutational status of mitochondrial ETC genes.
| Characteristic | No Mutations (n = 184) | Mutations (n = 16) | p-value |
|---|---|---|---|
| Age, median (years) | 57 | 68.5 | 0.001 |
| Young (<60) | 106 (57.6%) | 3 (18.8%) | 0.003 |
| Old (≥60) | 78 (42.4%) | 13 (81.2%) | |
| Sex | 0.30 | ||
| Female (n, %) | 87 (47.3%) | 5 (31.3%) | |
| Male (n, %) | 97 (52.7%) | 11 (68.7%) | |
| FAB | |||
| M0 (n, %) | 15 (8.2%) | 4 (26.7%) | 0.04 |
| M1 (n, %) | 41 (22.4%) | 5 (33.3%) | 0.35 |
| M2 (n, %) | 41 (22.4%) | 3 (20%) | >0.99 |
| M3 (n, %) | 19 (10.3%) | 1 (6.25%) | >0.99 |
| M4 (n, %) | 39 (21.3%) | 2 (13.3%) | 0.74 |
| M5 (n, %) | 22 (12.0%) | 0 | 0.38 |
| M6 (n, %) | 3 (1.64%) | 0 | >0.99 |
| M7 (n, %) | 3 (1.64%) | 0 | >0.99 |
| WB Count, median | 15.6 | 21.5 | 0.79 |
| ln (WB Count) | 2.60 | 2.74 | 0.75 |
| % BM Blast, median | 72 | 77 | 0.69 |
| % PB Blast, median | 34.5 | 49 | 0.99 |
| Risk Status | |||
| Poor (n, %) | 43 (23.4%) | 8 (50.0%) | 0.035 |
| Intermediate (n, %) | 99 (53.8%) | 7 (43.8%) | 0.44 |
| Good (n, %) | 38 (20.7%) | 1 (6.25%) | 0.20 |
| Cytogenetic Status | 0.80 | ||
| Normal (n, %) | 85 (47.5%) | 7 (43.8%) | |
| Abnormal (n, %) | 94 (52.5%) | 9 (62.5%) | |
| Transplant (Y/N) | 0.79 | ||
| No (n, %) | 102 (55.4%) | 10 (62.5%) | |
| Yes (n, %) | 82 (44.6%) | 6 (37.5%) | |
P-values calculated using non-parametric Mann-Whitney U or Fisher’s Exact test.
Next, we stratified the analysis by ETC complex to see if mutations in a specific complex were associated with patient’s clinical characteristics. Only Complex I, III and IV had enough patients with mutations to be included in the analysis, n = 4, n = 3 and n = 9, respectively. Patients were also grouped together by the presence of one or more mutations in either or both Complex I and IV (n = 12). Mutations in Complex I, alone, or in Complex III, alone, were not significantly associated with any clinical characteristics (Supplemental Tables S1 and S2. Mutations in Complex IV, alone, were significantly associated with age (median: 72 vs 57; p-value: 0.009) and the M0 FAB sub-classification of AML (%: 33.3 vs 8.47; p-value: 0.04; Supplemental Table S3). Mutations in Complex I and IV, either or both, were significantly associated with age (median: 71.5 vs 57; p-value: 0.008) and poor molecular risk (%: 58.3 vs 23.4; p-value: 0.015; Supplemental Table S4).
Association of ETC complex gene mutations with molecular characteristics
Patients with mutations in the ETC complex genes were more frequently mutated with TP53 (%: 31.3 vs 5.98; p-value: 0.004; Table 3). Mutations in Complex IV, alone, were also associated with mutations in TP53 (%: 33.3 vs 6.81; p-value: 0.026; Supplemental Table S5). When combined, either or both Complex I and IV mutations remained associated with TP53 mutation (%: 33.3 vs 6.38; p-value: 0.009; Supplemental Table S6; Fig. 1). No association was found between mutations in the ETC genes, Complex I alone or Complex III alone, and other mutations reported in AML (Supplemental Tables S7 and S8.
Table 3.
Molecular characteristics of patients with AML according to mutational status of mitochondrial ETC genes. P-values calculated using Fisher’s Exact test.
| Gene | No Mutations (n = 184) | Mutations (n = 16) | p-value |
|---|---|---|---|
| FLT3 | 53 (8.8%) | 3 (18.8%) | 0.56 |
| TP53 | 11 (5.98%) | 5 (31.3%) | 0.004 |
| DNMT3A | 45 (24.5%) | 4 (25%) | >0.99 |
| CEBPA | 11 (5.98%) | 2 (12.5%) | 0.28 |
| NRAS | 15 (8.15%) | 0 | 0.62 |
| TET2 | 16 (8.70%) | 1 (6.25%) | >0.99 |
| IDH1 | 18 (9.78%) | 1 (6.25%) | >0.99 |
| IDH2 | 18 (9.78%) | 2 (12.5%) | 0.67 |
| RUNX1 | 16 (8.70%) | 1 (6.25%) | >0.99 |
| NPM1 | 51 (27.7%) | 3 (18.8%) | 0.57 |
| WT1 | 12 (6.52%) | 0 | 0.60 |
Figure 1.
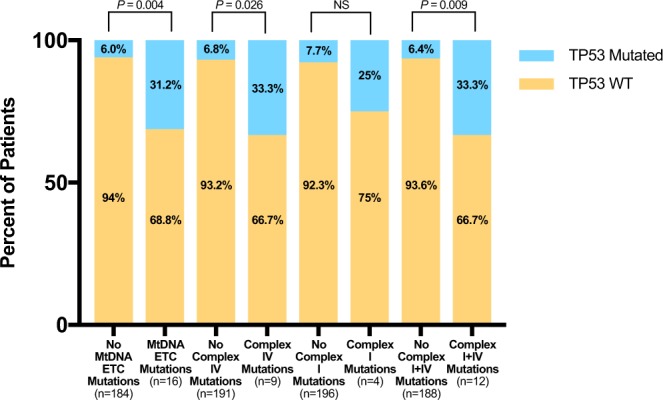
Distribution of TP53 in AML patients stratified by mtDNA ETC gene mutations. The percentage of patients with different types of mtDNA mutations that have TP53 mutations. Fisher Exact test was used for statistical analysis to determine if there was a statistically significant difference in proportion of patients with TP53 mutations between patients with or without mtDNA ETC mutations.
Association of mtDNA ETC gene mutations with clinical outcome
Patients with mtDNA ETC gene mutations had significantly shorter median overall survival than patients without mtDNA ETC mutations (median: 9.3 vs 20.1 months; p-value: 0.007; log-rank (Mantel-Cox) test, Fig. 2A). No significant difference in disease-free survival was detected between the ETC mutant and wild type patients (Fig. 2B). Because patients with APL or t(15;17) receive different treatment (ATRA) than other patients with AML and have better clinical outcome, we excluded these patients and reanalyzed the association between the presence of mtDNA ETC mutations and patients’ survival. Similarly, patients with mtDNA ETC gene mutations had shorter overall survival (median: 9.3 vs 15.8; p-value: 0.014) but not disease-free survival (Fig. 2C,D). After age-stratification, however, the difference in median overall survival was lost, likely due to the small number of patients (Supplemental Fig. S1).
Figure 2.

Association of ETC complex gene mutations with clinical outcome. Kaplan-Meier survival curves. (A) Overall Survival (OS) and (B) Disease-Free Survival (DFS) in 200 patients with AML stratified by mtDNA ETC gene mutation status. (C) Overall Survival and (D) Disease-Free Survival in 180 patients with AML (excluding M3 FAB classification, PML-RARA patients) stratified by mtDNA ETC gene mutation status.
In multivariable survival analysis using the Cox Proportional Hazards model—after adjustment for age, cytogenetic risk, transplant status and mutations in DNMT3A, RUNX1 and TP53—the association between ETC mutations and clinical outcome was not statistically significant (p-value: 0.229; Table 4).
Table 4.
Cox Proportional Hazards modeling for overall survival in patients with mutations in the mtDNA ETC genes (n = 194).
| Variables | Hazard Ratio | 95% CI | p-value | |
|---|---|---|---|---|
| Age | 1.02 | 1.00 | 1.03 | 0.037 |
| Cytogenetic Risk | ||||
| Intermediate | 3.22 | 1.65 | 6.28 | 0.001 |
| Poor | 5.35 | 2.42 | 11.8 | <0.001 |
| Transplant Status | 0.44 | 0.29 | 0.68 | <0.001 |
| DNMT3A | 1.47 | 0.96 | 2.25 | 0.080 |
| RUNX1 | 1.82 | 0.96 | 3.44 | 0.066 |
| TP53 | 2.00 | 1.02 | 3.94 | 0.044 |
| mtDNA ETC | 1.44 | 0.79 | 2.62 | 0.229 |
Patients with mutations in Complex I had a significantly shorter median overall survival than patients without mutations (5.85 vs 18.5 months; p-value: 0.009; Fig. 3A) compared with patients without the mutations. Patients with mutations in Complex III did not have a significant difference in overall survival (Fig. 3B). Patients with mutations in Complex IV had significantly shorter median overall survival than patients without mutations (median: 7.0 vs 18.5 months; p-value: 0.047; Fig. 3C). Thus, we performed survival analysis combining the presence of mutations in either or both Complex I and Complex IV. Patients with mutations in either or both Complex I and IV had significantly poorer overall survival than patients without mutations (median: 8.15 vs 19 months; p-value: 0.007; Fig. 3D).
Figure 3.

Kaplan-Meier survival curves for Complex I, III and IV of ETC genes in AML. (A) OS in patients with (n = 4) and without (n = 196) mutations in Complex I. (B) OS in patients with (n = 3) and without (n = 197) mutations in Complex III. (C) OS in patients with (n = 8) and without (n = 192) mutations in Complex IV. (D) OS in patients with (n = 12) and without (n = 188) mutations in either or both Complex I and IV.
We also stratified patients by whether they received transplant or not. We found a significant association between mtDNA ETC gene mutations and poorer overall survival in patients who did not receive transplant (median: 4.35 vs 9.9 months; p-value: 0.0123; Fig. 4A). In patients who received transplant, there was also a trend towards worse overall survival in patients with mtDNA ETC gene mutations compared with those without mutations but this trend did not reach statistical significance (Fig. 4B).
Figure 4.

Kaplan-Meier survival curves for patients with AML according to ETC gene mutations after stratification by whether patients received transplant or not. Overall survival of patients who (A) did not receive transplant or (B) did receive transplant stratified by mitochondrial ETC gene mutation status.
Because mutations in either or both Complex I and IV were associated with TP53 mutation, we also stratified patients according to TP53 mutational status. We found that among TP53-mutant patients, those with mutations in either or both Complex I and IV had significantly worse overall survival than those without mutations (median: 0.85 vs 9.4 months; p-value: 0.008; Fig. 5).
Figure 5.
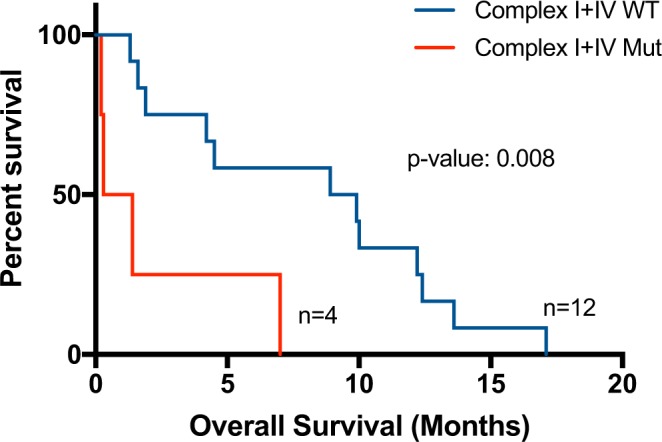
Kaplan-Meier survival curves of patients with AML according to mutations in either or both Complex I and IV of ETC genes in TP53 mutation positive patients. OS in TP53 mutated patients who had (n = 4) or did not have (n = 12) mutations in either or both Complex I and IV.
Using the Cox Proportional Hazards model we adjusted for age, cytogenetic risk, transplant status, DNMT3A, RUNX1 and TP53 mutations and found that in survival analysis, mutations in Complex I, alone, Complex III, alone, Complex IV, alone or either or both Complex I and IV were not significantly associated with overall survival.
Discussion
Acute myeloid leukemic cells are highly oxidized when compared with healthy hematopoietic stem cells; thus, mechanisms that contribute to this oxidation state represent an opportunity for targeted therapy14,15,21. Mutations in the mtDNA have been reported in several malignancies including AML20,22–24; the majority of these studies examined a limited number of patients and focused on targeted sequencing analysis. Thus, characterization of mtDNA mutations particularly those in genes involved in OXPHOS in a larger AML cohort is needed. Using the TCGA dataset, we identified 16 of 200 (8%) patients with AML had a mutation in one or more of their mitochondrial genes encoding ETC complexes. The high frequency of mtDNA ETC mutations in AML is similar to or greater than that of NRAS (8%), WT1 (6%), CEBPA (7%) and TP53 (8%) mutations—a few of the top 12 genes that are regularly mutated in AML, thus implicating these mutations in AML pathogenesis.
Complex I, or NADH dehydrogenase, binds NADH and transfers two electrons to ubiquinone (UQ) generating NAD+ and ubiquinol (UQH2) and pumps four H+ into the inner membrane space. Complex II, or succinate dehydrogenase, oxidizes succinate and transfers two electrons to quinone via FAD; electrons can also enter the ETC from this complex but this complex does not directly contribute to the proton gradient. Complex III, or cytochrome c oxidoreductase, pumps two H+ ions into the inner membrane space by oxidizing ubiquinone to ubiquinol25. Then the electrons are transferred through cytochrome bc1 to cytochrome c, which carries the electrons to complex IV25. At Complex IV, or cytochrome c oxidase, electrons carried over from cytochrome c are used to reduce molecular oxygen to two water molecules. Although mtDNA mutations were present in all complexes of the ETC, the majority of the mutations were found in Complex IV. Mutations in MT-CO1 and MT-CO2 of Complex IV have been previously reported in AML and were found to be associated with inferior disease-free survival23. Complex IV produces H2O from O2 and contributes protons to the proton gradient. The second most frequently mutated complex is Complex I, which is a major site of electron leakage that leads to the formation of superoxide anions. A previous study reported a potential association between the presence of mtDNA mutation in the ND1 gene of Complex I and the abnormal ROS production in blasts obtained from one patient with megacaryoblastic leukaemia26. Patients with mutations in either or both of these complexes had worse overall survival than patients without mutations in these complexes. While Complex III is also involved in electron transport and prone to electron leakage, its contribution to the production of superoxide is limited27,28. Nevertheless, the lack of association between mutations in Complex III and clinical outcome is likely due to the small number of patients carrying these mutations.
The role of mitochondrial dysfunction in aging is supported by several lines of evidence. Furthermore, previous studies have shown that somatic variation in mitochondrial DNA occur more frequently in aging tissues29. Our study further demonstrated the association between mutations in the mtDNA of ETC genes with age. This is also important since older patients with AML have significantly poorer clinical outcome compared with younger patients, and less than 10% of older patients achieve long term survival. Because ETC mutations occur more frequently in older patients and are potentially associated with worse survival, they present an opportunity for biomarker and drug development. Compared with normal hematopoietic cells, AML cells have greater mitochondrial mass without increasing respiratory chain complex activity. The lower spare reserve capacity in the respiratory chain makes it more susceptible to oxidative stress. Therefore, strategies that target the ETC chain may be effective against AML cells16.
The association between mitochondrial mutations in the ETC and mutations in TP53 is particularly interesting. p53 is known to translocate to the mitochondria during stress to activate apoptotic pathways30. p53 is also involved in regulating cellular respiration and energy metabolism, p53 has been found to induce expression and synthesis of cytochrome c oxidase (Complex IV)31,32 and possibly contributes to the Warburg effect. The Electron Transport Chain is a critical generator of reactive oxygen species in the mitochondria, and ROS has been shown to affect p53 stress sensitivity in leukemic cells33. A previous study demonstrated that translocation of p53 to the mitochondria to initiate apoptosis leads to decreased SOD2 activity and increased superoxide content34. Additionally, another study indicated an important role of p53 in maintaining genomic stability of mtDNA: via direct interaction between p53 and mtDNA polymerase γ. Conversely, loss of p53 can lead to increased susceptibility of mtDNA to mutations35. This could possibly explain the association between mtDNA ETC gene mutations and TP53 mutations. However, further mechanistic studies are needed to elucidate the exact relationship between TP53 mutations and mtDNA ETC gene mutations. Furthermore, patients with TP53 mutations are known to have inferior outcome in AML36. Here, we showed that further stratification of TP53 mutation positive patients by mutation status in either or both Complex I and IV was associated with even worse survival.
One limitation of this analysis, particularly those related to the association with clinical outcome would be the relatively small sample size of patients with mutations in each complex compared to patients without any mutation, especially after age-stratification. However, since this study analyzed the largest publically available dataset of AML, this triggers the need for including mtDNA in the mutational analysis of patients with AML.
Mitochondrial mutations have previously been proven to drive cancer pathogenesis in other cancers, thus expanding the research to study their role more critically in AML pathogenesis is of great interest. Here, we characterize mutations in mtDNA ETC genes in AML, we report their frequencies and association with age, TP53 mutations and patients’ clinical outcome. Future studies into the role of mitochondrial mutations in AML could further elucidate the mechanisms by which AML pathogenesis and progression occurs, as well as possibly identify new therapeutic targets.
Methods
Patient Data from The Cancer Genome Atlas (TCGA) dataset
Molecular and clinical patient data used for this analysis were retrieved from the Cancer Genome Atlas (TCGA) dataset from cBioPortal37,38, where scientists examined tumor specimens from 200 patients with AML39. Patients analyzed in the publically available TCGA dataset were enrolled in a single-institution tissue-banking protocol at Washington University. Patients from the provisional TCGA dataset with complete clinical (including National Comprehensive Cancer Network (NCCN) cytogenetic risk groups) and somatic mutation were included in the present analysis. We included 200 patients in our analysis for mitochondrial mutation status, clinical and molecular characteristics, as well as clinical outcome. These patient data were derived from an already published, publically available dataset and analyzed, thus ethical approval was not needed.
Statistical Analysis
Overall Survival was defined as time between initial diagnosis and death from any cause; Event-Free Survival was defined as time between initial diagnosis and withdrawal from study due to lack of complete remission, relapse or death. Survival curves were generated using the Kaplan-Meier survival curve between patients with and without mutations in their Electron Transport Chain complexes and ATP Synthase to delineate differences in survival due to the presence of mutations. Fisher Exact, for categorical variables, and the Mann-Whitney U test, for continuous variables, were used to assess the association between the presence of ETC mutations and clinical and molecular characteristics. ETC mutations were stratified into six groups: patients with mutations in any ETC complex, patients with mutations in Complex I, patients with mutations in Complex III, patients with mutations in Complex IV, patients with mutations in either or both Complex I and IV and patients with no mutations. Of the 13 ETC transport genes encoded in the mitochondrial genome, genes for Complex II are not mitochondrial-encoded, and thus any potential mutations that exist in Complex II were not included in these analyses. The Cox Proportional Hazards model was used to assess the association between various risk factors such as mitochondrial DNA mutation status, age, cytogenetic risk, etc. and overall and disease-free survival. We used a statistical cut-off of p-value ≤ 0.05 for inclusion of variables from univariate analysis to multivariate analysis. All statistical analyses were conducted using STATA 15.1 SE and GraphPad Prism.
Electronic supplementary material
Author Contributions
H.A. and S.W. conceived and designed the research project and wrote the manuscript. H.A. supervised the analysis. S.W. conducted the data analyses. M.A. provided clinical input on the data analysis. All authors reviewed the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-31489-0.
References
- 1.Engelking, L. R. Oxidative Phosphorylation in Textbook of Veterinary Physiological Chemistry (Third Edition) 219–224 (Academic Press, 2015).
- 2.Benov, L. How superoxide radical damages the cell. 217, 33–36 (2001). [DOI] [PubMed]
- 3.Kuo, C. F., Mashino, T. & Fridovich, I. alpha, beta-Dihydroxyisovalerate dehydratase. A superoxide-sensitive enzyme (1987). [PubMed]
- 4.Dhar SK, St Clair DK. Manganese superoxide dismutase regulation and cancer. Free Radic Biol Med. 2012;52:2209–2222. doi: 10.1016/j.freeradbiomed.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 5.Winterbourn CC. Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol Lett. 1995;82-83:969–974. doi: 10.1016/0378-4274(95)03532-X. [DOI] [PubMed] [Google Scholar]
- 6.Gaude E, Frezza C. Tissue-specific and convergent metabolic transformation of cancer correlates with metastatic potential and patient survival. Nat Commun. 2016;7:13041. doi: 10.1038/ncomms13041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ashton TM, McKenna WG, Kunz-Schughart LA, Higgins GS. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin Cancer Res. 2018;24:2482–2490. doi: 10.1158/1078-0432.CCR-17-3070. [DOI] [PubMed] [Google Scholar]
- 8.Yu M. Generation, function and diagnostic value of mitochondrial DNA copy number alterations in human cancers. Life Sci. 2011;89:65–71. doi: 10.1016/j.lfs.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 9.Chen XJ, Butow RA. The organization and inheritance of the mitochondrial genome. Nat Rev Genet. 2005;6:815–825. doi: 10.1038/nrg1708. [DOI] [PubMed] [Google Scholar]
- 10.Larman TC, et al. Spectrum of somatic mitochondrial mutations in five cancers. Proc Natl Acad Sci USA. 2012;109:14087–14091. doi: 10.1073/pnas.1211502109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hopkins JF, et al. Mitochondrial mutations drive prostate cancer aggression. Nat Commun. 2017;8:656. doi: 10.1038/s41467-017-00377-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsu CC, Lee HC, Wei YH. Mitochondrial DNA alterations and mitochondrial dysfunction in the progression of hepatocellular carcinoma. World J Gastroenterol. 2013;19:8880–8886. doi: 10.3748/wjg.v19.i47.8880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lowenberg B, Downing JR, Burnett A. Acute myeloid leukemia. N Engl J Med. 1999;341:1051–1062. doi: 10.1056/NEJM199909303411407. [DOI] [PubMed] [Google Scholar]
- 14.Ricciardi MR, et al. Targeting the leukemia cell metabolism by the CPT1a inhibition: functional preclinical effects in leukemias. Blood. 2015;126:1925–1929. doi: 10.1182/blood-2014-12-617498. [DOI] [PubMed] [Google Scholar]
- 15.Lagadinou ED, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12:329–341. doi: 10.1016/j.stem.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sriskanthadevan, S. et al. AML cells have low spare reserve capacity in their respiratory chain that renders them susceptible to oxidative metabolic stress. 10.1182/blood-2014-08-594408 (2015). [DOI] [PMC free article] [PubMed]
- 17.Latini, A. et al. Mitochondrial energy metabolism is markedly impaired by D-2-hydroxyglutaric acid in rat tissues. Mol Genet Metab86, 188–199, 10.1016/j.ymgme.2005.05.002 (2005). [DOI] [PubMed]
- 18.Figueroa, M. E. et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell18(6), 553–567 (2010). [DOI] [PMC free article] [PubMed]
- 19.Fu, X. et al. 2-Hydroxyglutarate Inhibits ATP Synthase and mTOR Signaling. Cell Metabolism22(3), 508–515 (2015). [DOI] [PMC free article] [PubMed]
- 20.Gattermann, N. Mitochondrial DNA. mutations in the hematopoietic system. Leukemia18, 18–22, 10.1038/sj.leu.2403209 (2004). [DOI] [PubMed]
- 21.Zhou FL, et al. Involvement of oxidative stress in the relapse of acute myeloid leukemia. J Biol Chem. 2010;285:15010–15015. doi: 10.1074/jbc.M110.103713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He L, et al. Somatic mitochondrial DNA mutations in adult-onset leukaemia. Leukemia. 2003;17:2487–2491. doi: 10.1038/sj.leu.2403146. [DOI] [PubMed] [Google Scholar]
- 23.Silkjaer T, et al. Characterization and prognostic significance of mitochondrial DNA variations in acute myeloid leukemia. Eur J Haematol. 2013;90:385–396. doi: 10.1111/ejh.12090. [DOI] [PubMed] [Google Scholar]
- 24.Chatterjee A, Mambo E, Sidransky D. Mitochondrial DNA mutations in human cancer. Oncogene. 2006;25:4663–4674. doi: 10.1038/sj.onc.1209604. [DOI] [PubMed] [Google Scholar]
- 25.Chaban Y, Boekema EJ, Dudkina NV. Structures of mitochondrial oxidative phosphorylation supercomplexes and mechanisms for their stabilisation. Biochim Biophys Acta. 2014;1837:418–426. doi: 10.1016/j.bbabio.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 26.Piccoli C, et al. MtDNA mutation associated with mitochondrial dysfunction in megakaryoblastic leukaemic cells. Leukemia. 2008;22:1938–1941. doi: 10.1038/leu.2008.69. [DOI] [PubMed] [Google Scholar]
- 27.Murphy M. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jastroch M, Divakaruni AS, Mookerjee S, Treberg JR, Brand MD. Mitochondrial proton and electron leaks. Essays Biochem. 2010;47:53–67. doi: 10.1042/bse0470053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cortopassi GA, Shibata D, Soong NW, Arnheim N. A pattern of accumulation of a somatic deletion of mitochondrial DNA in aging human tissues. Proc Natl Acad Sci USA. 1992;89:7370–7374. doi: 10.1073/pnas.89.16.7370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marchenko ND, Zaika A, Moll UM. Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J Biol Chem. 2000;275:16202–16212. doi: 10.1074/jbc.275.21.16202. [DOI] [PubMed] [Google Scholar]
- 31.Okamura S, et al. Identification of seven genes regulated by wild-type p53 in a colon cancer cell line carrying a well-controlled wild-type p53 expression system. Oncol Res. 1999;11:281–285. [PubMed] [Google Scholar]
- 32.Matoba S, et al. p53 regulates mitochondrial respiration. Science. 2006;312:1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- 33.Karawajew L, Rhein P, Czerwony G, Ludwig WD. Stress-induced activation of the p53 tumor suppressor in leukemia cells and normal lymphocytes requires mitochondrial activity and reactive oxygen species. Blood. 2005;105:4767–4775. doi: 10.1182/blood-2004-09-3428. [DOI] [PubMed] [Google Scholar]
- 34.Zhao, Y. et al. p53 Translocation to Mitochondria Precedes Its Nuclear Translocation and Targets Mitochondrial Oxidative Defense Protein-Manganese Superoxide Dismutase. 10.1158/0008-5472.CAN-04-3835 (2005). [DOI] [PubMed]
- 35.Achanta G, et al. Novel role of p53 in maintaining mitochondrial genetic stability through interaction with DNA Pol gamma. Embo j. 2005;24:3482–3492. doi: 10.1038/sj.emboj.7600819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kadia, T. M. et al. TP53 mutations in newly diagnosed acute myeloid leukemia: Clinicomolecular characteristics, response to therapy, and outcomes. Cancer, 18, 10.1002/cncr.30203 (2016). [DOI] [PMC free article] [PubMed]
- 37.Gao, J. et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Science Signaling6(269), pl1–pl1 (2013). [DOI] [PMC free article] [PubMed]
- 38.Cerami, E. et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data: Figure 1. Cancer Discovery2(5), 401–404 (2012). [DOI] [PMC free article] [PubMed]
- 39.Network, T. C. G. A. R. Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. 10.1056/NEJMoa1301689, doi:NJ201305303682205 (2013). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.