


Abstract
Variant interpretation is the key issue in molecular diagnosis. Spliceogenic variants exemplify this issue as each nucleotide variant can be deleterious via disruption or creation of splice site consensus sequences. Consequently, reliable in silico prediction of variant spliceogenicity would be a major improvement. Thanks to an international effort, a set of 395 variants studied at the mRNA level and occurring in 5′ and 3′ consensus regions (defined as the 11 and 14 bases surrounding the exon/intron junction, respectively) was collected for 11 different genes, including BRCA1, BRCA2, CFTR and RHD, and used to train and validate a new prediction protocol named Splicing Prediction in Consensus Elements (SPiCE). SPiCE combines in silico predictions from SpliceSiteFinder-like and MaxEntScan and uses logistic regression to define optimal decision thresholds. It revealed an unprecedented sensitivity and specificity of 99.5 and 95.2%, respectively, and the impact on splicing was correctly predicted for 98.8% of variants. We therefore propose SPiCE as the new tool for predicting variant spliceogenicity. It could be easily implemented in any diagnostic laboratory as a routine decision making tool to help geneticists to face the deluge of variants in the next-generation sequencing era. SPiCE is accessible at (https://sourceforge.net/projects/spicev2-1/).
INTRODUCTION
Since the advent of genome wide sequencing, interpretation of variants of unknown significance (VUS) has been recognized as the major bottleneck and challenge for clinical geneticists. Variants are usually classed within a 5-tiered scheme (1) from benign and likely benign variants (class 1 and 2, respectively) to likely pathogenic and pathogenic variants (class 4 and 5, respectively). The geneticist is on relatively solid ground in these four classes, where the biological impact is known or at least likely known. However, class 3 refers to the so called VUS where the effect of the sequence variation on the transcript and protein and thereby on the patient is simply not known. Clinical management logically stems from this knowledge (2) which is why variant classification is of utmost importance.
Hereditary breast and ovarian cancers are mainly due to BRCA1 (MIM #113705) and BRCA2 (MIM #600185) pathogenic variants. The BRCA genes embody the problem of variant interpretation due to their wide mutational spectrum, which is mostly devoid of specific hot spots. To exemplify this issue, over 30% of the variants in the Breast Cancer Information Core (BIC), ClinVar and BRCA Share databases are VUS (3–5).
Spliceogenic variants are probably the most challenging for the geneticists as each nucleotide variation, regardless of its location, can potentially affect pre-mRNA splicing and be pathogenic via disruption of 5′ or 3′ splice sites (5′/3′ ss), creation of new 5′/3′ ss or alteration of splicing regulatory elements. It is estimated that ∼15% of all point mutations causing human inherited disorders disrupt splice-site consensus sequences (6). Consequently, assessing the impact of variants on splicing is a mandatory task in molecular diagnosis. Toward this aim, several in silico prediction tools can be used either as stand-alone programs or as interfaces integrating multiple algorithms (see ‘Materials and Methods’ section). These tools are important to select variants that are worthy of expensive and time-consuming RNA analyses. This is why we published user’s guidelines from the splice network of French BRCA diagnostic laboratories within the Unicancer Genetic Group hereinafter named UGG, http://www.unicancer.fr/en/unicancer-group) (7), recommending the combined use of two bioinformatics variation scores MaxEntScan (MES) and Splice Site Finder-like (SSF-like) between the mutated and wild-type (WT) sequences. Two thresholds of relative decrease of scores at 15% for MES and 5% for SSF-like permitted to obtain a sensitivity of 96% and a specificity of 83%. While useful, these guidelines are prone to false-negative predictions (see below, ‘Results’ section) and could therefore be improved. Consequently, we developed a new prediction tool, called Splicing Predictions in Consensus Elements (SPiCE), to prioritize RNA studies to relevant variants that alter 5′ and 3′ splice consensus regions i.e. 11 bases for the 5′ splice site and 14 bases for the 3′ splice site. SPiCE uses logistic regression by running different combinations of in silico tools. Thanks to an international collaborative effort including the ENIGMA consortium (evidence-based network for the interpretation of germline mutant alleles, https://enigmaconsortium.org/) (8), we were able to collect 305 BRCA1 and BRCA2 variants occurring in 5′ and 3′ consensus regions with their corresponding splice study. SPiCE was developed using a training set of 142 BRCA1 and BRCA2 variants and validated on a further set of 163 BRCA1 and BRCA2 splice variants. Furthermore, and to demonstrate its versatility, SPiCE was successfully applied to another set of 90 variants occurring in 5′ and 3′ consensus regions of 9 non-cancer genes e.g. in CFTR (MIM#602421), CTRC (MIM#601405), HFE (MIM#613609), HJV (MIM#608374), LRP5 (MIM#603506), PDK1 (MIM#602524), RHD (MIM#111690), SLC40A1 (MIM#604653) and TFR2 (MIM#604250).
MATERIALS AND METHODS
Nomenclature
Nucleotide numbering is based on the cDNA sequence of BRCA1, BRCA2, CFTR, CTRC, HFE, HJV, LRP5, PKD1, RHD, SLC40A1, TFR2 (NCBI accession number NM_007294.2, NM_000059.3, NM_000492.3, NM_007272.2, NM_000410.3, NM_213653.3, NM_002335.3, NM_001009944.2, NM_016124.4, NM_014585.5, NM_003227.3, respectively), c.1 denoting the first nucleotide of the translation initiation codon, as recommended by the Human Genome Variation Society.
Definition of consensus splice site regions
Consensus splice site regions (5′ss and 3′ss) were defined according to Burge et al., (9), i.e. 11 bases for the 5′ splice site (from the 3 last exonic to the 8 first intronic bases) and 14 bases for the 3′ splice site (from the 12 last intronic to the first 2 exonic bases).
Datasets
Among this initiative, 395 variants occurring in the consensus 5′/3′ ss regions of 11 genes were collected, along with their respective RNA studies, and distributed between a training set and a validation set (Figure 1).
Figure 1.

Curated datasets and in vitro analyses methods used in this study.
The training set (Supplementary Table S1) comprises 142 BRCA1 and BRCA2 variants from the UGG network. We performed transcript analyses as previously described (7). Briefly, protocols for transcript analyses included (i) minigene-based splicing assays, (ii) RNA extracted from lymphoblastoid cell lines treated/untreated with puromycin. (iii) RNA extracted from blood collected into PAXgene tubes (Qiagen), (iv) RNA extracted from stimulated T lymphocytes. Controls (samples without variant) were always included in these experiments. No discordance was observed between invitro studies for the same variants.
To validate the SPiCE tool, we first gathered from the literature 208 transcript analyses from 163 distinct BRCA1 (n = 92) and BRCA2 (n = 71) variants reported in 56 publications. This curated collection of information was provided by members of the ENIGMA consortium as part of an ongoing data collection used for variant review (10,11) (Supplementary Table S2). Twelve of them (denoted by cross (†) in Supplementary Table S2) were analyzed at least twice and splicing alteration was constantly observed for 11 variants, with outcomes for different variants including exon skipping, use of cryptic splice site or combination of these events. Only one variant (c.518G>T in BRCA2) had contradictory reported and the reasons for this discordance remain unknown (12,13). Second, to extend the use of SPiCE to non-BRCA-genes, the second set of validation comprised 90 variants on CFTR (n = 44), CTRC (n = 2), HFE (n = 1), HJV (n = 1), LRP5 (n = 1), PKD1 (n = 1), RHD (n = 38), SLC40A1 (n = 1) and TFR2 (n = 1) with their splicing effect evaluated by minigene assay (Supplementary Table S3) (14). These variants were identified during the course of genetic counseling and thereby reflect clinical practice.
In silico tools
Five in silico prediction tools were tested: MES, (http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html) (15), SSF (16), Human Splicing Finder (HSF) (http://www.umd.be/HSF3/) (17), Neural Network Splice (NNS) (http://www.fruitfly.org/seq_tools/splice.html) (18) and GeneSplicer (GS) (http://www.cbcb.umd.edu/software/GeneSplicer/gene_spl.shtml) (19). Very briefly, the calculation of an MES score is based on maximum entropy of a nucleotide sequence with a set of constraints fixed by the MES model, including the variant’s neighboring bases. NNS also takes into account the variant’s neighboring position, but unlike MES, NNS is based on a machine learning technique i.e. artificial neural networks. For SSF and HSF, the score calculation is based on a position weight matrix and its homologous percentage with the tested sequence. We used SSF-like, a version of SSF, allowing calculation score of donor splice site with GT and GC canonical motifs, embedded in Alamut® and in SPiCE. At last, GS is based on a decision tree method. It captures potential strong dependencies between signal positions by dividing the dataset into subsets based on pairwise dependency between positions and modeling each subset separately (20). The outcomes of each of these tools were simultaneously obtained by using the commercial software (Alamut® Visual software version 2.8 rev. 1 and Alamut® Batch version 1.5.2., Interactive Biosoftware).
Logistic regression and model definition
First, we processed to descriptive analysis of bioinformatic prediction scores. We tested the discriminant capacity of these scores by receiver-operating characteristics (ROC) curves, representing the sensitivity as a function of 1-specificity (21), using the R package ROCR (22) and the correlation between variables by Pearson’s coefficient. Then, we used logistic regression to estimate the probability that a variant alters splicing. Parameter values were obtained by maximum likelihood, as objective function. This model was implemented in R software version 3.3.1 with the generalized linear model (glm) function. We considered that splicing alterations could correspond either to abnormal splicing events or to reinforcement of alternative splicing with partial or total effect. Splice event can be a single or multiple exon skipping and the use of exonic or intronic cryptic 5′ or 3′ splice sites. Selected variables to explain splicing alteration by a variant were (i) variation of prediction scores between WT and variant sequences, defined by Equation (1) and the score was annotated ΔMES, ΔSSF, ΔHSF, ΔNNS or ΔGS, (ii) localization in the invariant splice site positions (3′AG/5′GT), (iii) donor (5′) or acceptor (3′) splice sites, (iv) genes (e.g.: BRCA1 or BRCA2).
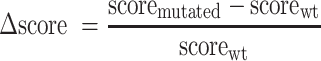 |
(1) |
To construct our final model we used a selection procedure based on a stepwise type approach with Akaike Information Criterion (AIC). Thereby AIC allows us to consider the likelihood of our model and the number of parameters in order to have the best model with a minimum of parameters. Models were compared by a likelihood ratio test. Cross-validation and other validation steps of the final model are described in the Supplementary Methods. AIC was considered more relevant than the Bayesian Information Criterion for a predictive approach. In any case, the two different criteria provided similar values (see Supplementary Table S7).
We developed SPiCE software, in the commonly utilized ‘R’ language to enable it to be freely applicable, information on this software are in Supplementary Material (see SPiCE handbook supplementary document). This software generates MES and SSF-like scores. For this purpose, the MES script was retrieved from the BurgeLab website (see in silico tools) and the SSF-like script was rewritten for SPiCE in R language according to its description under the original publication (16) and under the manual of the commercial Alamut software. Position weight matrices, used by SSF-like for scoring acceptor and donor splice sites, were obtained from SpliceDB which contains 28 468 pairs of splice site sequences (23).
In silico predictions using previously published guidelines
In order to compare SPiCE with our former guidelines, the BRCA1/2 validation set was assayed as previously described (7).
RESULTS
Aberrant splicing events were described for each dataset in Supplementary Table S4. Briefly we observed 76.7% (303/395) variants that alter splicing, with 44.6% of exon skipping, 10.9% use of 5′ alternative splice sites, 8.9% use of 3′ alternative splice site and 12.4% of multiple aberration.
BRCA1/BRCA2 training set
In total we performed 188 in vitro analyses on 142 variants including 37 unpublished variants on both BRCA1 (21 variants) and BRCA2 (16 variants). The variants from the training set were equally distributed between BRCA1 and BRCA2 genes, 50.7% (72/142) and 49.3% (70/142), respectively. Eighty-four variants (60%) were localized at the proximity of the 5′ ss and the 58 (40%) remainder at the proximity of the 3′ ss. Ninety-five variants altered splicing and were mainly (54.7%, 52/95) located outside the AG/GT dinucleotides (Table 1 and Figure 2A).
Table 1.
Distribution of variants in training and validation sets (n = 395)
| No. (%) of variants | No. (%) of variants altering splicing | ||||
|---|---|---|---|---|---|
| 5′/3′ splice site | 5′/3′ splice site | ||||
| Gene | 5′ | 3′ | Gene | 5′ | 3′ |
| Training set, n = 142 variants | n = 95 variants altering splicing | ||||
| BRCA1 | 42 (58.3) | 30 (41.7) | BRCA1 | 32 (66.7) | 16 (33.3) |
| BRCA2 | 42 (60.0) | 28 (40.0) | BRCA2 | 32 (68.1) | 15 (31.9) |
| Total | 84 (59.2) | 58 (40.8) | Total | 64 (67.4) | 31 (32.6) |
| BRCA1/BRCA2 validation set,n = 163 | n = 135 variants altering splicing | ||||
| BRCA1 | 54 (58.7) | 38 (41.3) | BRCA1 | 49 (64.5) | 27 (35.5) |
| BRCA2 | 40 (56.3) | 31 (43.7) | BRCA2 | 36 (61.0) | 23 (39.0) |
| Total | 94 (57.7) | 69 (42.3) | Total | 85 (63.0) | 50 (37.0) |
| Non-BRCA validation set,n = 90 | n = 73 variants altering splicing | ||||
| CFTR | 23 (52.3) | 21 (47.7) | CFTR | 23 (60.5) | 15 (39.5) |
| RHD | 26 (68.4) | 12 (31.6) | RHD | 22 (73.3) | 8 (26.7) |
| Other genesa | 4 (50.0) | 4 (50.0) | Other genesa | 3 (60.0) | 2 (40.0) |
| Total | 53 (58.9) | 37 (41.1) | Total | 48 (65.8) | 25 (34.2) |
a: LRP5, CTRC, HFE, HJV, PKD1, SLC40A1, TFR2.
Figure 2.

Localization and impact of variants according to distance from splice site. X-axis: variant altering splicing (black bar), variant without effect (gray bar). Y-axis: total number of variants for each position. Donor and acceptor splice sites were defined as −3 nt in exon to +8 nt in intron and −12 nt in intron to +2 nt in exon, respectively. (A) variants from training set. (B) variants from BRCA1/BRCA2 validation set. One single base deletion that affects the canonical AG splice site does not induce aberrant splicing. The reason is that the deletion removes a ‘A’ from the canonical ‘AG’ but without disrupting the consensus as the following neighboring nucleotide is another ‘A’ which in turn does preserve the consensus (C) variants from other genes validation set.
BRCA1/BRCA2 validation set
In the 163 variants collected from the literature, 92 (56.4%) variants were in BRCA1 and 71 variants in BRCA2. These variants were mainly localized on the donor sites compared to the acceptor sites, 58.3% (94/163) and 41.7% (69/163), respectively. Sixty of 135 (44.4%) variants that alter splicing were outside canonical dinucleotides (Table 1 and Figure 2B).
Non-BRCA validation set
We also selected 90 variants in nine non-BRCA genes, which were in CFTR (n = 44), CTRC (n = 2), HFE (n = 1), HJV (n = 1), LRP5 (n = 1), PKD1 (n = 1), RHD (n = 38), SLC40A1 (n = 1) and TFR2 (n = 1) (Supplementary Table S3). Fifty-three variants (58.9%) were in donor splice sites and 37 (41.1%) in acceptor sites. Seventy-three variants altered splicing in minigene assays. Half of these (n = 36; 49.3%) are in the AG/GT dinucleotides (Table 1 and Figure 2C). Some positions were poorly represented and this uneven distribution outside 5′/3′ ss can explain the imbalance between variants that do and do not affect splicing, 73 and 17 variants, respectively (Figure 2C).
Descriptive analyses of bioinformatics prediction score
To determine if prediction scores from different algorithms give similar information or not on our training set, we calculated Pearson coefficient correlation for each algorithm. The greatest correlations were between HSF and SSF-like (0.80) and between MES and NNS (0.87). GS score has the lowest correlation with the other prediction scores (ranging from 0.43 to 0.48). Excluding GS score, the lowest values were observed between SSF-like and MES (0.71) and between NNS and HSF (0.60) (Supplementary Table S5). The predictive capacity of each algorithm was measured by ROC curves. NNS and GS scores have the lowest area under the curve (AUC) values (0.907 and 0.736, respectively). MES and SSF-like scores have the best and similar AUC value (0.968 and 0.952, respectively) (Figure 3). As a result, MES and SSF-like provide high predictive capacity with distinct information.
Figure 3.

ROC curves of different bioinformatics scores from the training set (n = 142). GS: GeneSplicer; HSF: Human splicing finder; MES: MaxEntScan; NNS: Neural network splice; SSF: SpliceSite finder.
Model definition of SPiCE
Since our last large study in 2012 (7), we collected and analyzed in the UGG network a new set of 51 variants (37 unpublished variants). We applied our previous guidelines to identify variant that alter splicing and obtained a sensitivity equal to 74.3% (26/35), prompting us to develop SPiCE (Supplementary Table S6).
First, we performed univariate analysis for each variable (variation of prediction scores, localization in the invariant regions, donor (5′) or acceptor (3′) splice sites, genes). We observed that MES had a better Akaike Information Criterion (AIC) than the other variables (63.46) (Supplementary Table S7). Then, we performed multivariate analysis by adding other variables to MES. We found that only the combination of MES and SSF-like significantly improved the AIC with P-value of likelihood ratio test under 5% (Supplementary Table S7). The values for intercept, MES and SSF-like parameters are shown in Table 2. These three parameters were significantly different from 0 (P-value of Wald’s test < 0.05). Taken into account that MES and SSF-like do not score +7 and +8 position of the 5′ss, SPiCE should not be used at these positions.
Table 2.
Model parameters
| Parameters | Value | P-value of Wald’s test |
|---|---|---|
| β0 | −3.59 | 5.48e-6 |
| βMES | −8.21 | 4.28e-3 |
| βSSF | −32.30 | 6.37e-3 |
β0: Intercept; βMES: Parameter of MES score; βSSF : Parameter of SSF-like score.
We determined our thresholds by using ROC curve analyses on the training set (Figure 4A). The aim of these thresholds is to prioritize in vitro RNA studies of variants. Two probability thresholds were thus defined: optimal sensitivity threshold (ThSe) and optimal specificity threshold (ThSp), 0.115 and 0.749, respectively. As sensitivity is defined as the ratio of true positives divided by the sum of true positives and false negatives, ThSe is designed to give the highest detection rate while allowing false positives. On the other hand, specificity is the ratio of true negatives divided by the sum of true negatives and false positives, meaning ThSp is designed to minimize false positives while allowing false negatives. Sensitivity and specificity with ThSe are 100% (95/95) and 74.5% (35/47), respectively. Sensitivity and specificity with ThSp are 88.4% (84/95) and 95.7% (45/47), respectively. In both cases, accuracy was equal to 90.8% (data not shown). Our bootstrap analysis (Supplementary Table S8 and Figure S1) confirmed stability of model parameters and thresholds. We observed that cross-validation confirmed the pertinence of combined MES and SSF-like variation scores relative to the variation scores of MES or SSF-like alone (Supplementary Table S9 and Figure S2).
Figure 4.

ROC curve of the SPiCE logistic regression model. (A) on training set (n = 142), AUC = 0.986. (B) On BRCA1 and BRCA2 validation set (n = 160), AUC = 0.969. (C) On other genes validation set (n = 90), AUC = 0.989. Arrows correspond to decision thresholds for optimal sensitivity (0.115) and optimal specificity (0.749).
SPiCE performances on the BRCA1 and BRCA2 validation set
Following definition and training, SPiCE was validated on two independent sets of splice data. For each variant, the probability to have a splice effect was calculated and outcomes were predicted according to the previously determined thresholds (Table 3). To facilitate users’ interpretation, a graphical view was developed where decision thresholds are traced and variants spotted according to their values of their SSF-like and MES score variation (Figure 5). In-between thresholds, the area is thereby defined as the ‘gray area’ that includes only 16/160 variants. Optimal sensitivity threshold gave 99.3% sensitivity (134/135) and 68.0% (17/25) specificity. Optimal specificity threshold gave 92.6% sensitivity (125/135) and 92.0% (23/25) specificity (Figure 4B). Accuracy values were 94.4% (151/160) and 92.5% (148/160) for ThSe and ThSp, respectively, i.e. above accuracy obtained on the training set (90.8%).
Table 3.
SPiCE spliceogenicity prediction of variants in validation sets (n = 160 and n = 90)
| BRCA1 and BRCA2 validation set | Other genes validation set | |||
|---|---|---|---|---|
| With alteration | Without alteration | With alteration | Without alteration | |
| P > ThSp | 125 | 2 | 67 | 0 |
| ThSp < P > ThSe | 9 | 6 | 6 | 3 |
| P < ThSe | 1 | 17 | 0 | 14 |
P: Probability of variant to have splicing alteration; ThSe: Optimal sensitivity threshold; ThSp: Optimal specificity threshold.
Figure 5.

SPiCE graphical ouput results on BRCA1/BRCA2 validation set (n = 160). Representation of variants according to their SSF-like and MES scores variations in percentage. Blue area represents variants with probability of splicing alteration under decision threshold of optimal sensitivity, red area corresponds to probability upper decision threshold of optimal specificity and gray area is probability between these two thresholds. Blue points are variants without splicing effect and red points are variants altering splicing.
To further assess SPiCE efficiency, we compared the proportion of variants that affect splicing with their average SPiCE probability. Hence we subdivided our validation sets into groups according to their SPiCE probability. Ideally, the proportion of variants that affect splicing in any given group should be equal to the average SPiCE probability in this group. This is the case for our SPiCE model except for 4% (10/250) of variants with a probability between 0.115 and 0.432 (Supplementary Figure S3).
Then we studied a possible association between prediction accuracy and distance to canonical splice site (AG/GT). As shown in Supplementary Figure S4, SPiCE remains accurate throughout the consensus regions, even in the less conserved parts. However, we noted higher variability for polypyrimidine tract of 3′ ss (from −5 to −12).
SPiCE performances on the non-BRCA validation set
For CFTR and RHD for which we tested more than 35 variants, and 7 other genes for which we tested a few variants, SPiCE classification using ThSe gave a 100% sensitivity and a 82.3% specificity. ThSp gave 91.7% sensitivity and 100% specificity (Table 3 and Figure 4C). Combination of two thresholds of SPiCE protocol did not result in misclassified variants (0 false positive and 0 false negative). These results confirmed that the SPiCE protocol is pertinent in non-BRCA genes.
SPiCE performances with previous published guideline
We compared the performance of our previously published guidelines to SPiCE on validation sets, n = 250 (Table 4). Using ThSp, SPiCE improves the specificity to 95.2% (40/42) against 83% with previous guidelines whereas with ThSe SPiCE dramatically decreases the number of false negatives from 14 to 1 variant i.e. a sensitivity equals to 99.5%.
Table 4.
Contingency table on validation datasets (BRCA1/2 and other genes, (n = 250) with guidelines of Houdayer and coll (7)
| With alteration | Without alteration | |
|---|---|---|
| ΔMES > 15% and ΔSSF > 5% | 194 | 4 |
| ΔMES < 15% or ΔSSF < 5% | 14 | 38 |
Further quantitative aspects
We questioned the capability of SPiCE to predict the quantitative nature of the splice anomalies. To this aim, 232 analyses for which the semi-quantitative effect was known were selected from the training set and the non-BRCA validation set. These 232 analyses were for diagnostic purposes and the semi quantitative effect was taken into account for patient’s reporting. As a result, and despite the well-known difficulties in splice quantification, these data were considered reliable. Semi-quantitative effect was defined using the previously published classes i.e. 1S (no effect on splicing), 2S (partial effect) and 3S (complete effect) (7) and plotted against SPiCE probabilities (Supplementary Figure S5). A trend emerged as some partial effects led to lower probabilities as compared to complete effects but we were not able to define a prediction threshold between low/high intensity effects.
DISCUSSION
General considerations
This international effort represents the largest in silico study of splice variants with their corresponding in vitro/ex vivo transcript analyses conducted to date by a consortium. These international initiatives are needed to get results of wide scale relevance i.e. for the whole community. It enabled us to build SPiCE, a powerful prediction tool for variants occurring at splice site consensus regions, based on combination of MES and SSF-like by logistic regression. The reason is that among the five algorithms tested (GS, HSF, MES, NNS, SSF-like), we found that SSF-like and MES provide the best prediction on splicing effect of variants, as previously suggested by our group and others (24). Logistic regression analysis allows us to outperformed use of bioinformatics score variations of MES and SSF-like alone. SPiCE fulfills all the necessary criteria for model validation, e.g. stability of model, without bias. It has been validated on two replicative sets including 11 different genes and developed in the commonly utilized ‘R’ langage to ensure free and wide access.
SPiCE performs with high accuracy (95.6%) and sensitivity (99.5%) throughout the consensus sequences. The sole apparent false negative identified on BRCA1 and BRCA2 variants was c.5408G>C in the BRCA1 gene that leads to exon 23 skipping. The reason for this false-negative may be due to the complexity of splicing control i.e. due to another mechanism, such as the disruption of distal auxiliary splicing regulatory elements. This alternative explanation could be proven by dedicated minigene assays (12,25,26). Not surprisingly, there is a need for complementary prediction tools to complete our predictions and a fully comprehensive tool will eventually emerge from the combination of SPiCE and promising splicing regulatory element predictions (25,27,28). Moreover, by embedding comprehensive tools for exon definition, we would in turn be able to distinguish real exons from pseudoexons (29). Thanks to this international network of laboratories, these novel developments are planned to address this challenge.
Recommendations for routine analyses
SPiCE allows the user to know the risk of missing a true splice alteration according to the probability calculated. As sensitivity is a key issue in molecular diagnosis, we would recommend using the optimal sensitivity threshold (ThSe, probability above 0.115, i.e. including ‘gray area’) which in our hands gave only one false negative for BRCA1 while also a limiting number of false positives. On the other hand, depending on laboratory resources, the user can rely on the optimal specificity threshold (ThSp, probability above 0.749) which keeps false positives to a minimum as we observed only two false positives out of 42 variants without splice effect in our validation sets.
Previous prediction methods have been proposed for identifying variants that likely alter splicing. However, these methods were defined on small series thereby limiting their applicability (30–35). A recent work (36) on a large series of 272 variants in consensus regions suggested the used of a MES threshold of relative decrease of 10%, however this threshold leads to a specificity of 50% (21/42) on our validation datasets. The UGG network previously published a large series of splicing variants and accompanying guidelines for in silico predictions (7). Importantly SPiCE outperforms our previous results as demonstrated on the validation sets of variants from BRCA1, BRCA2 and other genes (Tables 3 and 4).
At this point in time, SPiCE predicts potential splicing alteration of variants at 5′ and 3′ ss but neither the type of the effect (exon skipping or use of alternative splice site) nor the importance of the effect (partial or total) are predicted, although the tool is able to detect a trend in the prediction severity of splicing defects (Supplementary Figure S5). This trend would allow to prioritize assays for those VUS predicted to have more severe effects on mRNA splicing. Importantly enough, SPiCE can be used beyond BRCA1 and BRCA2 and applied to other genes to guide geneticists in their daily practice. The majority of non-BRCA variants comes from two different genes (CFTR and RHD) but this should not create a bias as SPiCE runs MES and SSF which have been trained on our 20 000 protein-coding genes. Moreover we believe SPiCE versatility is demonstrated by testing these non-cancer genes i.e. involved in distinct pathways. This versatility is of special relevance as issues on misinterpretations and/or conflicting interpretations impact all fields of genetic diagnosis, leading to difficult situations for patients but also for health professionals. Given that 25% of clinical genetic results from commercial cancer panels had conflicting interpretation in ClinVar, the variant interpretation challenge is prone to erroneous medical decisions and eventually lawsuit as shown in Dravet syndrome (37). Without doubt, the development of reliable in silico tools is a major improvement toward reliable variant classification and patient’s management.
Overall, SPiCE has the potential of a widely used decision-making tool to guide geneticists toward relevant spliceogenic variants in the deluge of high-throughput sequencing data.
DEDICATION
This work is dedicated to the memory of our colleague Olga Sinilnikova.
DATA AVAILABILITY
SPiCE software is available at (https://sourceforge.net/projects/spicev2-1/).
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank Valentin Harter for biostatistics analysis advices and Emma Tudini for gathering data from the literature.
Notes
Present Address: Sophie Krieger, Laboratoire de biologie et génétique des cancers, Centre François Baclesse, Caen, France.
Present Address: Claude Houdayer, Service de Génétique, Institut Curie, Paris, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NHMRC Senior Research Fellowship [ID1061779 to A.B.S.]; The Cancer Council Queensland [ID1086286 to M.P., in part]; French National Cancer Institute Translational Research Grant; Direction Générale de l’Offre des Soins (INCa/DGOS). Funding for open access charge: Laboratoire de Biologie et Génétique du Cancer; Centre François Baclesse, Caen, France.
Conflict of interest statement. None declared.
REFERENCES
- 1. Plon S.E., Eccles D.M., Easton D., Foulkes W.D., Genuardi M., Greenblatt M.S., Hogervorst F.B.L., Hoogerbrugge N., Spurdle A.B., Tavtigian S.V.. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum. Mutat. 2008; 29:1282–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E. et al.. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015; 17:405–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Caputo S., Benboudjema L., Sinilnikova O., Rouleau E., Béroud C., Lidereau R.. Description and analysis of genetic variants in French hereditary breast and ovarian cancer families recorded in the UMD-BRCA1/BRCA2 databases. Nucleic Acids Res. 2012; 40:D992–D1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Landrum M.J., Lee J.M., Riley G.R., Jang W., Rubinstein W.S., Church D.M., Maglott D.R.. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014; 42:D980–D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Szabo C., Masiello A., Ryan J.F., Brody L.C.. The breast cancer information core: database design, structure, and scope. Hum. Mutat. 2000; 16:123–131. [DOI] [PubMed] [Google Scholar]
- 6. Baralle D., Lucassen A., Buratti E.. Missed threads. EMBO Rep. 2009; 10:810–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Houdayer C., Caux-Moncoutier V., Krieger S., Barrois M., Bonnet F., Bourdon V., Bronner M., Buisson M., Coulet F., Gaildrat P. et al.. Guidelines for splicing analysis in molecular diagnosis derived from a set of 327 combined in silico/in vitro studies on BRCA1 and BRCA2 variants. Hum. Mutat. 2012; 33:1228–1238. [DOI] [PubMed] [Google Scholar]
- 8. Spurdle A.B., Healey S., Devereau A., Hogervorst F.B.L., Monteiro A.N.A., Nathanson K.L., Radice P., Stoppa-Lyonnet D., Tavtigian S., Wappenschmidt B. et al.. ENIGMA—Evidence-based network for the interpretation of germline mutant alleles: an international initiative to evaluate risk and clinical significance associated with sequence variation in BRCA1 and BRCA2 genes. Hum. Mutat. 2012; 33:2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Burge C.B., Tuschi T., Sharp P.A.. Splicing of precursors to mRNAs by the spliceosomes. The RNA World II. 1999; NY: Cold Spring Harbor Laboratory Press; Oxford University Press; 525–560. [Google Scholar]
- 10. Vallée M.P., Di Sera T.L., Nix D.A., Paquette A.M., Parsons M.T., Bell R., Hoffman A., Hogervorst F.B.L., Goldgar D.E., Spurdle A.B. et al.. Adding in silico assessment of potential splice aberration to the integrated evaluation of BRCA gene unclassified variants. Hum. Mutat. 2016; 37:627–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Walker L.C., Whiley P.J., Houdayer C., Hansen T.V.O., Vega A., Santamarina M., Blanco A., Fachal L., Southey M.C., Lafferty A. et al.. Evaluation of a 5-Tier scheme proposed for classification of sequence variants using bioinformatic and splicing assay data: inter-reviewer variability and promotion of minimum reporting guidelines. Hum. Mutat. 2013; 34:1424–1431. [DOI] [PubMed] [Google Scholar]
- 12. Di Giacomo D., Gaildrat P., Abuli A., Abdat J., Frébourg T., Tosi M., Martins A.. Functional analysis of a large set of BRCA2 exon 7 variants highlights the predictive value of hexamer scores in detecting alterations of exonic splicing regulatory elements. Hum. Mutat. 2013; 34:1547–1557. [DOI] [PubMed] [Google Scholar]
- 13. Sanz D.J., Acedo A., Infante M., Durán M., Pérez-Cabornero L., Esteban-Cardeñosa E., Lastra E., Pagani F., Miner C., Velasco E.A.. A high proportion of DNA variants of BRCA1 and BRCA2 is associated with aberrant splicing in Breast/Ovarian cancer patients. Clin. Cancer Res. 2010; 16:1957–1967. [DOI] [PubMed] [Google Scholar]
- 14. Callebaut I., Joubrel R., Pissard S., Kannengiesser C., Gérolami V., Ged C., Cadet E., Cartault F., Ka C., Gourlaouen I. et al.. Comprehensive functional annotation of 18 missense mutations found in suspected hemochromatosis type 4 patients. Hum. Mol. Genet. 2014; 23:4479–4490. [DOI] [PubMed] [Google Scholar]
- 15. Yeo G., Burge C.B.. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004; 11:377–394. [DOI] [PubMed] [Google Scholar]
- 16. Shapiro M.B., Senapathy P.. RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res. 1987; 15:7155–7174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Desmet F.-O., Hamroun D., Lalande M., Collod-Béroud G., Claustres M., Béroud C.. Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009; 37:e67–e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Reese M.G., Eeckman F.H.. Searls GSD, Fickett J, Noordewier M. Novel neural network prediction systems for human promoters and splice sites. Gene-Finding and Gene Structure Prediction Workshop. 1995; Philadelphia, PA: 1–7. [Google Scholar]
- 19. Pertea M., Lin X., Salzberg S.L.. GeneSplicer: a new computational method for splice site prediction. Nucleic Acids Res. 2001; 29:1185–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jian X., Boerwinkle E., Liu X.. In silico tools for splicing defect prediction: a survey from the viewpoint of end users. Genet. Med. 2014; 16:497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zweig M.H., Campbell G.. Receiver-operating characteristic (ROC) plots: a fundamental evaluation tool in clinical medicine. Clin. Chem. 1993; 39:561–577. [PubMed] [Google Scholar]
- 22. Sing T., Sander O., Beerenwinkel N., Lengauer T.. ROCR: visualizing classifier performance in R. Bioinformatics. 2005; 21:3940–3941. [DOI] [PubMed] [Google Scholar]
- 23. Burset M., Seledtsov I.A., Solovyev V.V.. SpliceDB: database of canonical and non-canonical mammalian splice sites. Nucleic Acids Res. 2001; 29:255–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jian X., Boerwinkle E., Liu X.. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 2014; 42:13534–13544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ke S., Anquetil V., Zamalloa J.R., Maity A., Yang A., Arias M.A., Kalachikov S., Russo J.J., Ju J., Chasin L.A.. Saturation mutagenesis reveals manifold determinants of exon definition. Genome Res. 2018; 28:11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee Y., Rio D.C.. Mechanisms and regulation of alternative Pre-mRNA splicing. Annu. Rev. Biochem. 2015; 84:291–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Soukarieh O., Gaildrat P., Hamieh M., Drouet A., Baert-Desurmont S., Frébourg T., Tosi M., Martins A.. Exonic splicing mutations are more prevalent than currently estimated and can be predicted by using in silico tools. PLos Genet. 2016; 12:e1005756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Julien P., Miñana B., Baeza-Centurion P., Valcárcel J., Lehner B.. The complete local genotype–phenotype landscape for the alternative splicing of a human exon. Nat. Commun. 2016; 7:11558–11566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chasin L.A. Searching for splicing motifs. Adv. Exp. Med. Biol. 2007; 623:85–106. [DOI] [PubMed] [Google Scholar]
- 30. Holla Ø.L., Nakken S., Mattingsdal M., Ranheim T., Berge K.E., Defesche J.C., Leren T.P.. Effects of intronic mutations in the LDLR gene on pre-mRNA splicing: Comparison of wet-lab and bioinformatics analyses. Mol. Genet. Metab. 2009; 96:245–252. [DOI] [PubMed] [Google Scholar]
- 31. Houdayer C., Dehainault C., Mattler C., Michaux D., Caux-Moncoutier V., Pagès-Berhouet S., d’Enghien C.D., Laugé A., Castera L., Gauthier-Villars M. et al.. Evaluation of in silico splice tools for decision-making in molecular diagnosis. Hum. Mutat. 2008; 29:975–982. [DOI] [PubMed] [Google Scholar]
- 32. Théry J.C., Krieger S., Gaildrat P., Révillion F., Buisine M.-P., Killian A., Duponchel C., Rousselin A., Vaur D., Peyrat J.-P. et al.. Contribution of bioinformatics predictions and functional splicing assays to the interpretation of unclassified variants of the BRCA genes. Eur. J. Hum. Genet. 2011; 19:1052–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vreeswijk M.P.G., Kraan J.N., van der Klift H.M., Vink G.R., Cornelisse C.J., Wijnen J.T., Bakker E., van Asperen C.J., Devilee P.. Intronic variants in BRCA1 and BRCA2 that affect RNA splicing can be reliably selected by splice-site prediction programs. Hum. Mutat. 2009; 30:107–114. [DOI] [PubMed] [Google Scholar]
- 34. Whiley P.J., Guidugli L., Walker L.C., Healey S., Thompson B.A., Lakhani S.R., Da Silva L.M. Investigators, kConFab Investigators, kConFab Tavtigian S.V., Goldgar D.E. et al.. Splicing and multifactorial analysis of intronic BRCA1 and BRCA2 sequence variants identifies clinically significant splicing aberrations up to 12 nucleotides from the intron/exon boundary. Hum. Mutat. 2011; 32:678–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wimmer K., Roca X., Beiglböck H., Callens T., Etzler J., Rao A.R., Krainer A.R., Fonatsch C., Messiaen L.. Extensive in silico analysis of NF1 splicing defects uncovers determinants for splicing outcome upon 5′ splice-site disruption. Hum. Mutat. 2007; 28:599–612. [DOI] [PubMed] [Google Scholar]
- 36. Tang R., Prosser D.O., Love D.R.. Evaluation of bioinformatic programmes for the analysis of variants within splice site consensus regions. Adv. Bioinformatics. 2016; 2016:10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Levenson D. Lawsuit raises questions about variant interpretation and communication. Am. J. Med. Genet. 2017; 173:838–839. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
SPiCE software is available at (https://sourceforge.net/projects/spicev2-1/).