

ABSTRACT
The Notch signaling pathway is highly conserved and essential for animal development. It is required for cell differentiation, survival, and proliferation. Regulation of Notch signaling is a crucial process for human health. Ligands initiate a signal cascade by binding to Notch receptors expressed on a neighboring cell. Notch receptors interact with ligands through their epidermal growth factor-like repeats (EGF repeats). Most EGF repeats are modified by O-glycosylation with residues such as O-linked N-acetylglucosamine (O-GlcNAc), O-fucose, and O-glucose. These O-glycan modifications are important for Notch function. Defects in O-glycosylation affect Notch-ligand interaction, trafficking of Notch receptors, and Notch stability on the cell surface. Although the roles of each modification are not fully understood, O-fucose is essential for binding of Notch receptors to their ligands. We reported an EGF domain-specific O-GlcNAc transferase (EOGT) localized in the endoplasmic reticulum. Mutations in genes encoding EOGT or NOTCH1 cause Adams-Oliver syndrome. Dysregulation of Notch signaling because of defects or mutations in Notch receptors or Notch signal-regulating proteins, such as glycosyltransferases, induce a variety of congenital disorders. In this review, we discuss O-glycosylation of Notch receptors and congenital human diseases caused by defects in O-glycans on Notch receptors.
Key Words: Notch receptor, O-glycosylation, epidermal growth factor-like repeat, EGF domain-specific O-GlcNAc transferase, Adams-Oliver syndrome
INTRODUCTION
Notch signal transduction plays an important role in cell fate decisions in development and in homeostasis.1,2) During signal initiation, ligands interact with Notch receptors on the cell surface. The Notch signaling pathway is very well conserved in metazoans. In mammals, there are four Notch receptors (Notch 1, 2, 3, 4) and four ligands (Delta-like 1 (DLL1), Delta-like 4 (DLL4), Jagged 1, and Jagged 2). The expression patterns of Notch receptors and ligands are variable, and their combinations depend on the organ in which they are expressed and determine how their target genes are regulated.
Notch signaling begins when ligands bind to Notch receptors (Figure 1B). Internalization of the bound ligands into the signal-sending cell generates a pulling force.3) Simultaneously, the Notch extracellular region changes its conformation4) to expose a cleavage site for disintegrin and metalloproteinase domain-containing proteins (ADAM10 or ADAM17).5) The subsequent cleavage leads to release of the Notch receptor ectodomain and leaves a membrane-tethered extracellular fragment. γ-Secretase cleaves Notch in its transmembrane domain, resulting in release of the Notch intracellular domain (NICD) into the cytoplasm. The NICD translocates into the nucleus to coordinate the expression of target genes. Thus, Notch signaling cascades are well regulated at several stages.
Fig. 1.
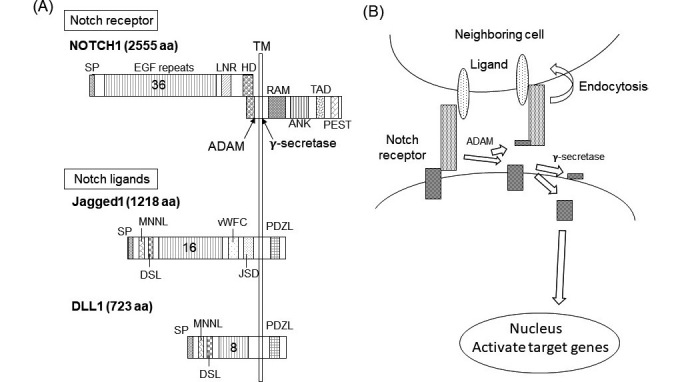
Notch signaling pathway (A) Structures of Notch receptors and ligands in mammals.
Numbers in the EGF repeats indicate the number of EGF domains in human NOTCH1, Jagged1 and DLL1. ANK, ankyrin repeats; DLL, Delta-like protein; DSL, Delta/Serrate/LAG-2 domain; EGF, Epidermal growth factor; HD, heterodimerization domain; JSD, Jagged Serrate domain; LNR, Lin-Notch repeats; PDZL, PDZ ligand domain; PDZ, post-synaptic density protein; PEST, proline, glutamic acid, serine, and threonine degradation domain; RAM, Rbp-associated molecule domain; SP, signal peptide; TAAD, transactivation domain; TM, transmembrane; vWFC, von Willebrand factor type C domain. (B) Notch activation pathway is illustrated.
O-GLYCOSYLATION OF NOTCH RECEPTORS
1. Epidermal growth factor-like repeats on Notch receptors
Both Notch receptors and their ligands are type I integral membrane proteins (Figure 1A). Their most characteristic common structures are epidermal growth factor-like repeats (EGF repeats), contained in their extracellular domains. An individual EGF domain consists of a 30–40-amino acid sequence. These sequences are similar to those of a small growth factor, EGF, which is composed of 53 amino acids.6) An EGF repeat contains six conserved cysteine residues that are essential for its three-dimensional structure because they form three disulfide bonds.6) EGF repeats are often found in secreted proteins and in the extracellular domains of transmembrane proteins. The major functions of EGF repeats are protein-protein interactions and trafficking.
Notch receptors possess 34–36 EGF repeats; there are far fewer in Notch ligands.1) For instance, human DLL4 contains 8 EGF repeats and human Jagged1 contains 16 EGF repeats. A large number of EGF repeats would make the three-dimensional structure quite complex. Notch receptors may use this to their advantage to protect the cleavage site, via a long domain consisting of many EGF repeats, during their maturation before Notch-ligand interaction.
Notch EGF11-12 is essential for interactions with both the DLL and Jagged ligands. Notch EGF11-13 is important for binding to DLL4, and Notch EGF8-12 is required for binding to Jagged1.7) Some Notch EGF repeats, not including EGF8-14, can bind calcium, which is required for their structural characteristics and protein-protein interactions. The roles for other EGF repeats are currently not well understood.
2. O-Glycans found on Notch receptors
The EGF repeats of Notch receptors are modified with a variety of glycans. These modifications include an N-glycan and three types of O-glycans that are initiated by O-fucose, O-glucose, or O-N-acetylglucosamine (GlcNAc) (Figure 2A). EGF repeats in the extracellular domain of mouse Notch receptors have been widely analyzed by mass spectrometry and most glycosylation sites have been thoroughly examined.8-10, 30, 59) Their consensus sequences for the addition of one or more specific O-glycans have been identified (Figure 2B). O-Glycans play important roles in the functions of Notch receptors. Both types of O-glycans regulate the transduction level of Notch signaling.11) Loss of O-fucose decreases binding of Notch ligands to Notch receptors and thus reduces Notch signaling,12) whereas loss of O-glucose compromises Notch signaling without affecting Notch-ligand binding.13)
Fig. 2.

O-Glycosylation found on Notch EGF repeats in mammalian cells.
(A) The structures of O-linked glycans found in mammalian Notch receptors are illustrated with each consensus sequence. C1 and C2 are the first and second conserved cysteine residues of the individual EGF domains, respectively. C3-C6 are represented in a similar manner. The consensus sequence for O-GlcNAc modification has not been defined, but was predicted to be C5XX(G/P/S)(Y/F/W)(T/S). Mutations in POFUT1 or POGLUT1 cause Dowling–Degos disease, whereas mutations in EOGT cause Adams-Oliver syndrome. The solid black line indicates the basic linkage in the O-glycan, whereas the thin gray line indicates the linkage once extension occurs. (B) Glycosylation sites on EGF repeats in the extracellular domain of mouse Notch1 receptors expressed in HEK293T cells were determined by mass spectrometry59): O-Fucose (triangle), O-Glucose (circle), O-GlcNAc (square).
3. O-Fucose and POFUT1
O-fucose modification of a Notch receptor was first discovered in a human urine sample in the 1970s, and later found on Notch EGF repeats from mice.14) The gene responsible for transferring O-fucose in mammals was identified as POFUT1, which encodes endoplasmic reticulum (ER)-localized soluble GDP-fucose protein O-fucosyltransferase 1 (POFUT1).15,16) The Drosophila homolog was named O-fut1.17) Importantly, POFUT1 adds O-fucose only to properly folded EGF repeats.15,18) Although consensus sequences for fucosyltransferases have been found in over 100 animal proteins,19) few of these proteins are O-fucosylated. For examples, urokinase plasminogen activators, which are proteolytic enzymes in fibrinolysis, are modified with O-fucose.
POFUT1 is required for Notch signaling. Loss of POFUT1 in mice is embryonic lethal because of a global loss of Notch signaling activity.20) Conditional loss of POFUT1 results in a phenotype very similar to that of conditional loss of Rbpj.21) Previous reports suggested that O-fucosylation by POFUT1 is essential for Notch receptor function. In contrast, O-fucose observed in Notch ligands does not appear to be essential. Drosophila POFUT1 (Ofut1) is not required for the function of ligands expressed in signal-sending cells.17) Similarly, mutations of O-fucosylation sites on DLL4 cause its intracellular accumulation, but it can still activate Notch signaling in neighboring cells.22) However, an O-fucosylation-defective DLL3 mutant failed to function in vivo, but the molecular mechanism has not yet been clarified.23)
In Drosophila, OFUT1 plays roles not only as a fucosyltransferase, but also as a chaperone to promote the folding of Notch and its exit from the ER.24) This non-enzymatic role of OFUT1 is important in flies grown at low temperatures.25) However, such activity has not been observed for POFUT in mammalian cells.
4. O-Glucose and POGLUT1
Nearly 30 years ago, O-glucose was discovered on EGF repeats of the bovine blood coagulation factors VII and IX.26)O-Glucose is attached to serine residues, then an elongated glycan is formed by addition of xylose. Currently two elongated forms have been identified, xylose-glucose and xylose-xylose-glucose. O-Glucose has been reported in mouse Notch1,14) Notch2,25) and Drosophila Notch.13) Not all consensus sequences in mammalian Notch1 are efficiently glucosylated.10) The efficiency of O-glycosylation is affected by the folding status of the EGF repeat and the amino acids near the consensus sequence.18)
The first O-glucosyltransferase to be identified was Rumi in Drosophila.27) Its mammalian homolog is designated O-glucosyltransferase I (POGLUT1). POGLUT1 is a soluble protein with a lysine-aspartic acid-glutamic acid-leucine motif that prevents secretion from the ER. Defects in Drosophila Rumi diminish Notch signaling in a temperature-dependent manner by affecting ADAM-dependent cleavage.13,27) In mammalian cells, O-glucose is required for activation of Notch signaling, although defects in POGLUT1 do not affect the binding of ligands to Notch receptors.13) The exact function of O-glucose modification is not well established, although its role has been discussed in terms of proper transport of the Notch receptor and quality control in the ER.18)
5. O-GlcNAc and EOGT
A decade ago, our group discovered O-GlcNAc on EGF20 by detecting O-glycans on the extracellular domain of Drosophila Notch using anti-O-GlcNAc antibodies (CDT110.6).28) Until this report, O-GlcNAc modification had not been detected for secreted or membrane proteins, but only for cytoplasmic or nuclear proteins. Later, O-GlcNAc modifications of the EGF repeats of other proteins such as Drosophila Delta, Serrate, and Dumpy were reported.28,29)
The gene responsible for extracellular O-GlcNAcylation of Drosophila EGF repeats was identified as Eogt by the Okajima group.29)Eogt encodes an ER-localized EGF domain-specific O-GlcNAc transferase (EOGT) which is expressed in various tissues of mice.30)
Mutations in Eogt did not show the characteristic phenotypes observed in Drosophila Notch mutants.29) Recently, mutations in EOGT were detected in patients with autosomal recessive Adams-Oliver syndrome (AOS).31,32) AOS is a rare congenital disorder characterized by aplasia cutis congenita and terminal transverse limb defects.33,34)
Our group further evaluated the mechanism of AOS and recently reported that Notch in EOGT-deficient cells exhibited decreased binding to DLL1 or DLL4. In contrast, binding to JAG1 was not affected. We introduced mutations into the O-GlcNAc sites of mouse Notch1, which resulted in decreased binding with DLL4. Our group suggested that EOGT functions in retinal angiogenesis by regulating Notch signaling in endothelial cells.29)
CONGENITAL DISORDERS CAUSED BY DYSREGULATION OF O-GLYCOSYLATION OF NOTCH RECEPTORS
1. Human diseases caused by mutations in Notch-related proteins
Mutations in genes encoding Notch-related proteins, such as Notch receptors, ligands, and glycosyltransferases, lead to variety of congenital disorders.35) For example, in humans, mutations in NOTCH2 or JAGGED1 cause Alagille syndrome, an autosomal dominant disorder characterized by abnormal development of the liver, eyes, kidney, pancreas, heart, vascular system, skeleton, or face.36,37) Mutations in NOTCH3 cause autosomal dominant cerebral arteriopathy and leukoencephalopathy, resulting in defects in small cerebral arteries and leading to subcortical infarcts and white matter damage.38) In this review, we focus on human diseases caused by mutations in O-glycosyltransferases that modify Notch receptors and Notch-related proteins. Mutations in POFUT1 or POGLUT1 cause Dowling–Degos disease, whereas mutations in EOGT causes AOS (Figure 2A). Interestingly, these diseases are tissue-specific with no apparent overlap. The status of O-glycan protein modifications may account for these diseases.
2. Dowling-Degos Disease
Dowling–Degos disease (DDD) is a rare autosomal dominant form of a reticulate pigmentary disorder. DDD causes postpubertal reticulate hyperpigmentation including small hyperkeratotic dark-brown papules.39) This hyperpigmentation is progressive and mainly affects flexures.
Initially, Keratin 5 (KRT5) was identified as a causative gene because its mutation was found in DDD patients from two families. However, KRT5 mutation was absent in some affected individuals. By genome-wide linkage analysis and exome sequencing, the Yao group reported a nonsense mutation (p.Glu144*) and a heterozygous deletion mutation (p.Lyc161Sefs*42) in POFUT1.40) Decreased expression of POFUT1 in HaCaT cells resulted in down-regulation of Notch1, Notch2, HES1 (a downstream target of Notch signaling), and KRT5.40) In addition, they found nine different heterozygous mutations in POGLUT1 from 13 patients, which included a nonsense mutation located at the beginning of the POGLUT1 protein (p.Trp4*) and point mutations (p.R279W).40) Thus, partial loss of POFUT1 or POGLUT1 activity causes DDD.
In contrast, heterozygous mice with mutations in Pofut1 or Poglut1 did not show the skin defects observed in human patients of DDD.20,40) Thus, it is possible that proteins besides Notch receptors play a role in DDD.41) Future studies are needed to clarify these points.
3. Adams-Oliver syndrome
The most characteristic feature of AOS is aplasia cutis congenita with terminal transverse limb defects.42) Patients show absent or scarred skin on their thin skulls. In addition, central nervous system anomalies or congenital heart defects are observed in 23% of AOS patients.42)
In 1945, Clarence Oaul Oliver and Forrest H. Adams reported a patient with anomalies in both feet and one hand, and a denuded surface of the scalp on the thin skull.43) Three generations of family members had similar symptoms, suggesting that this disease is inherited.43)
Currently, six mutated genes have been found in AOS patients: ARHGAP31,43) DOCK6,45) EOGT,31) RBPJ,46) NOTCH1,47,48) and DLL4.44,49) Among them, loss-of-function mutations in EOGT and DOCK6 were identified in patients with an autosomal recessive AOS.31) Homozygous mutations in EOGT were identified in three blood-related families with AOS.45) Homozygous mutations in DOCK6 were identified in two blood-related families with AOS.31)DOCK6 encodes a regulator of the Rho GTPase family, including CDC42 and Rac1, and is required for angiogenesis in hepatic microvascularization.50) Interestingly, 28% of AOS patients with DOCK6 mutations are sporadic, as there are no familial probands.42) Cardiac defects are more frequently seen in AOS patients with NOTCH1 mutations compared with patients having ARHGAP31 or DOCK6 mutations.42)
In contrast, autosomal dominant mutations in ARHGAP31 were observed in two families.51)ARHGAP31 encodes a GTPase-activating protein for Rac1 and Cdc42. ARHGAP31 regulates cycles of two forms of Ras-related C3 botulinum toxin substrate 1 (RAC1) between active GTP-bound and inactive GDP-bound forms. Mutation of ARHGAP31 resulted in constitutive GAP activity and consequent decreased Cdc42 signaling. In addition, autosomal dominant mutations in RBPJ, encoding recombination signal binding protein for the immunoglobulin kappa J region, were detected in two independent kindreds with autosomal dominant AOS.46) For DLL4, the mutations were reported as four non-familial probands, five familial probands, and 13 family members with clinical symptoms, among 91 families with AOS.44)
It is important to recognize that two distinctive gene families are responsible for AOS. Mutations in EOGT, RBPJ, NOTCH1, and DLL4 in the patients suggest that an impaired canonical Notch signaling pathway is associated with AOS. In contrast, loss-of-function mutations for DOCK6 or gain-of-function mutations for ARHGAP31 cause decreased Rac1/Cdc42 signaling. Further investigation of the contribution of disrupted canonical Notch signaling or Rac1/Cdc42 signaling to the abnormalities in these patients will aid understanding of the molecular pathogenesis of AOS.
4. Cancers
Breast cancers and their association with Notch signaling have been thoroughly studied.52) It has been reported that different types of cancers exhibit changes in the expression level of glycosyltransferase genes including POFUT1, POGLUT1, or Fringe.53) The expression level of POGLUT1 is elevated in brain tumors, hepatocellular carcinoma, colorectal cancers, and oral squamous cell carcinoma.54-56) An increase in POGLUT1 expression is observed in primary acute myelogenous leukemia and T-cell acute lymphoblastic leukemia.57) The roles of POFUT1 and POGLUT1 in initiating or supporting cancers require further examination.
CONCLUSION
In summary, O-glycosylation of Notch receptors plays an important role in their function. There are currently no effective therapies for the human diseases described in this review. Recently, a homozygous missense mutation (D233E) in POGLUT1, resulting in reduced Notch signaling, was identified in four siblings with autosomal recessive limb-girdle muscular dystrophy.58) Mutations in POGLUT1 were also detected in patients with DDD, which is symptomatically distinct from muscular dystrophy. Further studies are needed to identify the contributions of genetic background and any late effects of the underlying genetic cause that are not directly related to congenital anomalies.
ACKNOWLEDGMENTS
We thank all members who have done previous work in the Okajima lab.
REFERENCES
- 1).Haltom AR, Jafar-Nejadd H. The multiple roles of epidermal growth factor repeat O-glycans in animal development. Glycobiology, 2015; 25: 1027–1042. [DOI] [PMC free article] [PubMed]
- 2).Takeuchi H, Haltiwanger RS. Significance of glycosylation in Notch signaling. Biochem Biophys Res Commun, 2014; 453: 235–242. [DOI] [PMC free article] [PubMed]
- 3).Langridge PD, Struhl G. Epsin-dependent ligand endocytosis activates Notch by force. Cell, 2017; 171(6): 1383–1396. [DOI] [PMC free article] [PubMed]
- 4).Stephenson NL, Avis JM. Direct observation of proteolytic cleavage at the S2 site upon forced unfolding of the Notch negative regulatory region. Proc Natl Acad Sci U S A, 2012; 109(41): E2757–E2765. [DOI] [PMC free article] [PubMed]
- 5).Bozkulak EC, Weinmaster G. Selective use of ADAM10 and ADAM17 in activation of Notch1 signaling. Mol Cell Biol, 2009; 29(21): 5679–5695. [DOI] [PMC free article] [PubMed]
- 6).Savage CR Jr, Hash Jh, Cohen S. Epidermal growth factor. Location of disulfide bonds. J Biol Chem, 1973; 248: 7669–7672. [PubMed]
- 7).Luca VC, Jude KM, Pierce NW, Nachury MV, Fischer S, Garcia KC. Structural biology. Structural basis for Notch1 engagement of Delta-like 4. Science, 2015; 347: 847–853. [DOI] [PMC free article] [PubMed]
- 8).Harvey BM, Rana NA, Moss H, Leonardi J, Jafar-Neijad H, Haltiwanger RS. Mapping sites of O-glycosylation and Fringe elongation on Drosophila Notch. J Biol Chem, 2016; 291(31): 16348–16360. [DOI] [PMC free article] [PubMed]
- 9).Kakuda S, Haltiwanger RS. Deciphering the Fringe-mediated Notch code: identification of activating and inhibiting sites allowing discrimination between ligands. Developmental Cell, 2017; 40: 1–9. [DOI] [PMC free article] [PubMed]
- 10).Rana NA, Nita-Lazar A, Takeuchi H, Kakuda S, Luther KB, Haltiwanger RS. O-glucose trisaccharide is present at high but variable stoichiometry at multiple sites on mouse Notch1. J Biol Chem, 2011; 286: 31623–31637. [DOI] [PMC free article] [PubMed]
- 11).Haltom AR, Jafar-Nejad H. The multiple roles of epidermal growth factor repeat O-glycans in animal development. Glycobiology, 2015; 10: 2017–1042. [DOI] [PMC free article] [PubMed]
- 12).Stahl M, Uemura K, Ge C, Shi S, Tashima Y, Stanley P. Roles of Pofut1 and O-fucose in mammalian Notch signaling. J Biol Chem, 2008; 283(20): 13638–13651. [DOI] [PMC free article] [PubMed]
- 13).Fermandez-Valdivia R, Takeuchi H, Samagrghandi A, Lopez M, Leonardi J, Haltiwanger RS, et al. Regulation of mammalian Notch signaling and embryonic development by the protein O-glucosyltransferase Rumi. Development, 2011; 138(10): 1925–1934. [DOI] [PMC free article] [PubMed]
- 14).Moleny DJ, Shair LH, Lu FM, Xia J, Locke R, Matta KK, et al. Mammalian Notch1 is modified with two unusual forms of O-linked glycosylation found on epidermal growth factor-like modules. J Biol Chem, 2000; 275: 9604–9611. [DOI] [PubMed]
- 15).Wang Y, Spellman MW. Purification and characterization of a GDP-fucose:polypeptide fucosyltransferase from Chinese hamster ovary cells. J Biol Chem, 1998; 273: 8112–8118. [DOI] [PubMed]
- 16).Wang Y, Shao L, Shi S, Harris RJ, Spellman MW, Stanley P, et al. Modification of epidermal growth factor-like repeats with O-fucose. Molecular cloning and expression of a novel GDP-fucose protein O-fucosyltransferase. J Biol Chem, 2001; 276: 40338–40345. [DOI] [PubMed]
- 17).Okajima T, Irvine KD. Regulation of notch signaling by O-linked fucose. Cell, 2002; 111: 893–904. [DOI] [PubMed]
- 18).Takeuchi H, Kantharia J, Sethi MK, Bakker H, Haltiwanger RS. Site-specific O-glucosylation of the epidermal growth factor-like (EGF) repeats of notch: efficiency of glycosylation is affected by proper folding and amino acid sequence of individual EGF repeats. 2012, J Biol Chem, 2012; 287: 33934–433944. [DOI] [PMC free article] [PubMed]
- 19).Rampal R, Luther KB, Haltiwanger RS. Notch signaling in normal and disease states: possible therapies related to glycosylation. 2007, Curr Mol Med, 2007; 7(4): 427–425. [DOI] [PubMed]
- 20).Shi S, Stanley P. Protein O-fucosyltransferase 1 is an essential component of Notch signaling pathways. Proc Natl Acad Sci U S A, 2003; 100(9): 5234–5239. [DOI] [PMC free article] [PubMed]
- 21).Okamura Y, Saga Y. Pofut1 is required for the proper localization of the Notch receptor during mouse development. Mech Dev, 2008; 125(8): 663–673. [DOI] [PubMed]
- 22).Muller J, Rana NA, Serth K, Kakuda S, Haltiwanger RS, Gossler A. O-fucosylation of the notch ligand mDLL1 by POFUT1 is dispensable for ligand function. PLoS One, 2014; 9(2): e88571. [DOI] [PMC free article] [PubMed]
- 23).Serth K, Schuster-Gossler K, Kremmer E, Hansen B, Marohn-Kohn B, Goccler A. O-fucosylation of DLL3 is required for its function during somitogenesis. PLoS One, 2015; 10(4): e0123776. [DOI] [PMC free article] [PubMed]
- 24).Okajima T, Xu A, Lei L, Irvine KD. Chaperone activity of protein O-fucosyltransferase 1 promotes notch receptor folding. Science, 2005; 307(5715): 1599–1603. [DOI] [PubMed]
- 25).Ishio A, Sasamura T, Ayukawa T, Kuroda J, Ishikawa HO, Aoyama N, et al.O-fucose monosaccharide of Drosophila Notch has a temperature-sensitive function and cooperates with O-glucose glycan in Notch transport and Notch signaling activation. J Biol Chem, 2015, 290(1): 505–519. [DOI] [PMC free article] [PubMed]
- 26).Hase S, Kawabata S, Nishimura H, Takeya H, Sueyoshi T, Miyata T, et al. A new trisaccharide sugar chain linked to a serine residue in bovine blood coagulation factors VII and IX. J Biochem, 1988; 104(6): 867–868. [DOI] [PubMed]
- 27).Acar M, Jafar-Nejad H, Takeuchi H, Rajan S, Ibrani D, Rana NA, et al. Rumi is a CAP10 domain glycosyltransferase that modifies Notch and is required for Notch signaling. Cell, 2008; 132(2): 247–258. [DOI] [PMC free article] [PubMed]
- 28).Matsuura A, Ito M, Sakaidani Y, Kondo T, Murakami K, Furukawa K, et al.O-linked N-acetylglucosamine is present on the extracellular domain of notch receptors. J Biol Chem, 2008; 283(51): 35486–35495. [DOI] [PubMed]
- 29).Sakaidani Y, Nomura T, Matsuura A, Ito M, Suzuki E, Murakami K, et al.O-linked-N-acetylglucosamine on extracellular protein domains mediates epithelial cell-matrix interactions. Nat Commun, 2011;2: 583:1–9. [DOI] [PubMed]
- 30).Sawaguchi S, Varshney S, Ogawa M, Sakaidani Y, Yagi H, Takeshita K. O-GlcNAc on NOTCH1 EGF repeats regulates ligand-induced Notch signaling and vascular development in mammals. Elife, 2017; e24419. [DOI] [PMC free article] [PubMed]
- 31).Shaheen R, Aglan M, Keppler-Noreuil K, Fageih E, Ansari S, Horton K, et al. Mutations in EOGT confirm the genetic heterogeneity of autosomal-recessive Adams-Oliver syndrome. Am J Hum Genet, 2013; 92(4): 598–604. [DOI] [PMC free article] [PubMed]
- 32).Cohen I, Silberstein E, Perez Y, Landau D, Elbedour K, Langer Y, et al. Autosomal recessive Adams-Oliver syndrome caused by homozygous mutation in EOGT, encoding an EGF domain-specific O-GlcNAc transferase. Eur J Hum Genet, 2014; 22(3): 374–378. [DOI] [PMC free article] [PubMed]
- 33).Burton BK, Hauser L, Nadler HL. Congenital scallop defects with distal limb anomalies: report of a family. J Med Genet, 1976; 13(6): 466–468. [DOI] [PMC free article] [PubMed]
- 34).Bonafede RP, Beighton P. Autosomal dominant inheritance of scallop defects with ectrodactyly. Am J Med Genet, 1979; 3(1): 35–41. [DOI] [PubMed]
- 35).Masek J, Andresson ER. The developmental biology of genetic Notch disorders. Development, 2017; 144: 1743–1763. [DOI] [PubMed]
- 36).Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet, 1997; 16(3): 243–251. [DOI] [PubMed]
- 37).McDaniell R, Warthen DM, Sanchez-Lara PA, Pai A, Krants ID, Piccoli DA, et al. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am J Hum Genet, 2006; 79(1): 169–173. [DOI] [PMC free article] [PubMed]
- 38).Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature, 1996; 383: 707–710. [DOI] [PubMed]
- 39).Basmanav FB, Oprisoreanu AM, Pastermack SM, Thiele H, Fritz G, Wenzel J, et al. Mutations in POGLUT1, encoding protein O-glucosyltransferase 1, cause autosomal-dominant Dowling-Degos disease. Am J Hum Genet, 2014; 94(1): 135–143. [DOI] [PMC free article] [PubMed]
- 40).Li M, Cheng R, Liang J, Yan H, Zhang H, Yang L, et al. Mutations in POFUT1, encoding protein O-fucosyltransferase 1, cause generalized Dowling-Degos disease. Am J Hum Genet, 2013; 92(6): 895–903. [DOI] [PMC free article] [PubMed]
- 41).Haltom AR, Lee TV, Harvet BM, Leonardi J, Chen Y, Hong Y, et al. The protein O-glucosyltransferase Rumi modifies eyes shut to promote rhabdomere separation in Drosophila PLoS Genet, 2014; 10(11): e1004795. [DOI] [PMC free article] [PubMed]
- 42).Hassed S, Li S, Mulvhill J, Aston C, Palmer S. Adams-Oliver syndrome review of the literature:Refining the diagnostic phenotype. Am J Med Genet A, 2017; 173(3): 790–800. [DOI] [PubMed]
- 43).Adams FH, Oliver CP. Hereditary deformities in man. J Hered, 1945; 36: 3–7.
- 44).Meester JA, Southgate L, Stittrich AB, Venselaar H, Beekmans SJ, den Hollander N, et al. Heterozygous loss-of-function mutations in DLL4 cause Adams-Oliver syndrome. Am J Hum Genet, 2015; 97(3): 475–482. [DOI] [PMC free article] [PubMed]
- 45).Shaheen R, Faqeih E, Sunker A, Morsy H, AI-Sheddi T, Shamseldin HE, et al. Recessive mutations in DOCK6, encoding the guanidine nucleotide exchange factor DOCK6, lead to abnormal actin cytoskeleton organization and Adams-Oliver syndrome. Am J Hum Genet, 2011; 89(2): 328–333. [DOI] [PMC free article] [PubMed]
- 46).Hassed SJ, Willey GB, Wang S, Lee JY, Li S, Xu W, et al. RBPJ mutations identified in two families affected by Adams-Oliver syndrome. Am J Hum Genet, 2012; 91(2): 391–395. [DOI] [PMC free article] [PubMed]
- 47).Southgate L, Sukalo M, Karountzos ASV, Taylor EJ, Collinson CS, Ruddy D, et al. Haploinsufficiency of the NOTCH1 receptor as a cause of Adams-Oliver syndrome with variable cardiac anomalies. Circ Cardiovasc Genet, 2015; 8(4): 572–581. [DOI] [PMC free article] [PubMed]
- 48).Stittrich AB, Lehman A, Bodian DL, Ashworth J, Zong Z, Li H, et al. Mutations in NOTCH1 cause Adams-Oliver syndrome. Am J Hum Genet, 2014; 95(3): 275–284. [DOI] [PMC free article] [PubMed]
- 49).Aminkeng F. DLL4 loss-of-function heterozygous mutations cause Adams-Oliver syndrome. Clin Genet, 2015; 88(6): 532. [DOI] [PubMed]
- 50).Lehman A, Stittrich AB, Glusman G, Zong Z, Li H, Patrice E, et al. Diffuse angiopathy in Adams-Oliver syndrome associated with truncating DOCK6 mutations. Am J Med Genet A, 2014; 164(10): 2656–2662. [DOI] [PMC free article] [PubMed]
- 51).Southgate L, Machado RD, Snape KM, Primeau M, Dafou D, Ruddy DM, et al. Gain-of-function mutations of ARHGAP31, a Cdc42/Rac1 GTPase regulator, cause syndromic cutis aplasia and limb anomalies. Am J Hum Genet, 2011; 88(5): 574–585. [DOI] [PMC free article] [PubMed]
- 52).Brzozowa-Zasada M, Piecuch A, Michalski M, Segiet O, Kurek J, Harabin-Słowińska M. Notch and its oncogenic activity in human malignancies. Eur Surg, 2017; 49(5): 199–209. [DOI] [PMC free article] [PubMed]
- 53).Del Castillo Velasco-Herrera M, van der Weyden L, Nsengimana J, Speak AO, Sjöberg MK, Bishop DT, et al. Comparative genomics reveals that loss of lunatic fringe (LFNG) promotes melanoma metastasis. Mol Oncol, 2018; 12(2): 239–255. [DOI] [PMC free article] [PubMed]
- 54).Swaey ET, Chanrion M, Cai C, Wu G, Zhand J, Zender L, et al. Identification of a therapeutic strategy targeting amplified FGF19 in liver cancer by oncogenomic screening. Cancer Cell, 2011; 19(3): 347–358. [DOI] [PMC free article] [PubMed]
- 55).Loo LW, Tiirikainen M, Cheng I, Lum-Jones A, Seifried A, Chuch JM, et al. Integrated analysis of genome-wide copy number alterations and gene expression in microsatellite stable, CpG island methylator phenotype-negative colon cancer. Genes Chromosomes Cancer, 2013; 52(5): 450–466. [DOI] [PMC free article] [PubMed]
- 56).Yokota S, Ogawara K, Kimura R, Shimizu F, Baba T, Minakawa Y, et al. Protein O-fucosyltransferase 1: a potential diagnostic marker and therapeutic target for human oral cancer. Int J Oncol, 2013; 43(6): 1864–1870. [DOI] [PubMed]
- 57).Wang Y, Chang N, Zhang T, Liu H, Ma W, Chu Q, et al. Overexpression of human CAP10-like protein 46 KD in T-acute lymphoblastic leukemia and acute myelogenous leukemia. Gene Test Mol Biomarkers, 2010; 14(1): 127–133. [DOI] [PubMed]
- 58).Servian-Morilla E, Takeuchi H, Lee TV, Clarimon J, Mavillard F, Area-Gomez F, et al. A POGLUT1 mutation causes a muscular dystrophy with reduced Notch signaling and satellite cell loss. EMBO Mol Med, 2016; 8(11): 1289–1309. [DOI] [PMC free article] [PubMed]
- 59).Ogawa M, Senoo Y, Ikeda K, Takeuchi H, Okajima T. Structural divergence in O-GlcNAc glycans displayed on epidermal growth factor-like repeats of mammalian Notch1. Molecules, 2018; 23(7): 1745. [DOI] [PMC free article] [PubMed]