


Abstract
The detection of tumor-derived cell-free DNA in plasma is one of the most promising directions in cancer diagnosis. The major challenge in such an approach is how to identify the tiny amount of tumor DNAs out of total cell-free DNAs in blood. Here we propose an ultrasensitive cancer detection method, termed ‘CancerDetector’, using the DNA methylation profiles of cell-free DNAs. The key of our method is to probabilistically model the joint methylation states of multiple adjacent CpG sites on an individual sequencing read, in order to exploit the pervasive nature of DNA methylation for signal amplification. Therefore, CancerDetector can sensitively identify a trace amount of tumor cfDNAs in plasma, at the level of individual reads. We evaluated CancerDetector on the simulated data, and showed a high concordance of the predicted and true tumor fraction. Testing CancerDetector on real plasma data demonstrated its high sensitivity and specificity in detecting tumor cfDNAs. In addition, the predicted tumor fraction showed great consistency with tumor size and survival outcome. Note that all of those testing were performed on sequencing data at low to medium coverage (1× to 10×). Therefore, CancerDetector holds the great potential to detect cancer early and cost-effectively.
INTRODUCTION
Early detection of cancer - before it has had a chance to metastasize - presents the best strategy for increasing cancer survival. Recently, cancer detection using cell-free DNA (cfDNA) from blood has attracted significant interest due to its non-invasive nature. However, tumor cfDNA levels are very low in most early-stage and many advanced stage cancer patients (1,2). Therefore, the major challenge in cfDNA-based early cancer diagnostics is how to identify the tiny amount of tumor cfDNAs out of total cfDNAs in blood. The mainstream approach to address this challenge is mutation-based, i.e. using targeted deep sequencing (>5000× coverage), combined with error-suppression techniques, to call cfDNA mutations in a small gene panel (1–3). While this approach provides a sensitive way to monitor cancer recurrence when the mutations are known, a small gene panel could not serve diagnostic purposes because mutations can be wide-spread and very heterogeneous, even in the same type of cancer (4–7). However, enlarging the gene panel, while maintaining the sequencing depth, is cost-prohibitive. In this paper, we aim to address the challenge of detecting the trace amount of tumor cfDNA using a different approach, namely, using the cfDNA methylation patterns.
Several reasons motivate the methylation-based tumor cfDNA detection: (i) DNA methylation patterns are pervasive, meaning that the same methylation patterns (methylated or unmethylated) tend to spread throughout a genome region. This feature has been employed by Dennis Lo's team to evaluate DNA hypomethylation across large genome regions for cancer diagnosis (8). In addition, Lehmann-Werman et al. has experimentally confirmed that co-methylation across neighboring CpG sites can enhance distinction of disease-derived DNA in plasma cfDNA (9). (ii) Aberrant DNA methylation patterns occur early in the pathogenesis of cancer (10), therefore facilitating early cancer detection. In fact, DNA methylation abnormalities are one of the hallmarks of cancer and are associated with all aspects of cancer, from tumor initiation to cancer progression and metastasis (11–13). These nice properties inspired a number of recent approaches in using DNA methylation patterns for cancer diagnosis (14,15). Here, we aim to amplify aberrant cfDNA methylation signals at the resolution of single sequencing reads, therefore providing an ultra-sensitive detection of a tiny amount of tumor cfDNA even at a low sequencing coverage.
The key to our method is to focus on the joint methylation states of multiple adjacent CpG sites on an individual cfDNA sequencing read, in order to exploit the pervasive nature of DNA methylation for signal amplification. Traditional DNA methylation analysis focuses on the methylation rate of an individual CpG site in a cell population. This rate, often called the 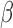 -value, is the proportion of cells in which the CpG site is methylated (see an example in Figure 1). However, such population-average measures are not sensitive enough to capture an abnormal methylation signal affecting only a small proportion of the cfDNAs. Figure 1 illustrates this point: the average methylation rates of the individual CpG sites are
-value, is the proportion of cells in which the CpG site is methylated (see an example in Figure 1). However, such population-average measures are not sensitive enough to capture an abnormal methylation signal affecting only a small proportion of the cfDNAs. Figure 1 illustrates this point: the average methylation rates of the individual CpG sites are 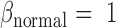 for normal plasma cfDNAs, and
for normal plasma cfDNAs, and 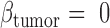 for tumor cfDNAs; assuming the presence of 1% tumor cfDNAs, the traditional measure yields
for tumor cfDNAs; assuming the presence of 1% tumor cfDNAs, the traditional measure yields 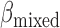 = 0.99, which is hard to differentiate from
= 0.99, which is hard to differentiate from 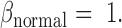 However, based on the pervasive nature of DNA methylation, we came up with a new way to differentiate disease-specific cfDNA reads from normal cfDNA reads. If we average the methylation values of all CpG sites in a given read (denoted
However, based on the pervasive nature of DNA methylation, we came up with a new way to differentiate disease-specific cfDNA reads from normal cfDNA reads. If we average the methylation values of all CpG sites in a given read (denoted 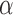 -value), we see a striking difference (0 and 1) between the abnormally methylated cfDNAs and the normal cfDNAs (
-value), we see a striking difference (0 and 1) between the abnormally methylated cfDNAs and the normal cfDNAs (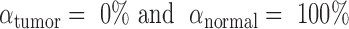 ). In other words, given the pervasive nature of DNA methylation, the joint methylation states of multiple adjacent CpG sites may easily distinguish cancer-specific cfDNA reads from normal cfDNA reads. Inspired by the
). In other words, given the pervasive nature of DNA methylation, the joint methylation states of multiple adjacent CpG sites may easily distinguish cancer-specific cfDNA reads from normal cfDNA reads. Inspired by the 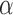 -value, we realized that the key to exploiting pervasive methylation is to estimate whether the joint probability of all CpG sites in a read follows the DNA methylation signature of a disease. We therefore propose a novel, read-based probabilistic approach, termed ‘CancerDetector’, that can sensitively identify a trace amount of tumor cfDNAs out of all cfDNAs in plasma.
-value, we realized that the key to exploiting pervasive methylation is to estimate whether the joint probability of all CpG sites in a read follows the DNA methylation signature of a disease. We therefore propose a novel, read-based probabilistic approach, termed ‘CancerDetector’, that can sensitively identify a trace amount of tumor cfDNAs out of all cfDNAs in plasma.
Figure 1.

Illustration of the rationale why the methylation value averaged across all CpG sites in a sequencing read (α-value) is more sensitive at detecting tumor-derived cfDNAs than the traditional methylation level of a CpG site averaged across all reads (β-value). Each line represents a sequencing read and each dot represents a CpG site.
We first evaluated CancerDetector on the simulated plasma samples that subsample and combine sequencing reads of a normal plasma cfDNA sample and a solid tumor sample at known mixing rates (or tumor fractions). The results showed that CancerDetector can achieve a Pearson's correlation coefficient (PCC) of 0.9974 (P-value 7.2E–8) between the predicted and true proportions of tumor cfDNAs at medium sequencing coverage (10×). And the prediction performance increases with the sequencing coverage—the higher the sequencing coverage, the closer the predicted tumor fraction is to the true value. Moreover, CancerDetector outperformed our previous method of cfDNA tumor fraction prediction, i.e. ‘CancerLocator’ (16), in terms of both prediction performance and robustness. We then tested CancerDetector on real plasma cfDNA samples and demonstrated its high performance across 10 experimental runs, i.e. sensitivity of 94.8 ± 3.6% (when specificity is 100%) for early-stage cancer patients; while CancerLocator has a sensitivity of 74.4 ± 10.0% (when specificity is 100%). In addition, the tumor fraction predicted by CancerDetector showed great consistency with clinical information, such as tumor size and survival outcome, in longitudinal samples. Note that we achieved these results based on real samples that the majority have low sequencing coverage (1×∼3×, averaged across all genome positions).
MATERIALS AND METHODS
Overview
The goal of this approach is to classify each read (in the methylation marker regions) into either the tumor-derived cfDNA class (abbreviated as class T) or the normal-plasma-derived cfDNA class (abbreviated as class N). In this paper, we focus on one type of cancer, liver cancer, but our method can be generalized to any cancer type. Our approach comprises three major steps: (i) Identifying the DNA methylation signatures of liver cancer. We derived the methylation markers of liver cancer based on DNA methylation data of liver tumors and their matched normal tissues as well as normal plasma cfDNA samples. The vast amount of methylation data was collected from the public database TCGA (The Cancer Genome Atlas (17)) and recent literatures (8,27). (ii) Calculating the likelihood for a read to harbor a methylation signature. Given a new patient, we performed the methylation sequencing on his/her plasma cfDNA sample. We obtained the sequencing reads of those cfDNA fragments that fall into the genomic regions of selected markers. To account for data uncertainty and inter-individual methylation variances in markers, we calculated the likelihood of each read to come from each class. (iii) Inferring cfDNA composition. The likelihood of each read to come from each class can be used to derive the tumor fraction in cfDNAs. Figure 2 gives an overall picture of our approach, and we detail individual steps in the sections below.
Figure 2.

Overview of the CancerDetector method. The color of cfDNA sequencing reads represents their origin: red (green) reads are from tumor (normal plasma) cfDNA fragments. These reads are from a hypomethylated marker (chr2:4050595–4050945).
Identify and characterize methylation markers specific to liver cancer
A methylation marker includes two kinds of information: its genomic region and methylation patterns in both solid tumor samples (class T) and normal plasma cfDNA samples (class N). To take advantage of the large amount of public methylation data from TCGA that were mainly generated by the microarray platform, we developed the following two-step procedure to obtain the liver-cancer-specific methylation markers:
Step 1: Identify genomic markers for liver cancer—Only genomic regions that are covered by sufficient microarray probes qualified as potential markers. Therefore, we use the definition of CpG clusters in our recent work (16) to identify all potential genomic regions. See the Results section for details. Among all potential regions, we select those regions whose methylation levels can differentiate most liver tumor samples from not only their matched normal liver tissues but also from normal plasma samples. This task inherently includes two steps: (i) Selecting those ‘frequently differential methylation regions (FDMR)’, in which the methylations are differential (greater than a cutoff) between matched tumor and normal tissues in more than half of the matched pairs. This step can remove markers specific to liver tissues, but retain markers specific to liver cancer. (ii) Selecting those FDMRs that can distinguish tumor samples from normal plasma samples, i.e. the difference between the medians of its methylation levels in two classes is greater than a cutoff. This step ensures that the tumor methylation signal can be identified in blood. Given a fixed sequencing coverage of cfDNAs, the more markers we use (that is, the larger the panel size), the lower quality these markers may have, but the more tumor-derived cfDNA reads we may identify. Therefore, there is a tradeoff between the markers’ quality and the amount of tumor cfDNA signals we can use. In this work, since all public plasma cfDNA samples have low sequencing coverages (1× to 3×), we chose the cutoff of the methylation difference in both steps as 0.2 in order to keep relatively good marker quality and maintain a large enough size for the methylation marker panel to capture sufficient tumor cfDNAs at this low sequencing coverage.
Step 2: Characterize methylation patterns—In each marker region identified in Step 1, we shall consider the inter-individual variance of methylation levels in each class (T and N). Given a region, we modelled the methylation levels of all samples in a class to follow a Beta distribution
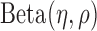 , which has been widely used in methylation data analyses (18–22) and our recent work (16). Specifically, a marker
, which has been widely used in methylation data analyses (18–22) and our recent work (16). Specifically, a marker 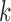 is associated with two methylation patterns, i.e.,
is associated with two methylation patterns, i.e., 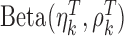 for the class T and
for the class T and 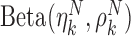 for the class N. Note that
for the class N. Note that 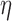 and
and 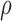 are two shape parameters (usually denoted
are two shape parameters (usually denoted 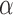 and
and 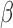 ) of a Beta distribution, but here we used the symbols
) of a Beta distribution, but here we used the symbols 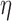 and
and 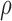 to avoid the confusion with
to avoid the confusion with 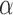 -value and
-value and 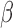 -values defined in Introduction section. The parameters of a Beta distribution can be easily learnt from the sample population of a class, using either the method of moments or maximum likelihood (23). To simplify notation, we denote the methylation pattern of marker k for class T as
-values defined in Introduction section. The parameters of a Beta distribution can be easily learnt from the sample population of a class, using either the method of moments or maximum likelihood (23). To simplify notation, we denote the methylation pattern of marker k for class T as 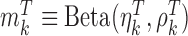 , and for class N as
, and for class N as  .
.
Calculate the class-specific likelihood of each cfDNA sequencing read
Our goal is to classify each cfDNA read as class T or N, based on the joint-methylation-status of multiple CpG sites on the read. The joint-methylation-status in a cfDNA read is denoted as 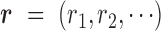 , where the binary value
, where the binary value 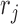 = 1 or 0 represents methylated or unmethylated status of the CpG site
= 1 or 0 represents methylated or unmethylated status of the CpG site 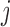 in read
in read 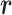 . We model this binary vector
. We model this binary vector 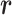 by the Beta-Bernoulli distribution (24). Specifically, given a methylation pattern
by the Beta-Bernoulli distribution (24). Specifically, given a methylation pattern 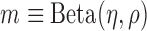 of the marker where read
of the marker where read 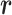 falls into, the methylation status
falls into, the methylation status 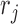 of each CpG site
of each CpG site 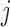 in the read is distributed as
in the read is distributed as 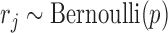 , where
, where 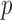 is the prior of average methylation rate of CpG sites within the marker and follows the Beta prior distribution
is the prior of average methylation rate of CpG sites within the marker and follows the Beta prior distribution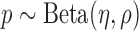 . Using this statistical model, the likelihood of the joint methylation status in read
. Using this statistical model, the likelihood of the joint methylation status in read 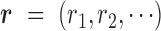 , given the methylation pattern
, given the methylation pattern 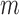 , can be calculated as below:
, can be calculated as below:
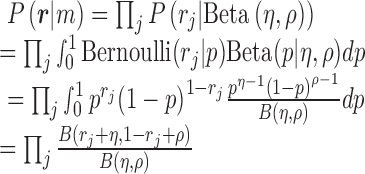 |
where 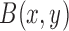 is the beta function. Therefore, for marker
is the beta function. Therefore, for marker 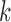 with methylation pattern
with methylation pattern 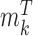 of class T and
of class T and 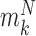 of class N, we can use the above formula to compute the class-specific likelihoods of read
of class N, we can use the above formula to compute the class-specific likelihoods of read 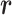 , i.e.,
, i.e., 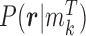 and
and 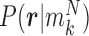 . Note that this likelihood calculation implements a probabilistic version of α-value for individual reads. An example is illustrated in Figure 3.
. Note that this likelihood calculation implements a probabilistic version of α-value for individual reads. An example is illustrated in Figure 3.
Figure 3.

Illustration of calculating the likelihood of a cfDNA sequencing read in a marker, given the methylation patterns of normal and tumor classes.
Predict tumor-derived cfDNA fraction
As illustrated in Figure 2, we develop a probabilistic framework to infer the tumor-derived cfDNA fraction (i.e. tumor fraction), denoted as 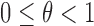 , by classifying cfDNA reads into two classes (class T for tumor-derived DNAs and class N for normal plasma cfDNAs), based on a set of markers associated with the methylation patterns of two classes. We denote the methylation patterns of all
, by classifying cfDNA reads into two classes (class T for tumor-derived DNAs and class N for normal plasma cfDNAs), based on a set of markers associated with the methylation patterns of two classes. We denote the methylation patterns of all 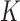 markers as
markers as 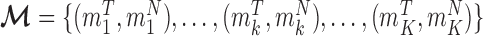 . We also denote the methylation sequencing data of a patient's cfDNAs as a set of
. We also denote the methylation sequencing data of a patient's cfDNAs as a set of 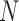 reads
reads 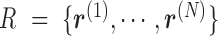 that in total cover
that in total cover 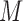 CpG sites. For a read that is aligned to the region of marker
CpG sites. For a read that is aligned to the region of marker 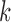 , we assume that it can come from one of two classes with the class-specific likelihood
, we assume that it can come from one of two classes with the class-specific likelihood 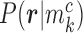 , where
, where 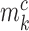 is the methylation pattern of class
is the methylation pattern of class 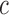 . Let
. Let 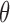 be the tumor-derived cfDNA fraction, so the fraction of normal cfDNA is
be the tumor-derived cfDNA fraction, so the fraction of normal cfDNA is 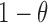 . We want to estimate
. We want to estimate 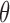 by maximizing the log-likelihood
by maximizing the log-likelihood 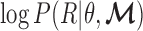 . This is a maximum likelihood estimation problem. Assuming the independence of each read (as widely adopted in literatures (25,26)),
. This is a maximum likelihood estimation problem. Assuming the independence of each read (as widely adopted in literatures (25,26)), 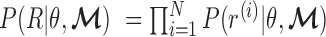 . We can then expand the likelihood
. We can then expand the likelihood 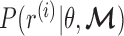 of read
of read 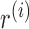 as follows:
as follows:
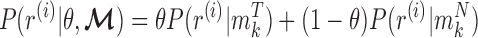 |
Since 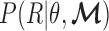 has only one parameter
has only one parameter 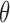 to be estimated, we can simply apply a grid search to exhaustively enumerate all 1000 fraction values which are uniformly distributed between 0% and 100%, i.e., 0%, 0.1%, …, 99.9% and 100%. This method can get the global optimization at the precision of 0.1%, which we think is sufficient for capturing the tiny amount of tumor-derived cfDNAs. Since the grid search is computationally fast, we can easily refine the steps to determine
to be estimated, we can simply apply a grid search to exhaustively enumerate all 1000 fraction values which are uniformly distributed between 0% and 100%, i.e., 0%, 0.1%, …, 99.9% and 100%. This method can get the global optimization at the precision of 0.1%, which we think is sufficient for capturing the tiny amount of tumor-derived cfDNAs. Since the grid search is computationally fast, we can easily refine the steps to determine 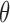 at higher resolutions.
at higher resolutions.
Removal of ‘confounding’ markers
Above, we estimated a global tumor fraction (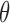 ) across all cancer-specific markers. The tumor fraction (
) across all cancer-specific markers. The tumor fraction (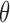 ) can also be estimated only for a single marker. Ideally, for an early-stage cancer patient, the estimated
) can also be estimated only for a single marker. Ideally, for an early-stage cancer patient, the estimated 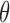 should be a small number (e.g., <20%), either across all markers or in individual markers. However, in real cancer patient data, we observed a number of markers with individually estimated tumor fractions far larger than the global tumor fraction. Therefore, cfDNA fragments harboring aberrant methylation in these ‘outlier’ markers obviously do not come from cancerous cells, but likely from normal cells (e.g. white blood cells) due to inter-individual variance (e.g. age, environment exposure, or other diseases the person may have). Consequently, including these ‘confounding’ markers would impair the accuracy of tumor fraction estimation. We therefore design an iterative algorithm to adjust the global tumor fraction after identifying and removing ‘germline’ markers. We denote
should be a small number (e.g., <20%), either across all markers or in individual markers. However, in real cancer patient data, we observed a number of markers with individually estimated tumor fractions far larger than the global tumor fraction. Therefore, cfDNA fragments harboring aberrant methylation in these ‘outlier’ markers obviously do not come from cancerous cells, but likely from normal cells (e.g. white blood cells) due to inter-individual variance (e.g. age, environment exposure, or other diseases the person may have). Consequently, including these ‘confounding’ markers would impair the accuracy of tumor fraction estimation. We therefore design an iterative algorithm to adjust the global tumor fraction after identifying and removing ‘germline’ markers. We denote 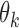 as the tumor fraction at the marker
as the tumor fraction at the marker 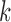 ,to distinguish from the global fraction
,to distinguish from the global fraction 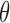 obtained using all markers. The procedure of this algorithm is presented below:
obtained using all markers. The procedure of this algorithm is presented below:
Initialization—Let
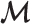 denote the set of markers used for
denote the set of markers used for 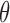 estimation. Initially, we put all markers into
estimation. Initially, we put all markers into 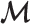 .
.Step 1: Remove ‘confounding’ markers – Estimate
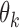 of each marker
of each marker 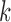 in
in 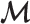 and calculate the standard deviation of all
and calculate the standard deviation of all 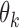 , denoted as
, denoted as 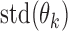 . Remove from
. Remove from 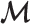 those markers whose
those markers whose 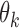 >
>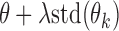 , where
, where 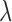 is an input fixed parameter.
is an input fixed parameter.Step 2: Update
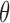 —Estimate the global fraction
—Estimate the global fraction 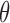 using all markers of
using all markers of 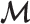 updated in Step 1.
updated in Step 1.Step 3: Iterate Steps 1 and 2, until
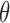 converges.
converges.
The output 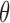 is the adjusted global tumor fraction after removing ‘confounding’ markers. The parameter
is the adjusted global tumor fraction after removing ‘confounding’ markers. The parameter 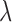 of this algorithm controls how far the
of this algorithm controls how far the 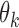 of those ‘confounding’ markers deviates from the average
of those ‘confounding’ markers deviates from the average 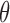 . We can estimate this parameter using normal plasma cfDNA samples, because it is expected that the optimal
. We can estimate this parameter using normal plasma cfDNA samples, because it is expected that the optimal 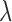 should be able to adjust the global
should be able to adjust the global 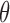 of the normal samples to be close to zero.
of the normal samples to be close to zero.
Methylation data collection, generation and processing
Data collection
We collected the methylation profiles of 49 solid liver tumor samples and their matched adjacent solid liver tissue samples from the TCGA database. All of these samples were assayed using the Infinium HumanMethylation450 microarray. For the plasma cfDNA samples, we used the methylation sequencing data from Chan et al. (8) with European Genome-Phenome Archive database (abbreviated as EGA) accession number EGAS00001000566 and Sun et al. (27) with EGA accession number EGAS00001001219. They include the Whole Genome Bisulfite Sequencing (WGBS) data of plasma samples taken from 32 healthy people, 8 patients infected with chronic hepatitis B virus (HBV) and 29 liver cancer patients.
Data generation
Since the public WGBS data of plasma cfDNA samples have very low sequencing coverage (1×∼3×), we generated WGBS data of plasma cfDNA samples from four healthy people, at higher coverage (∼10× on average); and generated WGBS data of solid tumor samples from two cancer patients, in order to simulate higher-coverage cfDNA WGBS data from cancer patients. We also generated WGBS data of plasma samples collected from 4 liver cancer patients. Blood samples were centrifuged at 1600 × g for 10 min and then the plasma was transferred into new microtubes and centrifuged at 16 000 × g for another 10 min. The plasma was collected and stored at −80°C. cfDNA was extracted from 5 ml plasma using the Qiagen QIAamp Circulating Nucleic Acids Kit and quantified using a Qubit 3.0 Fluoromter (Thermo Fisher Scientific). Bisulfite conversion of cfDNA was performed using the EZ-DNA-Methylation-GOLD kit (Zymo Research). After that, an Accel-NGS Methy-Seq DNA library kit (Swift Bioscience) was used to prepare the sequencing libraries. The DNA libraries were then sequenced with 150bp paired-end reads. For the solid tumor samples, bisulfite conversion was performed with the EZ-DNA-Methylation-GOLD kit (Zymo Research), and the sequencing libraries were prepared using the TruSeq DNA Methylation Kit. The DNA libraries were then sequenced with 150-bp paired-end reads using HiSeq X (Illumina). In total, 10 WGBS data have been deposited to the EGA database (https://www.ega-archive.org) with accession number EGAS00001002728 for public research use.
Human subjects
The blood samples of four healthy people and four liver tumor patients were collected with informed consent for research use. This project was approved by the Institutional Review Boards (IRBs) of University of Southern California (IRB #HS-15-00740). Two solid liver tumor tissues were purchased from OriGene Technologies, Inc.
Processing methylation microarray data
The microarray data (level 3 in TCGA database) provide the methylation levels of individual CpG sites. We define the methylation level of a CpG cluster as the average methylation level of all CpG sites in the cluster. A cluster's methylation level is marked as ‘not available’ (NA) if more than half of its CpG sites do not have methylation measurements.
Processing WGBS data
We used Bismark (28) to align the reads to the reference genome hg19 and call the methylated cytosines. After the removal of PCR duplicates, the numbers of methylated and unmethylated cytosines were counted for each CpG site. The methylation level of a CpG cluster is calculated as the ratio between the number of methylated cytosines and the total number of cytosines within the cluster. However, if the total number of cytosines in the reads aligned to the CpG cluster is <30, the methylation level of this cluster is treated as NA (Not Available). This WGBS data processing procedure is used for calculating the average methylation level of a CpG cluster in normal plasma samples that are used for identifying methylation markers. When a plasma cfDNA sample is used as test data, we extracted the joint-methylation-status of all CpG sites of individual sequencing reads that are aligned to the regions of the marker panel from Bismark's output, then fed this information into CancerDetector as its input data. Since the sequencing coverage of real data is low, in this work, we used all reads covering at least one CpG site. For the cfDNA methylation data with high coverage, we can filter out those reads covering <3 CpG sites to improve the input data quality.
RESULTS
Identify methylation markers specific to liver cancer
Defining all genomic regions eligible to serve as methylation markers
Our training data are from the TCGA solid tissues, measured by the Infinium HumanMethylation450 microarray with ∼450 000 CpGs. However, the majority of our testing data (8,27) are WGBS data with very low sequencing coverage. Therefore, we grouped the CpG sites into CpG clusters in order to use more mappable reads from the testing data. For a CpG site covered by a probe on the microarray, we define the region 100 bp up- and down-stream as its flanking region and assume that all CpG sites located within this region have the same average methylation level as the CpG sites covered by probes. Two adjacent CpG sites are grouped into a CpG cluster if their flanking regions overlap. Finally, only those CpG clusters containing at least three CpGs covered by microarray probes are used, in order to achieve robust measurement of methylation levels. This procedure yielded 42 374 CpG clusters, which together include about one-half of all the CpG sites on the Infinium HumanMethylation450 microarray. Most of these clusters are each associated with only one gene. These CpG clusters are used for subsequent feature selection. The definitions of all CpG clusters are listed in Supplementary Table S1.
Selecting liver-cancer-specific markers and characterizing their patterns in normal and tumor classes
Given the 42 374 CpG clusters, we selected the cancer-specific markers by using the method described in Materials and Methods section on the training data: (i) 49 pairs of solid liver tumors and their matched normal liver tissues and (ii) 75% of all the 32 healthy plasma samples. Note that the remaining 25% of healthy plasma samples are used as test data, and we randomly partitioned the healthy plasma samples in the ratio of 75/25 as the training/test data, respectively, 10 times. This indicates that we will have 10 different sets of training/test data and each set can yield different selected markers and tumor fraction estimation. Each set of training/test data and its result is called an experimental run. In each of the 10 runs, we identified an average of 3,214 liver-cancer-specific markers (CpG clusters), and the majority of these markers were shared by all runs. The biomarkers for each run are listed in Supplementary Table S2. We then characterized the methylation patterns for each marker in the normal and tumor classes as two Beta distributions, with learnt shape parameters that can capture the inter-individual variance of methylation levels within a class. In addition, the adjacent CpG sites within the selected marker show significant correlation in their methylation statuses. The average sample-wise correlation between the adjacent probes within each selected CpG cluster (∼3200 among 10 runs) is 0.626 with the median of P-values 2.4e–55, across a large cohort of 711 normal samples of 18 tissue types collected from the TCGA database.
Simulation experiments demonstrated the ultrasensitivity of CancerDetector in detecting tumor cfDNAs
We simulated the methylation data of a plasma cfDNA sample by sampling and mixing the methylation sequencing reads of two real samples, a normal plasma cfDNA sample and a solid tumor sample, at a variety of tumor fractions (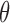 ) and different sequencing coverages (
) and different sequencing coverages (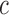 ). This strategy can allow us to mimic real data and precisely control the tumor fraction and sequencing coverage in the mixture samples, in order to test the power and requirement of the CancerDetector method, e.g. at what tumor fraction and sequencing coverage can tumor-derived cfDNAs be detected. We compare CancerDetector with another probabilistic cfDNA deconvolution method, ‘CancerLocator’ (16), that we recently developed and is so far the only method aimed at deconvoluting cancer signals from cfDNA methylation data. While CancerDetector is a read-based method, CancerLocator is based on traditional
). This strategy can allow us to mimic real data and precisely control the tumor fraction and sequencing coverage in the mixture samples, in order to test the power and requirement of the CancerDetector method, e.g. at what tumor fraction and sequencing coverage can tumor-derived cfDNAs be detected. We compare CancerDetector with another probabilistic cfDNA deconvolution method, ‘CancerLocator’ (16), that we recently developed and is so far the only method aimed at deconvoluting cancer signals from cfDNA methylation data. While CancerDetector is a read-based method, CancerLocator is based on traditional 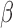 -values by deconvolving
-values by deconvolving 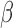 -values of markers in the cfDNAs as a linear combination of the
-values of markers in the cfDNAs as a linear combination of the 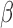 -values of two classes (tumor or normal cfDNAs).
-values of two classes (tumor or normal cfDNAs).
To compare the sensitivity of the two methods in identifying a minor trace of tumor cfDNAs (i.e., 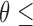 5%), we simulated plasma cfDNA samples at 8 different tumor fractions (
5%), we simulated plasma cfDNA samples at 8 different tumor fractions (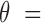 0, 0.1%, 0.3%, 0.5%, 0.8%, 1%, 3% and 5%), and 3 different sequencing coverages (
0, 0.1%, 0.3%, 0.5%, 0.8%, 1%, 3% and 5%), and 3 different sequencing coverages (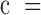 2, 5, 10). The real samples used in the simulation procedure are the WGBS data of two normal plasma samples (N1L and N2L) and of two solid liver tumor samples (HCC1 and HCC2). This experimental setting results in
2, 5, 10). The real samples used in the simulation procedure are the WGBS data of two normal plasma samples (N1L and N2L) and of two solid liver tumor samples (HCC1 and HCC2). This experimental setting results in 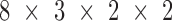 = 96 mixed samples. Figure 4 demonstrates the sensitivity of the two methods in detecting tumor cfDNAs, where scatter plots are shown for the predicted tumor fractions averaged over 10 experimental runs of the simulated samples with eight given tumor fractions at three given sequencing coverages (2×, 5× and 10×). As clearly shown in Figure 4, the blood tumor fractions predicted by CancerDetector are highly consistent with the true values and have very low prediction variances: e.g., when using the highest sequencing coverage 10×, CancerDetector achieved a Pearson's Correlation Coefficient (PCC) of 0.9974±0.0012 (P-value = 7.2E–8), averaged over 10 runs. The consistency increased with the sequencing coverage, i.e. average PCC = 0.9811, 0.9926, 0.9974 and their associated P-values 2.5E–5, 5.3E–6, 7.2E–8 for the sequencing coverages of 2×, 5×, 10×, respectively. More importantly, it can be observed that CancerDetector can (i) detect tumor cfDNAs with a low tumor fraction (
= 96 mixed samples. Figure 4 demonstrates the sensitivity of the two methods in detecting tumor cfDNAs, where scatter plots are shown for the predicted tumor fractions averaged over 10 experimental runs of the simulated samples with eight given tumor fractions at three given sequencing coverages (2×, 5× and 10×). As clearly shown in Figure 4, the blood tumor fractions predicted by CancerDetector are highly consistent with the true values and have very low prediction variances: e.g., when using the highest sequencing coverage 10×, CancerDetector achieved a Pearson's Correlation Coefficient (PCC) of 0.9974±0.0012 (P-value = 7.2E–8), averaged over 10 runs. The consistency increased with the sequencing coverage, i.e. average PCC = 0.9811, 0.9926, 0.9974 and their associated P-values 2.5E–5, 5.3E–6, 7.2E–8 for the sequencing coverages of 2×, 5×, 10×, respectively. More importantly, it can be observed that CancerDetector can (i) detect tumor cfDNAs with a low tumor fraction (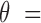 1%) at low sequencing coverage (2×), and (ii) improve the detection limit from 1% to 0.3% when increasing the sequencing coverages (5× and 10×). On the other hand, the
1%) at low sequencing coverage (2×), and (ii) improve the detection limit from 1% to 0.3% when increasing the sequencing coverages (5× and 10×). On the other hand, the 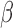 -value-based method, CancerLocator, cannot detect any tumor DNAs when the tumor fraction
-value-based method, CancerLocator, cannot detect any tumor DNAs when the tumor fraction 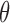 is <5% and 2× sequencing coverage, or
is <5% and 2× sequencing coverage, or 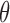 <3% with 5× coverage. Even with 10× sequencing coverage, its predictions are not stable (there is high prediction variance) and deviate strongly from the true tumor fractions. In summary, this result demonstrates that the read-based CancerDetector method can sensitively detect a small amount of tumor cfDNAs, even at low sequencing coverage, and the prediction accuracy and stability increase with higher sequencing coverage.
<3% with 5× coverage. Even with 10× sequencing coverage, its predictions are not stable (there is high prediction variance) and deviate strongly from the true tumor fractions. In summary, this result demonstrates that the read-based CancerDetector method can sensitively detect a small amount of tumor cfDNAs, even at low sequencing coverage, and the prediction accuracy and stability increase with higher sequencing coverage.
Figure 4.

Predicted blood tumor fractions (averaged over 10 runs) for the liver cancer cfDNA samples, simulated by subsampling and mixing sequencing reads from a real healthy cfDNA sample (N1L or N2L) and a solid liver tumor sample (HCC1 or HCC2) at eight different tumor fractions: 0, 0.1%, 0.3%, 0.5%, 0.8%, 1%, 3%, 5%, and at 3 different sequencing coverages (2×, 5× and 10×). In each log-log plot, a blue point represents a simulated sample with error bars (standard deviation of predicted tumor fraction), the x-axis is its true tumor fraction and the y-axis is its predicted tumor fraction. When the predicted tumor fraction is out of range (>5%), we draw the point above the box.
Testing on real data confirmed the high sensitivity of CancerDetector in deconvoluting tumor cfDNA
We compiled a collection of public plasma samples (32 healthy people, 8 HBV carriers and 29 liver cancer patients) from Chan et al. (8) and Sun et al. (27). These data have low sequencing coverages (1× to 3×). We randomly split the 32 healthy plasma samples into training set (75%) and test set (25%) 10 times (runs). In each run, using the combined training set and TCGA microarray data of solid liver tumors and matched normal tissues, we identified the liver-cancer-specific methylation markers and then predicted tumor fractions in the test set: the plasma samples from 8 HBV carriers, 33 liver cancer patients (i.e. 29 of them were collected from Chan et al. (8) and Sun et al. (27), and four of them are collected in our lab), and the remaining 25% of healthy subjects (i.e., 8 healthy people). In summary, in each run, the training data for selecting markers and characterizing their methylation patterns include 24 healthy plasma samples and all TCGA liver samples; and the test data for classification include cancer positives (33 liver cancer patients) and cancer negatives (16 non-cancer patients). The performance of predicting tumor fractions can be measured by the Receiver Operating Characteristic (ROC) curve, where the sensitivity (a.k.a. true positive rate) and specificity (a.k.a. one minus false positive rate) of separating cancer and non-cancer samples are calculated and plotted by using different tumor fraction cutoffs. As shown in Figure 5A and B, the average ROC curve of CancerDetector outperforms that of CancerLocator in terms of both prediction performance and robustness (i.e., much lower standard deviations). For example, when we chose the point of the top-left corner in the ROC curve for determining the tumor fraction threshold, at the specificity of 100% our method can achieve an average sensitivity of 94.9% across 10 runs with standard deviation 2.7%, where the standard deviation of sensitivity is calculated among 10 runs for the fixed specificity (e.g. 100%); while the 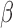 -value based CancerLocator method achieved on average a sensitivity of 77.3% with standard deviation 9.4% at the specificity of 100%. Note that there are at least 25 early-stage (Barcelona Clinic Liver Cancer stage A) patients among the 33 liver cancer patients. Testing only on the 25 early-stage cancer patients and healthy/HBV samples, at the specificity of 100% our method can also achieve an average sensitivity of 94.8% with a standard deviation of 3.6%; while CancerLocator obtained a sensitivity of 74.4% with a standard deviation 10.0%. Summarizing the performance comparison using the Area Under Curve (AUC), our method can achieve an AUC of 0.990 averaged over 10 runs with standard deviation 0.004 for all real samples and an average AUC of 0.988 with standard deviation 0.005 for early-stage cancer patients; while CancerLocator obtained a lower average AUC of 0.982 with standard deviation 0.014 for real samples and an average AUC of 0.979 with standard deviation 0.0143 for early-stage cancer patients. We also compared our method with the methylated haplotype load based method (14), using WGBS data of low sequencing coverage on cfDNAs from non-cancer individuals and liver cancer patients. As shown in Figure 5C, the competing method achieved the sensitivity of 71.2 ± 13.0% at the specificity of 100%, which is less accurate and robust than our method (achieved sensitivity of 94.9 ± 2.7% at the specificity of 100%). We also compared with the average-methylation-rate-based method (14) that achieved the sensitivity of 76.5 ± 17.0% at the specificity of 100% and also has lower performance than our method. For details see the Supplementary Materials. We also note that CancerDetector correctly predicted the cfDNA tumor fractions of all eight chronic hepatitis B virus (HBV) samples to be the same range of the normal samples (i.e. close to zero) that are well distinguished from cancer samples. These results demonstrated that CancerDetector can go beyond distinguishing healthy samples from cancer samples and handle more sophisticated scenarios, such as differentiating HBV carriers from cancer patients. Therefore, using real plasma samples we also demonstrated that the read-based CancerDetector method can more sensitively detect tumor cfDNAs.
-value based CancerLocator method achieved on average a sensitivity of 77.3% with standard deviation 9.4% at the specificity of 100%. Note that there are at least 25 early-stage (Barcelona Clinic Liver Cancer stage A) patients among the 33 liver cancer patients. Testing only on the 25 early-stage cancer patients and healthy/HBV samples, at the specificity of 100% our method can also achieve an average sensitivity of 94.8% with a standard deviation of 3.6%; while CancerLocator obtained a sensitivity of 74.4% with a standard deviation 10.0%. Summarizing the performance comparison using the Area Under Curve (AUC), our method can achieve an AUC of 0.990 averaged over 10 runs with standard deviation 0.004 for all real samples and an average AUC of 0.988 with standard deviation 0.005 for early-stage cancer patients; while CancerLocator obtained a lower average AUC of 0.982 with standard deviation 0.014 for real samples and an average AUC of 0.979 with standard deviation 0.0143 for early-stage cancer patients. We also compared our method with the methylated haplotype load based method (14), using WGBS data of low sequencing coverage on cfDNAs from non-cancer individuals and liver cancer patients. As shown in Figure 5C, the competing method achieved the sensitivity of 71.2 ± 13.0% at the specificity of 100%, which is less accurate and robust than our method (achieved sensitivity of 94.9 ± 2.7% at the specificity of 100%). We also compared with the average-methylation-rate-based method (14) that achieved the sensitivity of 76.5 ± 17.0% at the specificity of 100% and also has lower performance than our method. For details see the Supplementary Materials. We also note that CancerDetector correctly predicted the cfDNA tumor fractions of all eight chronic hepatitis B virus (HBV) samples to be the same range of the normal samples (i.e. close to zero) that are well distinguished from cancer samples. These results demonstrated that CancerDetector can go beyond distinguishing healthy samples from cancer samples and handle more sophisticated scenarios, such as differentiating HBV carriers from cancer patients. Therefore, using real plasma samples we also demonstrated that the read-based CancerDetector method can more sensitively detect tumor cfDNAs.
Figure 5.

Predicted blood tumor fractions for the real data in all 10 runs: (A) average ROC curve with standard deviation bars for CancerDetector, (B) average ROC curve with standard deviation bars for our previous method CancerLocator, (C) average ROC curve with standard deviation bars for the methylated haplotype load based method (14) and (D) the relationship between the tumor size and average blood tumor fraction predicted by CancerDetector.
In general, the predicted tumor fraction correlates well with tumor size. As shown in Figure 5D, among the 26 liver cancer patients with tumor size information, the PCC between the predicted tumor fraction and tumor size is 0.87 (P-value = 7.37e-09). Even after removing the three patients with the largest tumors (size>6cm), we still get a relatively good PCC of 0.42 (P-value = 4.61e–02).
CancerDetector can also be used for monitoring the cancer progression and treatment. We used two cancer liver patients from Chan et al. (8) whose plasma samples were obtained before surgical tumor resection and at multiple time points after the surgery. The first patient survived beyond 12 months, while the second patient died of metastatic disease after the operation (8). As shown in Figure 6, the predicted blood tumor fractions are consistent with the treatment effects: the first patient's tumor fractions quickly fall into the normal range; while those of the second retain relatively high values after the surgery.
Figure 6.

Average predicted blood tumor fractions for longitudinal data of two liver cancer patients before and after tumor resections in all 10 runs. The second patient passed away after surgery.
DISCUSSION
The success of early cancer detection largely relies on (i) the high-quality cancer-specific methylation markers, and (ii) the computational method for the ultra-sensitive detection of tiny amounts of tumor cfDNAs (usually <5%, or even <0.5% in early-stage cancer patients). In this work, we propose a novel method to deconvolute the tumor cfDNA out of total cfDNA at the resolution of individual reads. Compared with traditional cancer detection methods, our method has two advantages in identifying subtle tumor signals: (i) Exploit the pervasive nature of DNA methylation to significantly amplify aberrant cfDNA signals: As demonstrated in Figure 1 and in our experimental results, the joint methylation status (α-value) of multiple CpG sites in a read carries more sensitive tumor signals than the average methylation rate (β-value) of an individual CpG site. Our probabilistic method based on α-value is particularly advantageous when tumor fractions and sequencing coverages are low. (ii) Jointly deconvolute tumor fraction across all markers. Existing methods often focus on detecting tumor signals in one or several tumor markers, not aggregating signals from a large set of markers (9,29,30). Alternatively, our method holds the belief that subtle tumor signals should occur at multiple places in the genome. In practice, if using low-coverage sequencing to profile cfDNA with low tumor fraction, tumor-specific reads may not be observed in all markers (for details see Supplementary Materials). Since our method can detect tumor cfDNA at the read level, we combine all possible signals to provide a robust and sensitive estimate of the tumor fraction. The key considerations of (i) and (ii) promised, as demonstrated, that our method could do well for extremely low tumor fractions (<1%) at low or medium sequencing coverage (5× and 10×). Therefore, our approach holds the potential to largely reduce the cost of cancer detection.
In the field of methylation studies, our method shares some common concepts with previous studies (14,25,26,31–35), but is distinct in several major points. The joint methylation status of individual sequencing reads, often referred to as epialleles (epigenetic alleles), captures the ‘phased information’ of CpG sites, and can represent the methylation haplotype (9,14,32). With the advancement of base-resolution sequencing techniques (such as WGBS), epialleles have recently been studied in several major lines of DNA methylation research (14,25,26,31–35), such as tumor clones and their phylogeny (26,33,34), intratumor heterogeneity (25,35), solid tissue studies (31), and tissue deconvolution of cfDNAs (14). Most of these studies proposed new measures based on epialleles, such as proportion of discordant reads (PDR) (25), Epipolymorphism (31), methylation entropy (32), and methylated haplotype load (MHL) (14). These measures are intrinsically population-averaged metric at the marker level, designed for assessing discordancy, diversity, and co-methylation level of joint methylation states over a pile of reads, not for classifying individual reads. Therefore, it is challenging for those methods to search for tiny tumor signals in cfDNA data with low tumor fraction and sequencing coverage.
Although a number of targeted sequencing assays that use error-suppressing technologies have been developed for detecting point mutations or tissue/tumor-specific DNA methylation in plasma cfDNA samples, our method is fundamentally different from them in two aspects: (i) We address a different challenge. Targeted sequencing assays address the challenge of how to detect extremely low-allele-frequency genetic variant or tissue/tumor-specific DNA methylation alteration in individual hotspot loci that are provided from a small panel, which needs the (methylation-specific) PCR amplification and/or sequencing depth up to 10 000×∼100 000× (9,36–38). However, due to the large genetic/epigenetic landscape of diverse cancer etiologies and the presence of very few tumor DNA fragments in blood at early stages, a small panel may possibly miss many tumor signals. On the contrary, our method aims to estimate an overall value of the cfDNA tumor fraction based on a large marker set, which can be achieved even for low-coverage data. (ii) Sequencing reads are used in different ways. The variant detection algorithms for targeted sequencing data usually analyze the reads that cover individual hotspot loci; while our method scans all reads covering all markers and then aggregates those probabilistically tumor-like reads to derive an overall tumor fraction.
Our method can be further enhanced with increased size and quality of data, in the following ways: (i) Improve the quality and quantity of cancer methylation markers: Due to the limited number of methylation data samples for paired liver cancer and non-cancer samples, in this work, we identified the liver cancer methylation markers using the microarray data of only 49 pairs of liver samples and the low-coverage WGBS data of 24 normal plasma samples. We expect that the growing amount of high-resolution methylation data will significantly improve the quality and quantity of the methylation marker panel. In addition, in principle we can improve the quality of cancer methylation markers by filtering out those markers that could be potentially contributed by different blood cells or different tissues (such as liver, lung, colon, kidney, pancreas, etc). Although this strategy can improve the quality of markers, it reduces the number of markers and therefore the number of reads falling into the marker regions, eventually compromising the diagnostic performance if the sequencing coverage is very low, as confirmed by our results using the low coverage sequencing data in this study. Therefore, there is a tradeoff between the quality and quantity of markers. On the other hand, by identifying and removing patient-specific ‘constitutive markers’, our approach can maximally use all good-quality markers. If given the blood or tissue methylation data of the same patient, our approach shall identify ‘confounding markers’ even more specifically. (ii) Improve the quality of cfDNA bisulfite sequencing data: Although our results have demonstrated the cfDNA methylation can be used for sensitive non-invasive cancer diagnosis, there are still experimental and technical limitations that generally exist for all bisulfite-sequencing data analyses. For example, the bisulfite conversion can degrade more than 45% of DNA (up to 90%) (39). It results in much less input cfDNA for the subsequent experiment, although PCR amplification is adopted for this issue. However, the PCR amplification procedure can inevitably bring in both PCR errors and biases. Therefore, future work shall focus on alleviating these limitations in both experimental and computational procedures. (iii)Transform the tumor fraction estimation into a cancer diagnostic decision: Generally, the higher the tumor fraction 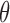 in plasma cfDNAs, the more likely an individual may get cancer. In practice, we should perform the cross validation on a large number of non-cancer samples to build a reliable upper-limit of a non-cancer
in plasma cfDNAs, the more likely an individual may get cancer. In practice, we should perform the cross validation on a large number of non-cancer samples to build a reliable upper-limit of a non-cancer 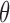 threshold, i.e.,
threshold, i.e., 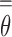 , so that any individual with cfDNA tumor fraction
, so that any individual with cfDNA tumor fraction 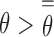 may be predicted as a cancer carrier. In this current work, due to the very limited number of non-cancer plasma samples available, we can only assess the performance of the estimated
may be predicted as a cancer carrier. In this current work, due to the very limited number of non-cancer plasma samples available, we can only assess the performance of the estimated 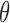 by using ‘ROC AUC’ that does not depend on a specific
by using ‘ROC AUC’ that does not depend on a specific 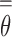 cutoff. However, in future work, when sufficient data of non-cancer plasma samples are available, the threshold
cutoff. However, in future work, when sufficient data of non-cancer plasma samples are available, the threshold 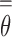 can be determined for making diagnostics decisions.
can be determined for making diagnostics decisions.
DATA AVAILABILITY
The source code of CancerDetector is available online https://zhoulab.dgsom.ucla.edu/pages/CancerDetector.
Supplementary Material
ACKNOWLEDGEMENTS
The authors greatly acknowledge Dr Yuk Ming Dennis Lo and his circulating nucleic acids research group in the Chinese University of Hong Kong for his cfDNA data (8,27).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [NHLBI MAPGEN U01HL108634 to X.J.Z.]; National Science Foundation Graduate Research Fellowship [DGE-1418060 to M.S.]. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. University of California at Los Angeles has a patent pending for CancerDetector.
REFERENCES
- 1. Bettegowda C., Sausen M., Leary R.J., Kinde I., Wang Y., Agrawal N., Bartlett B.R., Wang H., Luber B., Alani R.M. et al. . Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014; 6:224ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Newman A.M., Bratman S. V, To J., Wynne J.F., Eclov N.C.W., Modlin L.A., Liu C.L., Neal J.W., Wakelee H.A., Merritt R.E. et al. . An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014; 20:548–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Newman A.M., Lovejoy A.F., Klass D.M., Kurtz D.M., Chabon J.J., Scherer F., Stehr H., Liu C.L., Bratman S. V, Say C. et al. . Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 2016; 34:547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Burrell R.A., McGranahan N., Bartek J., Swanton C.. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013; 501:338–345. [DOI] [PubMed] [Google Scholar]
- 5. Greenman C., Stephens P., Smith R., Dalgliesh G.L., Hunter C., Bignell G., Davies H., Teague J., Butler A., Stevens C. et al. . Patterns of somatic mutation in human cancer genomes. Nature. 2007; 446:153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schmitt M.W., Prindle M.J., Loeb L.A.. Implications of genetic heterogeneity in cancer. Ann. N. Y. Acad. Sci. 2012; 1267:110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Turner N.C., Reis-Filho J.S.. Genetic heterogeneity and cancer drug resistance. Lancet Oncol. 2012; 13:e178–e185. [DOI] [PubMed] [Google Scholar]
- 8. Chan K.C.A., Jiang P., Chan C.W.M., Sun K., Wong J., Hui E.P., Chan S.L., Chan W.C., Hui D.S.C., Ng S.S.M. et al. . Noninvasive detection of cancer-associated genome-wide hypomethylation and copy number aberrations by plasma DNA bisulfite sequencing. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:18761–18768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lehmann-Werman R., Neiman D., Zemmour H., Moss J., Magenheim J., Vaknin-Dembinsky A., Rubertsson S., Nellgård B., Blennow K., Zetterberg H. et al. . Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:201519286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Baylin S.B., Esteller M., Rountree M.R., Bachman K.E., Schuebel K., Herman J.G.. Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum. Mol. Genet. 2001; 10:687–692. [DOI] [PubMed] [Google Scholar]
- 11. Cheishvili D., Boureau L., Szyf M.. DNA demethylation and invasive cancer: implications for therapeutics. Br. J. Pharmacol. 2015; 172:2705–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Plass C., Pfister S.M., Lindroth A.M., Bogatyrova O., Claus R., Lichter P.. Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat. Rev. Genet. 2013; 14:765–780. [DOI] [PubMed] [Google Scholar]
- 13. Roy D.M., Walsh L.A., Chan T.A.. Driver mutations of cancer epigenomes. Protein Cell. 2014; 5:265–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guo S., Diep D., Plongthongkum N., Fung H.-L., Zhang K., Zhang K.. Identification of methylation haplotype blocks aids in deconvolution of heterogeneous tissue samples and tumor tissue-of-origin mapping from plasma DNA. Nat. Genet. 2017; 49:635–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xu R.-H., Wei W., Krawczyk M., Wang W., Luo H., Flagg K., Yi S., Shi W., Quan Q., Li K. et al. . Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat. Mater. 2017; 16:1155–1161. [DOI] [PubMed] [Google Scholar]
- 16. Kang S., Li Q., Chen Q., Zhou Y., Park S., Lee G., Grimes B., Krysan K., Yu M., Wang W. et al. . CancerLocator: non-invasive cancer diagnosis and tissue-of-origin prediction using methylation profiles of cell-free DNA. Genome Biol. 2017; 18:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cancer Genome Atlas Research Network Weinstein J.N., Collisson E.A., Mills G.B., Shaw K.R.M., Ozenberger B.A., Ellrott K., Shmulevich I., Sander C., Stuart J.M.. The cancer genome atlas Pan-Cancer analysis project. Nat. Genet. 2013; 45:1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Houseman E.A., Christensen B.C., Yeh R.-F., Marsit C.J., Karagas M.R., Wrensch M., Nelson H.H., Wiemels J., Zheng S., Wiencke J.K. et al. . Model-based clustering of DNA methylation array data: a recursive-partitioning algorithm for high-dimensional data arising as a mixture of beta distributions. BMC Bioinformatics. 2008; 9:365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kuan P.F., Wang S., Zhou X., Chu H.. A statistical framework for Illumina DNA methylation arrays. Bioinformatics. 2010; 26:2849–2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang L., Meng J., Liu H., Huang Y.. A nonparametric Bayesian approach for clustering bisulfate-based DNA methylation profiles. BMC Genomics. 2012; 13(Suppl. 6):S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hebestreit K., Dugas M., Klein H.-U.. Detection of significantly differentially methylated regions in targeted bisulfite sequencing data. Bioinformatics. 2013; 29:1647–1653. [DOI] [PubMed] [Google Scholar]
- 22. Wen Y., Chen F., Zhang Q., Zhuang Y., Li Z.. Detection of differentially methylated regions in whole genome bisulfite sequencing data using local Getis-Ord statistics. Bioinformatics. 2016; 32:btw497. [DOI] [PubMed] [Google Scholar]
- 23. Bowman K.O., Shenton L.R.. The beta distribution, moment method, Karl Pearson and RA Fisher. Far East J. Theor. Stat. 2007; 23:133–164. [Google Scholar]
- 24. Shah A., Knowles D.A., Ghahramani Z.. An empirical study of stochastic variational algorithms for the Beta Bernoulli process. Proceedings of the 32nd International Conference on Machine Learning. 2015; 37:1594–1603. [Google Scholar]
- 25. Landau D.A., Clement K., Ziller M.J., Boyle P., Fan J., Gu H., Stevenson K., Sougnez C., Wang L., Li S. et al. . Locally disordered methylation forms the basis of intratumor methylome variation in chronic lymphocytic leukemia. Cancer Cell. 2014; 26:813–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yuan K., Sakoparnig T., Markowetz F., Beerenwinkel N.. BitPhylogeny: a probabilistic framework for reconstructing intra-tumor phylogenies. Genome Biol. 2015; 16:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sun K., Jiang P., Chan K.C.A., Wong J., Cheng Y.K.Y., Liang R.H.S., Chan W., Ma E.S.K., Chan S.L., Cheng S.H. et al. . Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:201508736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Krueger F., Andrews S.R.. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011; 27:1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sharma S. Tumor markers in clinical practice: General principles and guidelines. Indian J. Med. Paediatr. Oncol. 2009; 30:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sturgeon CM, Diamandis EP. Use of tumor markers in clinical practice: quality requirements American Association for Clinical Chemistry. 2009; American Association for Clinical Chemistry. [Google Scholar]
- 31. Landan G., Cohen N.M., Mukamel Z., Bar A., Molchadsky A., Brosh R., Horn-Saban S., Zalcenstein D.A., Goldfinger N., Zundelevich A. et al. . Epigenetic polymorphism and the stochastic formation of differentially methylated regions in normal and cancerous tissues. Nat. Genet. 2012; 44:1207–1214. [DOI] [PubMed] [Google Scholar]
- 32. Li S., Garrett-Bakelman F., Perl A.E., Luger S.M., Zhang C., To B.L., Lewis I.D., Brown A.L., D’Andrea R.J., Ross M.E. et al. . Dynamic evolution of clonal epialleles revealed by methclone. Genome Biol. 2014; 15:472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li S., Garrett-Bakelman F.E., Chung S.S., Sanders M.A., Hricik T., Rapaport F., Patel J., Dillon R., Vijay P., Brown A.L. et al. . Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat. Med. 2016; 22:792–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Swanton C., Beck S.. Epigenetic noise fuels cancer evolution. Cancer Cell. 2014; 26:775–776. [DOI] [PubMed] [Google Scholar]
- 35. Zheng X., Zhao Q., Wu H.-J., Li W., Wang H., Meyer C.A., Qin Q., Xu H., Zang C., Jiang P. et al. . MethylPurify: tumor purity deconvolution and differential methylation detection from single tumor DNA methylomes. Genome Biol. 2014; 15:419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Herman J.G., Graff J.R., Myöhänen S., Nelkin B.D., Baylin S.B.. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:9821–9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Distler J. Quantification of methylated DNA by HeavyMethyl duplex PCR. Methods Mol. Biol. 2009; 507:339–346. [DOI] [PubMed] [Google Scholar]
- 38. Zhang Q., Hu G., Yang Q., Dong R., Xie X., Ma D., Shen K., Kong B.. A multiplex methylation-specific PCR assay for the detection of early-stage ovarian cancer using cell-free serum DNA. Gynecol. Oncol. 2013; 130:132–139. [DOI] [PubMed] [Google Scholar]
- 39. Leontiou C.A., Hadjidaniel M.D., Mina P., Antoniou P., Ioannides M., Patsalis P.C.. Bisulfite conversion of DNA: performance comparison of different kits and methylation quantitation of epigenetic biomarkers that have the potential to be used in non-invasive prenatal testing. PLoS One. 2015; 10:e0135058. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The source code of CancerDetector is available online https://zhoulab.dgsom.ucla.edu/pages/CancerDetector.