


Abstract
With expanding applications of next-generation sequencing in medical genetics, increasing computational methods are being developed to predict the pathogenicity of missense variants. Selecting optimal methods can accelerate the identification of candidate genes. However, the performances of different computational methods under various conditions have not been completely evaluated. Here, we compared 12 performance measures of 23 methods based on three independent benchmark datasets: (i) clinical variants from the ClinVar database related to genetic diseases, (ii) somatic variants from the IARC TP53 and ICGC databases related to human cancers and (iii) experimentally evaluated PPARG variants. Some methods showed different performances under different conditions, suggesting that they were not always applicable for different conditions. Furthermore, the specificities were lower than the sensitivities for most methods (especially, for the experimentally evaluated benchmark datasets), suggesting that more rigorous cutoff values are necessary to distinguish pathogenic variants. Furthermore, REVEL, VEST3 and the combination of both methods (i.e. ReVe) showed the best overall performances with all the benchmark data. Finally, we evaluated the performances of these methods with de novo mutations, finding that ReVe consistently showed the best performance. We have summarized the performances of different methods under various conditions, providing tentative guidance for optimal tool selection.
INTRODUCTION
In recent decades, next-generation sequencing (NGS) represented by whole-exome sequencing (WES) rapidly promoted the understanding of the genetic mechanisms of human diseases (1,2). Numerous sequence variants in the human genome can be detected by WES, most of which are missense variants that cause amino acid changes in proteins (3,4). However, only a small subset of missense variants may be involved in human diseases, including cancers, Mendelian diseases, and complex and undiagnosed diseases (5). Experimental validation of many missense variants is infeasible because it would waste tremendous manpower and resources. To address these limitations, a growing number of in silico computational methods have been developed based on sequence homology, protein structure and evolutionary conservation (6–26). In general, these methods can be classified into three kinds: (i) function-prediction methods that predict the likelihood of a given missense variant causing pathogenic changes in protein function, (ii) conservation methods that use multiple alignments to measure the degree of conservation at a given nucleotide site and (iii) ensemble methods that integrate information from multiple-component methods.
These computational methods have been widely used to predict potentially deleterious variants in human diseases (27,28) and were described in our previous studies (29,30). To facilitate the interpretation of human genomic variants (31,32), we integrated the functional consequences of different computational methods, allele frequencies, and other genetic and clinical information related to all possible coding variants into a database, referred to as VarCards (33). However, it is unclear how the performances of these computational methods vary under different conditions. Although some previous studies compared the performances of existing computational methods (34–39), limited benchmark datasets and no experimentally evaluated data were used in these comparative studies. In addition, these studies mainly focused on measuring the area under the curve (AUC) with receiver operating characteristic (ROC) curves, and other important measures (such as the accuracy at 95% sensitivity or specificity) were not fully evaluated. For example, clinicians and geneticists may adopt computational methods to estimate the pathogenicity of missense variants in genetic counseling for known disease-causing genes (40), which would be expected to distinguish pathogenic variants with a high sensitivity (a true positive rate [TPR] over 95%) (25). Furthermore, some recently developed tools, such as REVEL (23), VEST3 (16) and M-CAP (25), were not fully evaluated in previous studies. Therefore, a more comprehensive and systematic analysis of pathogenicity-computation methods is highly needed to meet the demands of different users and assist them in selecting appropriate methods.
We did not involve in developing or investing (scientifically or otherwise) in any of pathogenicity-computation methods, hence we independently evaluated 12 performance measures of 23 methods based on three classes of benchmark datasets. These methods showed different performances using different data sources and under different conditions. REVEL, VEST3 and the combination of both tools showed the best overall performance under most conditions. Based on our findings, clinicians and researchers can choose appropriate methods or use several tools simultaneously to interpret the pathogenicity of missense variants.
MATERIALS AND METHODS
Pathogenicity-computation methods
We compared 23 pathogenicity-computation methods, including (i) 10 function-prediction methods: FATHMM (26), fitCons (19), LRT (8), MutationAssessor (14), MutationTaster (13), PolyPhen2-HDIV (11), PolyPhen2-HVAR (11), PROVEAN (15), SIFT (10) and VEST3 (16); (ii) four conservation methods: GERP++ (12), phastCons (6), phyloP (7) and SiPhy (9); and (iii) nine ensemble methods: CADD (17), DANN (21), Eigen (24), FATHMM-MKL (22), GenoCanyon (20), M-CAP (25), MetaLR (18), MetaSVM (18) and REVEL (23) (Supplementary Table S1). The predicted pathogenicity scores of the 23 methods were directly downloaded from the dbNSFP database v3.3 (41). These scores have been widely used in medical genetics for distinguishing deleterious and tolerable missense variants. All of the prediction scores and other genetic and clinical information were integrated into the online database, VarCards (33), which we recently developed. The cutoff values used for distinguishing deleterious missense variants were based on the dbNSFP database (41), ANNOVAR (42) or the original studies (Supplementary Table S1).
Benchmark datasets of missense variants
Three independent data sources were used as benchmark datasets to compare the performances of the 23 computational methods. Both deleterious missense variants and benign missense variants were included in each benchmark dataset (Supplementary Table S2). The first benchmark dataset was sourced from the ClinVar database (43), which compiles clinically observed genetic variants (https://www.ncbi.nlm.nih.gov/clinvar/). As recommended in the American College of Medical Genetics and Genomics (ACMG) guidelines (40), terms such as ‘pathogenic’, ‘likely pathogenic’, ‘benign’, ‘likely benign’ and ‘uncertain significance’ were adopted to describe the variants in ClinVar database. We only selected ‘pathogenic’ and ‘benign’ missense variants deposited in the ClinVar database after 4 March 2015 to ensure the accuracy of benchmark dataset and to avoid any overlaps between our tested benchmark data and the training data used for the 23 computational methods.
As TP53 is the most commonly mutated gene in human cancers, the second dataset was downloaded from the IARC TP53 database (44,45), which contains various types of clinical and genetic data on human TP53 variants related to different cancers (http://p53.iarc.fr/). To our knowledge, IARC TP53 database includes the maximum number of somatic missense variants whose pathogenicity have been characterized. The defined functional and non-functional somatic missense variants detected in patients with various cancers in the IARC TP53 database were used as benchmark dataset for further analysis. Non-functional variants in the IARC TP53 database are variants that significantly changed the expression level of the TP53 proteins, whereas functional variants did not. The term of ‘non-functional variant’ in the IARC TP53 database is analogous to the pathogenicity measure of ‘pathogenic’ reported in ClinVar. The COSMIC database included somatic variants across several cancers; however, functional effects or clinical significance of all collected variants were unknown. We then sourced missense variants from the ICGC database (http://icgc.org/), and only 811 somatic missense variants with clinical significance (pathogenic/likely pathogenic and benign/likely benign mutations) were collected for further analysis.
The third benchmark dataset was sourced from a large-scale experimentally evaluated study (46), wherein a complementary DNA library was constructed that consisted of all possible amino acid substitutions in the peroxisome proliferator activated receptor γ (PPARG) gene. In this study, a pooled functional assay was developed and experimentally evaluated with all possible protein variants. The pathogenicity for any missense variants of the PPARG gene was sourced from the Missense InTerpretation by Experimental Response (MITER) database (http://miter.broadinstitute.org/).
Measures used for performance evaluation
We evaluated the performances of the 23 computational methods based on following 12 criteria: (i) The positive predictive value (PPV) represents the conditional probability that deleterious variants in the benchmark data are correctly classified as deleterious variants by the computational method. (ii) The negative predictive value (NPV) represents the conditional probability that benign variants in the benchmark data are correctly classified as benign variants by the computational method. (iii) The false negative rate (FNR) represents the proportion of deleterious variants that are incorrectly predicted to be benign variants. (iv) The sensitivity (also referred to as the TPR) represents the proportion of deleterious variants in the benchmark data that are correctly predicted to be deleterious variants by the computational method. The FNR and sensitivity are paired measures whose sum = 100%. (v) The false positive rate (FPR) represents the proportion of benign variants in the benchmark data that are incorrectly predicted to be deleterious variants by the computational method. (vi) The specificity (also referred to as the true negative rate) represents the proportion of benign variants in the benchmark data that are correctly predicted to be benign variants by the computational method. The FPR and specificity are paired measures whose sum = 100%. (vii) The accuracy represents the proportion of benign variants and deleterious variants in the benchmark data that are correctly predicted to be benign variants and deleterious variants, respectively. (viii) The Mathew correlation coefficient (MCC) represents the correlation coefficient between the observed and predicted classifications, within a range of −1 to 1. A coefficient of 1 indicates a perfect prediction, 0 indicates a random prediction and −1 indicates complete disagreement between the prediction and the true classification. (ix) The ROC curve reflects the sensitivity and specificity at different thresholds for each computational method. (x) For each ROC, the AUC is a single scalar value, which reduces the complexity of the ROC curve. Generally, the higher the AUC, the better the performance of computational method. In this study, the ‘pROC’ package (47) was used to plot the ROC curves. (xi) The high-sensitivity regional AUC (hser-AUC) is defined as the area under the ROC curve corresponding to high sensitivity (TPR > 95%). (xii) The high-specificity regional AUC (hspr-AUC) is defined as the area under the ROC curve corresponding to high specificity (FPR < 5%). The hser-AUC and hspr-AUC were evaluated to meet the needs of some users who expect to distinguish deleterious variants with a high sensitivity or specificity. Some of the above measures are derived from the parameters of true positive (TP), true negative (TN), false positive (FP) and false negative (FN), as shown below.
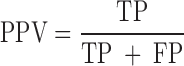 |
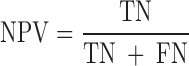 |
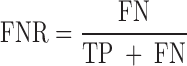 |
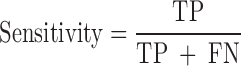 |
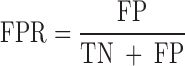 |
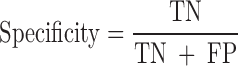 |
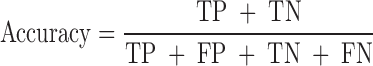 |
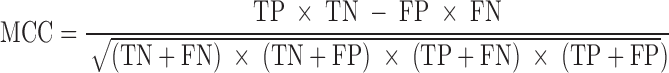 |
Combination of REVEL and VEST3
Because the REVEL and VEST3 methods showed the best overall performances with all three sets of benchmark data, we combined the predictive scores of both methods and named the new method as ReVe. The predictive score of both the REVEL and VEST3 methods represented the probability that a missense variant was classified as pathogenic. Because the distribution of the predicted scores of all possible missense variants in the whole exome of the two methods are obviously different, we cannot combine these methods simply by calculating the average predictive score. To combine the two computational methods, we separately ranked the predictive scores of all possible missense variants in the whole exome obtained with the REVEL and VEST3 methods, and calculated the percentile value of each missense variant in each method. The individual percentile values of the REVEL and VEST3 methods were uniformly distributed. Then, for each missense variant, we calculated the mean percentile values based on both of these two methods. Finally, we ranked the mean values for all missense variants in the whole exome. To facilitate interpretation, we present the final predictive scores of ReVe as percentiles that reflect the relative rank of pathogenicity for missense variants, with the lowest score (i.e. 0.00) being the most benign variant and the highest score (i.e. 1.00) being the most deleterious variant.
De novo mutations (DNMs) from the Simons Simplex Collection (SSC)
DNMs identified in 2508 autism spectrum disorder (ASD) patients and 1911 unaffected siblings were sourced from the SSC (48,49) and had been previously catalogued in the NP de novo database that we developed (30). The VarCards (33) and ANNOVAR (42) were used to annotate these DNMs as done in our previous studies (30,33), including functional effects at the transcription level (stop-gain, stop-loss, splicing, frameshift, non-frameshift, missense, synonymous) and functional consequences of the 23 computational methods and ReVe. We then compared differences in the mutation rates for predicted deleterious missense variants, as well as benign missense variants by the computational methods in the ASD and sibling groups.
RESULTS
We performed an extensive investigation of the core model, training data, testing data and updated information of the 23 computational methods (Supplementary Table S1), and found that most of these computational methods trained their models based on variants from HGMD and UniProt, and the genomic differences among mammals, and that none of these methods trained their model using variants from the ClinVar, TP53 or MITER databases. In addition, most of these methods (except MutationAssessor, VEST3, phastCons and PhyloP) have not been updated since 4 March 2015. To avoid potential overlaps between benchmark data in this study and training data in the computational methods, only the ‘pathogenic’ and ‘benign’ missense variants recorded after 4 March 2015 in the ClinVar database were used for further analysis. Furthermore, although the benchmark data we used in present study and the training data of computational methods were sourced from different database, potential circularity may exist, as previously reported (35). We therefore removed all variants from ClinVar benchmark data that overlapped with the training data of the computational methods including variants from HGMD and UniProt, finally resulting in 2098 pathogenic variants and 2782 benign variants (Supplementary Table S2).
Performance evaluations based on germline variants in human genetic diseases
Based on the clinically observed genetic variants from the ClinVar database (37), we noted that the predicted values of 2141 (43.87%), 995 (20.39%), 357 (7.32%), 299 (6.13%) and 299 (6.13%) of the 4880 testing variants were not available for the M-CAP, LRT, MutationAssessor, Eigen and fitCons methods, respectively. The evaluation of 12 performance measures for all the 23 methods is summarized in Table 1. We found that the PPV ranged from 51.61% to 83.03% (median, 64.47%), and four methods (MetaSVM, REVEL, M-CAP and MetaLR) had a PPV > 80% (Supplementary Figure S1a). The NPV values of the 23 methods were generally higher than those of the PPV values, ranging from 60.64% to 94.93% (median, 87.07%), and seven methods (MutationTaster, FATHMM-MKL, CADD, Eigen, GERP++, VEST3 and PolyPhen2-HDIV) had NPV values > 90% (Supplementary Figure S2a). In addition, we found that the specificities (100% – the FPR) ranged from 34.80% to 89.95% (median, 64.75%, Supplementary Figures S3 and S4) and that the sensitivities (100% – the FNR) ranged from 50.52% to 96.07% (median, 88.24%, Supplementary Figures S5 and S6). The overall specificities were much lower than the sensitivities, suggesting that some predicted deleterious variants were actually benign variants and that some methods require more rigorous cutoff values when distinguishing deleterious variants. We noted that only two methods (REVEL and VEST3) showed >80% sensitivity and specificity. Generally, only two methods [REVEL (84.43%) and VEST3 (84.27%)] presented an accuracy >80% (Supplementary Figure S7a). Furthermore, we found that REVEL and VEST3 consistently showed the highest MCC scores (Supplementary Figure S8a).
Table 1.
Performance evaluation based on ClinVar benchmark data
| Methods | Missing | PPV (%) | NPV (%) | Specificity (%) | FPR (%) | Sensitivity (%) | FNR (%) | Accuracy (%) | MCC | AUC | hser-AUC | hspr-AUC |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Class one: function prediction methods | ||||||||||||
| FATHMM | 195 | 63.48 | 67.35 | 77.84 | 22.16 | 50.52 | 49.48 | 66.02 | 0.296 | 0.694 | 0.536 | 0.586 |
| fitCons | 299 | 51.61 | 64.47 | 62.23 | 37.77 | 54.01 | 45.99 | 58.72 | 0.162 | 0.611 | 0.505 | 0.509 |
| LRT | 995 | 72.28 | 87.52 | 67.63 | 32.37 | 89.75 | 10.25 | 78.35 | 0.586 | 0.789 | 0.631 | 0.521 |
| MutationAssessor | 357 | 68.42 | 83.15 | 69.66 | 30.34 | 82.32 | 17.68 | 75.28 | 0.518 | 0.850 | 0.584 | 0.668 |
| MutationTaster | 34 | 62.01 | 94.93 | 55.56 | 44.44 | 96.07 | 3.93 | 72.99 | 0.542 | 0.610 | 0.560 | 0.504 |
| PolyPhen2-HDIV | 52 | 61.84 | 90.26 | 56.58 | 43.42 | 92.02 | 7.98 | 71.93 | 0.503 | 0.839 | 0.644 | 0.551 |
| PolyPhen2-HVAR | 52 | 68.50 | 88.47 | 68.97 | 31.03 | 88.24 | 11.76 | 77.32 | 0.571 | 0.865 | 0.657 | 0.627 |
| PROVEAN | 134 | 73.03 | 83.45 | 76.99 | 23.01 | 80.32 | 19.68 | 78.45 | 0.569 | 0.858 | 0.617 | 0.628 |
| SIFT | 190 | 67.11 | 86.78 | 67.90 | 32.10 | 86.36 | 13.64 | 75.86 | 0.541 | 0.860 | 0.633 | 0.597 |
| VEST3 | 16 | 77.95 | 90.33 | 81.03 | 18.97 | 88.55 | 11.45 | 84.27 | 0.689 | 0.929 | 0.729 | 0.756 |
| Class two: conservation methods | ||||||||||||
| GERP++ | 16 | 52.48 | 91.12 | 34.80 | 65.20 | 95.50 | 4.50 | 60.90 | 0.364 | 0.739 | 0.598 | 0.507 |
| phastCons | 0 | 63.60 | 87.58 | 61.86 | 38.14 | 88.37 | 11.63 | 73.26 | 0.507 | 0.767 | 0.642 | 0.517 |
| phyloP | 0 | 61.13 | 88.96 | 56.51 | 43.49 | 90.71 | 9.29 | 71.21 | 0.486 | 0.848 | 0.619 | 0.594 |
| SiPhy | 17 | 63.65 | 82.12 | 64.75 | 35.25 | 81.40 | 18.60 | 71.93 | 0.460 | 0.776 | 0.625 | 0.515 |
| Class three: ensemble methods | ||||||||||||
| CADD | 1 | 63.96 | 93.19 | 59.99 | 40.01 | 94.18 | 5.82 | 74.69 | 0.556 | 0.877 | 0.683 | 0.584 |
| DANN | 1 | 64.47 | 83.32 | 65.71 | 34.29 | 82.55 | 17.45 | 72.95 | 0.480 | 0.807 | 0.649 | 0.544 |
| Eigen | 299 | 65.05 | 91.97 | 62.88 | 37.12 | 92.64 | 7.36 | 75.59 | 0.563 | 0.871 | 0.669 | 0.595 |
| FATHMM-MKL | 1 | 56.54 | 93.70 | 44.36 | 55.64 | 96.04 | 3.96 | 66.57 | 0.451 | 0.822 | 0.663 | 0.552 |
| GenoCanyon | 0 | 56.10 | 80.99 | 50.22 | 49.78 | 84.37 | 15.63 | 64.90 | 0.358 | 0.683 | 0.561 | 0.509 |
| M-CAP | 2141 | 80.51 | 60.64 | 35.00 | 65.00 | 92.20 | 7.80 | 77.58 | 0.335 | 0.814 | 0.562 | 0.622 |
| MetaLR | 31 | 80.28 | 75.98 | 88.21 | 11.79 | 63.24 | 36.76 | 77.44 | 0.538 | 0.874 | 0.703 | 0.670 |
| MetaSVM | 31 | 83.03 | 77.09 | 89.95 | 10.05 | 64.77 | 35.23 | 79.09 | 0.574 | 0.858 | 0.547 | 0.693 |
| REVEL | 30 | 81.12 | 87.07 | 85.27 | 14.73 | 83.33 | 16.67 | 84.43 | 0.684 | 0.920 | 0.689 | 0.756 |
AUC, area under the curve; FNR, false negative rate; FPR, false positive rate; hser-AUC, high-sensitivity regional area under the curve; hspr-AUC, high-specificity regional area under the curve; MCC, Mathew correlation coefficient; NPV, negative predictive value; PPV, positive predictive value. ClinVar is an open database that aggregates clinically observed genetic variants. We obtained 4880 missense variants from this database, including 2098 pathogenic variants and 2782 benign variants.
We then evaluated the performances of 23 computational methods by measuring their AUCs. Compared to the other methods, the VEST3 (AUC = 0.929) and REVEL (AUC = 0.920) methods showed the best overall performance, followed by CADD (AUC = 0.877), MetaLR (AUC = 0.874) and Eigen (AUC = 0.871) (Table 1 and Figure 1A). The AUCs of other computation methods ranged from 0.610 to 0.865. Popularly used methods, such as SIFT (AUC = 0.860), PolyPhen2-HDIV (AUC = 0.839) and PolyPhen2-HVAR (AUC = 0.865) performed medially, but were superior to conservation-only methods. Given that clinicians and geneticists sometimes require that computational methods present with a high accuracy at a high sensitivity or specificity (typically >95%), we further compared the hser-AUC and hspr-AUC values, which were important for genetic testing (25), but were ignored in previous comparative studies (34–39). As a result, we found that VEST3 (0.729), MetaLR (0.703), REVEL (0.689) and CADD (0.683) showed the best performances with hser-AUC values > 0.68 (Table 1 and Figure 1B). In addition, REVEL (0.756), VEST3 (0.756) and MetaSVM (0.693) showed the best performances in terms of the hspr-AUC (Table 1 and Figure 1C). It is worth noting that VEST3 and REVEL showed better performances for all AUC, hser-AUC and hspr-AUC measures. Both methods have a FPR < 30% at a TPR of 95% (Figure 1B) and a TPR close to 70% at a FPR of 5% (Figure 1C).
Figure 1.

Overall performance of the computational methods with the three sets of benchmark data. The AUC, hser-AUC and hspr-AUC of all computational methods are shown, based on germline variants of human genetic diseases from the ClinVar database (A–C), somatic variants of human cancers from the IARC TP53 database (D–F) and experimentally validated PPARG variants (G–I). The AUC, hser-AUC and hspr-AUC values for each computational method are shown in the figures. The solid lines represent function-prediction methods, the dashed lines represent conservation methods and the dotted lines represent ensemble methods. The performance measures of AUC, hser-AUC and hspr-AUC does not rely on the cutoff values. This figure is online available interactively at http://159.226.67.237/sun/roc/.
Most pathogenic variants of human genetic disease were extremely rare, we therefore characterized the allele frequency (AF) of ClinVar benchmark data and found that over 99% of pathogenic variants have AFs < 0.1% and over 80% of them were not observed in GnomAD database. Therefore, our results can provide users guidance for distinguishing rare pathogenic variants.
Performance evaluations based on somatic variants of human cancers
We then collected numerous function-defined somatic missense variants from the IARC TP53 database (44,45), which comprises TP53 gene variations related to different human cancers. This collection contained in 477 non-functional missense variants and 537 functional missense variants (Supplementary Table S2). The performance evaluation of the 23 methods is summarized in Table 2. Generally, most of the 23 methods presented higher NPVs (ranging from 64.66% to 100%, median value of 90.78%, Supplementary Figure S2b) and sensitivities (ranging from 50.73% to 100%, median value of 94.76%, Supplementary Figure S5b), but lower PPVs (ranging from 47.04% to 76.64%, median value of 63.71%, Supplementary Figure S1b) and specificity (ranging from 0.00% to 80.07%, median value of 51.96%, Supplementary Figure S3b). These data suggested that some computational methods need to employ more rigorous cutoff values for predicting deleterious somatic missense mutations. In general, the PROVEAN, SIFT and VEST3 methods presented the highest accuracies (Supplementary Figure S7b) and MCC scores (Supplementary Figure S8b).
Table 2.
Performance evaluation based on TP53 benchmark data
| Methods | Missing | PPV (%) | NPV (%) | Specificity (%) | FPR (%) | Sensitivity (%) | FNR (%) | Accuracy (%) | MCC | AUC | hser-AUC | hspr-AUC |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Class one: function prediction methods | ||||||||||||
| FATHMM | 0 | 47.04 | NA | 0.00 | 100.00 | 100.00 | 0.00 | 47.04 | NA | 0.877 | 0.612 | 0.638 |
| fitCons | 0 | 47.04 | NA | 0.00 | 100.00 | 100.00 | 0.00 | 47.04 | NA | 0.521 | 0.521 | 0.501 |
| LRT | 29 | 70.38 | 82.73 | 66.67 | 33.33 | 85.05 | 14.95 | 75.53 | 0.524 | 0.731 | 0.556 | 0.513 |
| MutationAssessor | 1 | 64.49 | 92.56 | 53.36 | 46.64 | 95.18 | 4.82 | 73.05 | 0.526 | 0.880 | 0.634 | 0.674 |
| MutationTaster | 0 | 60.92 | 90.81 | 46.00 | 54.00 | 94.76 | 5.24 | 68.93 | 0.459 | 0.568 | 0.538 | 0.502 |
| PolyPhen2-HDIV | 0 | 58.48 | 93.30 | 38.92 | 61.08 | 96.86 | 3.14 | 66.17 | 0.430 | 0.865 | 0.655 | 0.556 |
| PolyPhen2-HVAR | 0 | 61.01 | 92.45 | 45.62 | 54.38 | 95.81 | 4.19 | 69.23 | 0.471 | 0.883 | 0.665 | 0.595 |
| PROVEAN | 0 | 76.64 | 87.77 | 76.16 | 23.84 | 88.05 | 11.95 | 81.76 | 0.643 | 0.898 | 0.688 | 0.666 |
| SIFT | 0 | 67.68 | 90.78 | 60.52 | 39.48 | 93.08 | 6.92 | 75.84 | 0.560 | 0.879 | 0.691 | 0.606 |
| VEST3 | 0 | 65.61 | 92.86 | 55.68 | 44.32 | 95.18 | 4.82 | 74.26 | 0.545 | 0.912 | 0.663 | 0.727 |
| Class two: conservation methods | ||||||||||||
| GERP++ | 0 | 57.67 | 81.23 | 41.90 | 58.10 | 89.10 | 10.90 | 64.10 | 0.347 | 0.732 | 0.577 | 0.541 |
| phastCons | 0 | 67.43 | 78.92 | 65.55 | 34.45 | 80.29 | 19.71 | 72.49 | 0.461 | 0.751 | 0.591 | 0.517 |
| phyloP | 0 | 64.73 | 78.69 | 60.52 | 39.48 | 81.55 | 18.45 | 70.41 | 0.427 | 0.802 | 0.561 | 0.584 |
| SiPhy | 4 | 65.45 | 66.84 | 71.48 | 28.52 | 60.38 | 39.62 | 66.24 | 0.321 | 0.731 | 0.537 | 0.531 |
| Class three: ensemble methods | ||||||||||||
| CADD | 0 | 63.71 | 92.08 | 51.96 | 48.04 | 94.97 | 5.03 | 72.19 | 0.512 | 0.841 | 0.679 | 0.577 |
| DANN | 0 | 64.78 | 76.85 | 61.82 | 38.18 | 79.04 | 20.96 | 69.92 | 0.412 | 0.752 | 0.638 | 0.520 |
| Eigen | 0 | 69.14 | 85.78 | 65.18 | 34.82 | 87.84 | 12.16 | 75.84 | 0.540 | 0.849 | 0.632 | 0.618 |
| FATHMM-MKL | 0 | 57.54 | 84.86 | 39.66 | 60.34 | 92.03 | 7.97 | 64.30 | 0.367 | 0.804 | 0.601 | 0.539 |
| GenoCanyon | 0 | 69.34 | 64.66 | 80.07 | 19.93 | 50.73 | 49.27 | 66.27 | 0.324 | 0.679 | 0.501 | 0.520 |
| M-CAP | 11 | 47.49 | 100.00 | 0.95 | 99.05 | 100.00 | 0.00 | 47.76 | 0.067 | 0.803 | 0.614 | 0.560 |
| MetaLR | 0 | 47.94 | 100.00 | 3.54 | 96.46 | 100.00 | 0.00 | 48.92 | 0.130 | 0.898 | 0.628 | 0.694 |
| MetaSVM | 0 | 48.13 | 100.00 | 4.28 | 95.72 | 100.00 | 0.00 | 49.31 | 0.144 | 0.578 | 0.562 | 0.562 |
| REVEL | 0 | 56.20 | 94.54 | 32.22 | 67.78 | 97.90 | 2.10 | 63.12 | 0.391 | 0.901 | 0.663 | 0.664 |
AUC, area under the curve; FNR, false negative rate; FPR, false positive rate; hser-AUC, high-sensitivity regional area under the curve; hspr-AUC, high-specificity regional area under the curve; MCC, Mathew correlation coefficient; NA, not available; NPV, negative predictive value; PPV, positive predictive value. The IARC TP53 Database compiles TP53 mutation data that have been reported in the peer-reviewed literature. We obtained 1014 somatic missense mutations that have been reported in this database, including 477 non-functional mutations and 537 functional mutations.
We found that two methods had AUCs > 0.90 (VEST3 = 0.912, REVEL = 0.901) and that seven methods (PROVEAN, MetaLR, PolyPhen2-HVAR, MutationAssessor, SIFT, FATHMM, PolyPhen2-HDIV) had AUCs > 0.85 (Figure 1D and Table 2). We note that none of these nine methods with high AUCs were conservation-only methods. In addition, we found that the SIFT (0.691), PROVEAN (0.688), CADD (0.679), PolyPhen2-HVAR (0.665), VEST3 (0.663), REVEL (0.663) and PolyPhen2-HDIV (0.655) methods had relative hser-AUC values > 0.65 (Figure 1E and Table 2). Furthermore, the VEST3 (0.727), MetaLR (0.694), MutationAssessor (0.674), PROVEAN (0.666) and REVEL (0.664) methods had relative hspr-AUCs > 0.65 (Figure 1F and Table 2). Together, only three methods (VEST3, REVEL and PROVEAN) showed both hser-AUCs and hspr-AUCs > 0.65, based on the TP53 benchmark data.
To further evaluate their performance using somatic variants, we collected missense variants from the ICGC database. The functional effects of most of the collected variants in the ICGC database were unknown, only 763 missense variants were classified into ‘pathogenic/likely pathogenic mutations’ and 48 variants were classified into ‘benign/likely benign mutations’. Based on these variants, we re-evaluated the performances of the 23 computational methods and found that most showed similar performances, based on the IARC TP53 benchmark data (Supplementary Table S3). The REVEL (AUC = 0.896) and VEST3 (AUC = 0.890) methods once again exhibited the best overall performances.
Performance evaluations based on experimentally validated variants
To further evaluate the performance of these 23 methods, we collected benchmark data containing 147 pathogenic variants and 2386 benign variants in the PPARG gene that were experimentally validated in a pooled functional assay (46) (Supplementary Table S2). Unlike ClinVar datasets which were directly submitted by various researchers, all entries in the PPARG dataset have been experimentally validated and fewer false positive variants exist in the PPARG datasets than in the ClinVar datasets. We found that most methods showed relatively high NPVs (ranging from 94.20% to 100%, median value of 98.92%, Supplementary Figure S2c), FPRs (ranging from 33.29% to 90.65%, median value of 70.52%, Supplementary Figure S4c), and sensitivities (ranging from 65.31% to 100%, median value of 96.6%, Supplementary Figure S5c), but relatively low PPVs (ranging from 6.16% to 13.63%, media value of 7.62%, Supplementary Figure S1c), specificities (ranging from 9.35% to 66.71%, median value of 29.49%, Supplementary Figure S3c), and FNRs (ranging from 0.00% to 34.69%, median value of 3.4%, Supplementary Figure S6c) (Table 3). Similar to the above two benchmark datasets, these results suggested that most deleterious variants predicted by some computational methods were actually benign variants. In addition, we found that the fitCons, MutationAssessor, PROVEAN and FATHMM methods had the highest accuracies (Supplementary Figure S7c) and that the PROVEAN, MutationAssessor, REVEL, SIFT and VEST3 methods had the highest MCC scores (Supplementary Figure S8c).
Table 3.
Performance evaluation based on PPARG benchmark data
| Methods | Missing | PPV (%) | NPV (%) | Specificity (%) | FPR (%) | Sensitivity (%) | FNR (%) | Accuracy (%) | MCC | AUC | hser-AUC | hspr-AUC |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Class one: function prediction methods | ||||||||||||
| FATHMM | 0 | 7.43 | 95.89 | 49.87 | 50.13 | 65.31 | 34.69 | 50.77 | 0.071 | 0.700 | 0.626 | 0.584 |
| fitCons | 0 | NA | 94.20 | 100.00 | 0.00 | 0.00 | 100.00 | 94.20 | NA | 0.539 | 0.550 | 0.512 |
| LRT | 0 | 7.84 | 99.44 | 29.59 | 70.41 | 97.28 | 2.72 | 33.52 | 0.140 | 0.642 | 0.505 | 0.623 |
| MutationAssessor | 7 | 13.63 | 98.63 | 66.71 | 33.29 | 85.03 | 14.97 | 67.78 | 0.252 | 0.852 | 0.625 | 0.599 |
| MutationTaster | 0 | 6.37 | 100.00 | 9.47 | 90.53 | 100.00 | 0.00 | 14.73 | 0.078 | 0.580 | 0.580 | 0.502 |
| PolyPhen2-HDIV | 0 | 7.78 | 99.56 | 28.46 | 71.54 | 97.96 | 2.04 | 32.49 | 0.139 | 0.755 | 0.630 | 0.523 |
| PolyPhen2-HVAR | 0 | 8.16 | 98.83 | 35.41 | 64.59 | 93.20 | 6.80 | 38.77 | 0.141 | 0.809 | 0.617 | 0.577 |
| PROVEAN | 0 | 13.47 | 98.92 | 65.00 | 35.00 | 88.44 | 11.56 | 66.36 | 0.257 | 0.855 | 0.654 | 0.593 |
| SIFT | 0 | 8.95 | 99.57 | 39.02 | 60.98 | 97.28 | 2.72 | 42.40 | 0.176 | 0.827 | 0.658 | 0.538 |
| VEST3 | 0 | 8.56 | 100.00 | 34.16 | 65.84 | 100.00 | 0.00 | 37.98 | 0.171 | 0.880 | 0.719 | 0.646 |
| Class two: conservation methods | ||||||||||||
| GERP++ | 0 | 6.16 | 97.81 | 9.35 | 90.65 | 96.60 | 3.40 | 14.41 | 0.049 | 0.596 | 0.533 | 0.533 |
| phastCons | 0 | 6.85 | 98.92 | 19.11 | 80.89 | 96.60 | 3.40 | 23.61 | 0.095 | 0.580 | 0.565 | 0.502 |
| phyloP | 0 | 6.69 | 98.09 | 19.36 | 80.64 | 93.88 | 6.12 | 23.69 | 0.080 | 0.744 | 0.525 | 0.538 |
| SiPhy | 0 | 6.95 | 98.09 | 23.64 | 76.36 | 92.52 | 7.48 | 27.64 | 0.090 | 0.618 | 0.568 | 0.568 |
| Class three: ensemble methods | ||||||||||||
| CADD | 0 | 8.02 | 100.00 | 29.38 | 70.62 | 100.00 | 0.00 | 33.48 | 0.154 | 0.773 | 0.692 | 0.531 |
| DANN | 0 | 7.30 | 97.65 | 31.31 | 68.69 | 87.76 | 12.24 | 34.58 | 0.097 | 0.666 | 0.609 | 0.519 |
| Eigen | 0 | 7.02 | 99.38 | 20.03 | 79.97 | 97.96 | 2.04 | 24.56 | 0.107 | 0.821 | 0.610 | 0.543 |
| FATHMM-MKL | 0 | 6.42 | 97.83 | 15.13 | 84.87 | 94.56 | 5.44 | 19.74 | 0.064 | 0.647 | 0.533 | 0.508 |
| GenoCanyon | 0 | 6.71 | 99.00 | 16.64 | 83.36 | 97.28 | 2.72 | 21.32 | 0.089 | 0.569 | 0.546 | 0.502 |
| M-CAP | 20 | 6.46 | 100.00 | 9.97 | 90.03 | 100.00 | 0.00 | 15.24 | 0.080 | 0.817 | 0.650 | 0.582 |
| MetaLR | 0 | 8.07 | 98.60 | 35.54 | 64.46 | 91.84 | 8.16 | 38.81 | 0.135 | 0.731 | 0.605 | 0.594 |
| MetaSVM | 0 | 9.12 | 98.86 | 43.63 | 56.37 | 91.84 | 8.16 | 46.43 | 0.168 | 0.719 | 0.613 | 0.522 |
| REVEL | 0 | 9.54 | 99.80 | 42.37 | 57.63 | 98.64 | 1.36 | 45.64 | 0.196 | 0.849 | 0.722 | 0.585 |
AUC, area under the curve; hser-AUC, high-sensitivity regional area under the curve; hspr-AUC, high-specificity regional area under the curve; FNR, false negative rate; FPR, false positive rate; MCC, Mathew correlation coefficient; NA, not available; NPV, negative predictive value; PPV, positive predictive value. We obtained experimentally validated 2533 missense mutations including 147 pathogenic mutations and 2386 benign mutations of PPARG gene from MITER database.
Compared with the other methods, the VEST3 method had the best overall performance in PPARG dataset with an AUC of 0.880, followed by PROVEN with an AUC of 0.855, MutationAssessor with an AUC of 0.852 and REVEL with an AUC of 0.849 (Table 3 and Figure 1G). In addition, we found that REVEL (0.722) and VEST3 (0.719) had hser-AUCs > 0.70, followed by CADD with a hser-AUC of 0.692 and SIFT with a hser-AUC of 0.658 (Table 3 and Figure 1H). Furthermore, VEST3 had an hspr-AUC of 0.646, which was higher than the hspr-AUC values of 0.623 for LRT, 0.599 for MutationAssessor, 0.594 for MetaLR, 0.593 for PROVEN and 0.585 for REVEL (Table 3 and Figure 1I). These results indicated that REVEL and VEST3 showed the best overall performances based on the experimentally validated benchmark data.
Correlation of computational methods
Based on the ClinVar benchmark dataset, we calculated the Spearman rank correlation coefficient between any two computational methods and found that fitCons, MutationTaster, FATHMM, M-CAP and GenoCanyon were lowly to moderately correlated with other methods and the rest of methods were moderately to highly correlated with each other (Supplementary Figure S10a). We then respectively investigated consistencies between computational methods based on ClinVar deleterious data or benign data. In particular, we found that M-CAP, REVEL, MetaLR and MetaSVM were highly correlated with each other and the rest of methods were almost lowly correlated based on the ClinVar deleterious data (Supplementary Figure S10b). In addition, MutationTaster, fitCons and GenoCanyon were lowly correlated with other methods, Eigen, GERP++, phastCons, phyloP and FATHMM-MKL were highly correlated with each other and the rest of methods were moderately correlated with other methods based on ClinVar benign data (Supplementary Figure S10c). We also analyzed their consistencies based on the somatic TP53 mutation benchmark data (Supplementary Figure S10d–f) and the experimentally PPARG validated benchmark data (Supplementary Figure S10g–i) and observed similar results, compared with the ClinVar benchmark data. Generally, most of methods exhibited lower correlation based on deleterious data than benign data for all benchmark data, suggesting that these methods have high consistencies for distinguishing benign variants but low consistencies for distinguishing deleterious variants.
To further investigate the correlations between the computational methods, we then characterized the respective ratios of overlapping TP, TN, FP or FN variants among computational methods, based on the ClinVar benchmark dataset. Specifically, most methods moderately to highly shared TP and TN variants (Supplementary Figure S11a and b), but lowly to moderately shared FP and FN variants (Supplementary Figure S11c and d), suggesting the correctly predicted variants were obviously shared, but that the incorrectly predicted variants were not obviously consistent among the different methods. In addition, we found that TP variants exhibited more consistencies than the TN variants and that the FP variants exhibited more consistencies than the FN variants. Together, these results can provide researches guidance in further study for combining different methods and obtaining better performance, compared to standalone methods.
Combining the REVEL and VEST3 methods
Based on the three sets of benchmark data, we found that the REVEL and VEST3 methods had the best overall performance. We then evaluated whether the combination of both methods (i.e. ReVe) could achieve better performance than either method individually with the three benchmark datasets (Supplementary Figure S12), based on measures of AUC, hser-AUC and hspr-AUC. For the ClinVar benchmark dataset, the AUC of the combined ReVe method was 0.942, which was higher than that of VEST3 (AUC = 0.929), REVEL (AUC = 0.920) or any other method (Figure 1A). In addition, we found that the hser-AUC for ReVe (0.742) was also higher than that of VEST3 (0.729) and REVEL (0.689) (Figure 1B). Similarly, the hspr-AUC for ReVe (0.793) was higher than that for REVEL (0.756) and VEST3 (0.756), respectively (Figure 1C). Generally, the ReVe method showed the best overall performance, achieving a specificity of 71.60% at a sensitivity of 95% (Figure 1B) and a sensitivity of 76.17% at a specificity of 95% (Figure 1C). Consistently, for the benchmark datasets of somatic variants and experimentally validated variants, the ReVe method showed a similar or better performance than REVEL, VEST3 and other methods (Figure 1D–I). Particularly, ReVe achieved the second highest AUC and hspr-AUC after VEST3, and the third highest hspr-AUC higher than VEST3 based on the somatic benchmark dataset (Figure 1D–F). For the experimentally validated PPARG benchmark dataset, ReVe outperformed all other methods in terms of the AUC and hser-AUC, and showed the second highest hspr-AUC after VEST3 (Figure 1G–I). All the predictive scores of missense variants of ReVe method in the whole genome were freely searched, browsed, and downloaded from the VarCards database (33), which we recently developed. We encourage further studies can develop new tool by combining more computational methods based our guidance and test their performance.
De novo missense mutations in autism
Previous WES studies have demonstrated that de novo mutations (DNMs) play important roles in the pathogenesis of ASD (49–51). We then sourced 1651 and 1107 de novo missense mutations (DNMMs) from 2508 ASD subjects and 1911 unaffected siblings from a previous study (49), and applied the 23 computational methods and ReVe to identify deleterious DNMMs and evaluate the performances of the methods. The better methods would be expected to show higher odds ratios (ORs) and more predicted deleterious DNMMs in ASD subjects compared to their siblings. Based on the results of computational methods, we found that the rate of deleterious DNMMs in ASD subjects was significantly higher than that in control subjects (Figure 2A and Supplementary Table S4). The ReVe method achieved the highest OR value and lowest P-value (OR = 1.42, P = 0.0002), followed by FATHMM (OR = 1.41, P = 0.0004), MetaSVM (OR = 1.34, P = 0.0006), MetaLR (OR = 1.34, P = 0.0006), REVEL (OR = 1.27, P = 0.0004) and VEST3 (OR = 1.21, P = 0.0009). The quantity of deleterious DNMMs predicted by ReVe was 313, similar to that for FATHMM (n = 300), MetaSVM (n = 372) and MetaLR (n = 380). Although REVEL and VEST3 exhibited lower OR values than ReVe, MetaSVM and MetaLR, but predicted more deleterious DNMMs (REVEL = 539, VEST3 = 806). As a negative control, we found that the rate of benign DNMMs predicted by most computation methods including ReVe in subjects with ASD was not significantly higher than that in control subjects (Figure 2B and Supplementary Table S4).
Figure 2.

Performance evaluations based on DNMs. The OR, 95% confidence interval and P-values were calculated by Poisson's ratio test. The area of each ball is proportional to the number of missense variants predicted to be deleterious or benign. The orange balls represent function-prediction methods, the dark gray balls represent conservation methods, and the green balls represent ensemble methods. A given missense variant with a predictive score of ReVe greater than 0.86 was regarded as a deleterious variant. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
DISCUSSION
In silico computational methods have been widely employed in investigating the pathogenicity of missense variants (5,40,52). The selection of appropriate methods is strongly needed for prioritizing candidate variations and genes in human diseases. We evaluated 12 performance measures of 23 computational methods based on three independent benchmark datasets that represented different genetic aspects: the ClinVar dataset represented germline variants in human genetic diseases, the TP53 and the ICGC datasets represented somatic variants of human cancers and the PPARG dataset from MITER database represented experimentally validated variants. All these dataset have not been directly used as training data for the 23 computational methods.
According to the results of ClinVar benchmark dataset, we preliminarily found that the ensemble methods performed better than the function prediction methods and conservation-only methods. Based on the somatic variants and experimentally validated variant benchmark dataset, we found that function prediction methods and ensemble methods have comparable performances and were superior to conservation-only methods. Although, some methods (such as REVEL) with high AUCs do employ the conservation score as an important component for their predictions, we think that conservation is likely still an important predictor. In addition, we found that computational methods within a class exhibited various performances for different benchmark datasets, possibly because they employed different training models and training data. For example, PROVEAN showed better performance for somatic variants and experimentally validated variants benchmark data than the germline variants benchmark data. Furthermore, REVEL (23) and VEST3 (16) stably achieved the highest discriminatory power under most conditions based on all the sets of benchmark data. We noted that both REVEL and VEST3 were trained using random forest model, which is different from the rest of 21 computational methods. However, whether it is a main factor need to be further validated. Although the fitCons (19), MutationTaster (13), GERP++ (12), phastCons (6), phyloP (7), SiPhy (9), CADD (17), DANN (21), Eigen (24), FATHMM-MKL (22) and GenoCanyon (20) methods did not perform better than VEST3 (16) or REVEL (23), they offer important advantage in genome-wide NGS applications because they provide predictive scores for non-coding and regulatory variants.
It has been reported that a large degree of circularity exists between the training data and the testing data of computational methods (35). To reduce the potential circularity, the following strategies were adopted in this study. First, we selected benchmark datasets that have not been used as training data for any computational method. Second, only variants recorded after 4 March 2015 in the ClinVar database were selected in the analysis. Third, we evaluated the degree of circularity and removed any overlaps between the benchmark datasets and the publicly available training data of computational methods. Although we could not remove all potential redundancy since not all training data of computational methods were publicly available, we encourage further developers to publish all training and testing data used in supplementary files. In this study, the TP53 gene variants and ICGC somatic variations related to human cancers, experimentally evaluated PPARG variants, and DNMs in autism served as additional benchmark data without any overlaps between training data of computational methods, strengthening the confidence of our conclusion that REVEL and VEST3 showed the best overall performances.
For all three sets of benchmark data, most computational methods exhibited higher sensitivities than specificities, suggesting that some predicted deleterious variants were actually benign variants. Therefore, clinicians and geneticists should be careful with predicted deleterious variants when prioritizing candidate variants and genes of human diseases. Stricter cutoff values and the combination of multiple methods were previously recommended for predicting deleterious variants (41). To provide researchers and clinicians more guidance, we summarized the recommended cutoff values and corresponding sensitivities and specificities based on the benchmark data shown in Supplementary Table S5. In particular, FATHMM, MutationTaster, Eigen, M-CAP, MetaLR and MetaSVM were most affected by the recommended cutoff values. Furthermore, we compared the performance measures of hser-AUC and hspr-AUC to provide tentative guidance for clinicians and geneticists in selecting high-sensitivity or high-specificity computational methods with different applications in mind (25). Our results indicated that REVEL and VEST3, as well ReVe outperformed the other computational methods in testing with all three benchmark datasets and with the DNMs. We encourage further study to select appropriate methods for predicting the pathogenicity of missense variants.
Although previous studies have been conducted to compare the performances of some computational methods (34–39), major differences are evident between earlier studies and this study. First, we collected different independent benchmark datasets covering clinically observed genetic variants, cancer somatic variants, and experimentally validated variants, in order to systematically evaluate their performances. Second, our analysis included several recently developed methods such as REVEL (23), M-CAP (25) and Eigen (24), which were all reported first in 2016. The best performing computational methods (such as REVEL and VEST3) identified in this study were not included in previous comparative studies. We strongly recommend employing these two methods, as well as ReVe, in further studies and genetic testing, rather than other well-known computation methods (such as SIFT, Polyphen2 and MutationTaster). Third, a total of 12 performance measures of the computational methods were systematically compared in light of the different needs of users. For example, the hser-AUC performance measure was essential for distinguishing deleterious variants from variants of uncertain significance, and hspr-AUC was useful for accurately identifying disease-causing variants and genes.
We noted that the computational methods exhibited an overall better performance for the ClinVar than the experimentally evaluated PPARG benchmark data. One possible reason for this outcome is that various researchers submitted the ClinVar data, most of which was not experimentally validated, and false-positive data may be included in the ClinVar data. We strongly recommend that scientists develop new methods of functional prediction based on more strict training data, especially for the experimentally validated data. Additionally, to interpret whether a missense variant is involved in human disease, more systematic evaluations regarding its pathogenicity are urgently needed (5,33,40,52). Users should also investigate the AF of variants and the gene-level mutation rate in the general population, as well as the gene function and protein domain (3,33). Together, our findings could help clinicians and researchers choose appropriate methods or use several tools simultaneously to interpret the pathogenicity of missense variants.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the members of the Beijing Institute of Life Science, Chinese Academy of Sciences for their valuable discussions regarding this work.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Key R&D Program of China [2016YFC0900400]; National Natural Science Foundation of China [31571301]. Funding for open access charge: National Natural Science Foundation of China [31571301].
Conflict of interest statement. None declared.
REFERENCES
- 1. Rabbani B., Tekin M., Mahdieh N.. The promise of whole-exome sequencing in medical genetics. J. Hum. Genet. 2014; 59:5–15. [DOI] [PubMed] [Google Scholar]
- 2. Goodwin S., McPherson J.D., McCombie W.R.. Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016; 17:333–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B. et al. . Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016; 536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boycott K.M., Vanstone M.R., Bulman D.E., MacKenzie A.E.. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat. Rev. Genet. 2013; 14:681–691. [DOI] [PubMed] [Google Scholar]
- 5. MacArthur D.G., Manolio T.A., Dimmock D.P., Rehm H.L., Shendure J., Abecasis G.R., Adams D.R., Altman R.B., Antonarakis S.E., Ashley E.A. et al. . Guidelines for investigating causality of sequence variants in human disease. Nature. 2014; 508:469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S.. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005; 15:1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Siepel A., Pollard K.S., Haussler D.. Apostolico A, Guerra C, Istrail S, Pevzner PA, Waterman M. New methods for detecting lineage-specific selection. RECOMB 2006. LNCS (LNBI). 2006; 3909:Heidelberg: Springer; 190–205. [Google Scholar]
- 8. Chun S., Fay J.C.. Identification of deleterious mutations within three human genomes. Genome Res. 2009; 19:1553–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Garber M., Guttman M., Clamp M., Zody M.C., Friedman N., Xie X.. Identifying novel constrained elements by exploiting biased substitution patterns. Bioinformatics. 2009; 25:i54–i62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kumar P., Henikoff S., Ng P.C.. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009; 4:1073–1081. [DOI] [PubMed] [Google Scholar]
- 11. Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R.. A method and server for predicting damaging missense mutations. Nat. Methods. 2010; 7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Davydov E.V., Goode D.L., Sirota M., Cooper G.M., Sidow A., Batzoglou S.. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 2010; 6:e1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schwarz J.M., Rodelsperger C., Schuelke M., Seelow D.. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 2010; 7:575–576. [DOI] [PubMed] [Google Scholar]
- 14. Reva B., Antipin Y., Sander C.. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011; 39:e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Choi Y., Sims G.E., Murphy S., Miller J.R., Chan A.P.. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012; 7:e46688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Carter H., Douville C., Stenson P.D., Cooper D.N., Karchin R.. Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genomics. 2013; 14:S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kircher M., Witten D.M., Jain P., O’Roak B.J., Cooper G.M., Shendure J.. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014; 46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dong C., Wei P., Jian X., Gibbs R., Boerwinkle E., Wang K., Liu X.. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015; 24:2125–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gulko B., Hubisz M.J., Gronau I., Siepel A.. A method for calculating probabilities of fitness consequences for point mutations across the human genome. Nat. Genet. 2015; 47:276–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lu Q., Hu Y., Sun J., Cheng Y., Cheung K.H., Zhao H.. A statistical framework to predict functional non-coding regions in the human genome through integrated analysis of annotation data. Sci. Rep. 2015; 5:10576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Quang D., Chen Y., Xie X.. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics. 2015; 31:761–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shihab H.A., Rogers M.F., Gough J., Mort M., Cooper D.N., Day I.N., Gaunt T.R., Campbell C.. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics. 2015; 31:1536–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ioannidis N.M., Rothstein J.H., Pejaver V., Middha S., McDonnell S.K., Baheti S., Musolf A., Li Q., Holzinger E., Karyadi D. et al. . REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 2016; 99:877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ionita-Laza I., McCallum K., Xu B., Buxbaum J.D.. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat. Genet. 2016; 48:214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jagadeesh K.A., Wenger A.M., Berger M.J., Guturu H., Stenson P.D., Cooper D.N., Bernstein J.A., Bejerano G.. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 2016; 48:1581–1586. [DOI] [PubMed] [Google Scholar]
- 26. Shihab H.A., Gough J., Cooper D.N., Stenson P.D., Barker G.L., Edwards K.J., Day I.N., Gaunt T.R.. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 2013; 34:57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Biesecker L.G., Green R.C.. Diagnostic clinical genome and exome sequencing. N. Engl. J. Med. 2014; 370:2418–2425. [DOI] [PubMed] [Google Scholar]
- 28. Cheng F., Zhao J., Zhao Z.. Advances in computational approaches for prioritizing driver mutations and significantly mutated genes in cancer genomes. Brief. Bioinform. 2016; 17:642–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li J., Jiang Y., Wang T., Chen H., Xie Q., Shao Q., Ran X., Xia K., Sun Z.S., Wu J.. mirTrios: an integrated pipeline for detection of de novo and rare inherited mutations from trios-based next-generation sequencing. J. Med. Genet. 2015; 52:275–281. [DOI] [PubMed] [Google Scholar]
- 30. Li J., Cai T., Jiang Y., Chen H., He X., Chen C., Li X., Shao Q., Ran X., Li Z. et al. . Genes with de novo mutations are shared by four neuropsychiatric disorders discovered from NPdenovo database. Mol. Psychiatry. 2016; 21:290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Johansen Taber K.A., Dickinson B.D., Wilson M.. The promise and challenges of next-generation genome sequencing for clinical care. JAMA Intern. Med. 2014; 174:275–280. [DOI] [PubMed] [Google Scholar]
- 32. Wright C.F., FitzPatrick D.R., Firth H.V.. Paediatric genomics: diagnosing rare disease in children. Nat. Rev. Genet. 2018; 19:253–268. [DOI] [PubMed] [Google Scholar]
- 33. Li J., Shi L., Zhang K., Zhang Y., Hu S., Zhao T., Teng H., Li X., Jiang Y., Ji L. et al. . VarCards: an integrated genetic and clinical database for coding variants in the human genome. Nucleic Acids Res. 2017; 46:D1039–D1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Thusberg J., Olatubosun A., Vihinen M.. Performance of mutation pathogenicity prediction methods on missense variants. Hum. Mutat. 2011; 32:358–368. [DOI] [PubMed] [Google Scholar]
- 35. Grimm D.G., Azencott C.A., Aicheler F., Gieraths U., MacArthur D.G., Samocha K.E., Cooper D.N., Stenson P.D., Daly M.J., Smoller J.W. et al. . The evaluation of tools used to predict the impact of missense variants is hindered by two types of circularity. Hum. Mutat. 2015; 36:513–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wei P., Liu X., Fu Y.X.. Incorporating predicted functions of nonsynonymous variants into gene-based analysis of exome sequencing data: a comparative study. BMC Proc. 2011; 5:S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gnad F., Baucom A., Mukhyala K., Manning G., Zhang Z.. Assessment of computational methods for predicting the effects of missense mutations in human cancers. BMC Genomics. 2013; 14:S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rodrigues C., Santos‐Silva A., Costa E., Bronze‐Da‐Rocha E.. Performance of in silico tools for the evaluation of UGT1A1 missense variants. Hum. Mutat. 2015; 36:1215–1225. [DOI] [PubMed] [Google Scholar]
- 39. Konig E., Rainer J., Domingues F.S.. Computational assessment of feature combinations for pathogenic variant prediction. Mol. Genet. Genomic Med. 2016; 4:431–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E. et al. . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015; 17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu X., Wu C., Li C., Boerwinkle E.. dbNSFP v3.0: A One-Stop database of functional predictions and annotations for human nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016; 37:235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang K., Li M., Hakonarson H.. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Landrum M.J., Lee J.M., Benson M., Brown G., Chao C., Chitipiralla S., Gu B., Hart J., Hoffman D., Hoover J. et al. . ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016; 44:D862–D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kato S., Han S.Y., Liu W., Otsuka K., Shibata H., Kanamaru R., Ishioka C.. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:8424–8429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bouaoun L., Sonkin D., Ardin M., Hollstein M., Byrnes G., Zavadil J., Olivier M.. TP53 variations in human Cancers: New lessons from the IARC TP53 database and genomics data. Hum. Mutat. 2016; 37:865–876. [DOI] [PubMed] [Google Scholar]
- 46. Majithia A.R., Tsuda B., Agostini M., Gnanapradeepan K., Rice R., Peloso G., Patel K.A., Zhang X., Broekema M.F., Patterson N. et al. . Prospective functional classification of all possible missense variants in PPARG. Nat. Genet. 2016; 48:1570–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Frédérique L., Natalia T., Alexandre H., Natacha T., Xavier R., Jean-Charles S., Markus M.. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. 2011; 12:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fischbach G.D., Lord C.. The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron. 2010; 68:192–195. [DOI] [PubMed] [Google Scholar]
- 49. Iossifov I., O’Roak B.J., Sanders S.J., Ronemus M., Krumm N., Levy D., Stessman H.A., Witherspoon K.T., Vives L., Patterson K.E. et al. . The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014; 515:216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li J., Wang L., Guo H., Shi L., Zhang K., Tang M., Hu S., Dong S., Liu Y., Wang T. et al. . Targeted sequencing and functional analysis reveal brain-size-related genes and their networks in autism spectrum disorders. Mol. Psychiatry. 2017; 22:1282–1290. [DOI] [PubMed] [Google Scholar]
- 51. Li J., Wang L., Yu P., Shi L., Zhang K., Sun Z.S., Xia K.. Vitamin D-related genes are subjected to significant de novo mutation burdens in autism spectrum disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2017; 174:568–577. [DOI] [PubMed] [Google Scholar]
- 52. Biesecker L.G., Green R.C.. Diagnostic clinical genome and exome sequencing. N. Engl. J. Med. 2014; 371:1170. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.