

Supplemental Digital Content is available in the text
Keywords: adaptation, clustering, epidemic, HIV, human leukocyte antigen, immune escape, phylogenetics, RT-I135X, Saskatchewan
Abstract
Objective:
HIV incidence in the Canadian province of Saskatchewan, where Indigenous persons make up 80% of those infected, are among the highest on the continent. Reports of accelerated HIV progression, associated with carriage of certain human leukocyte antigen (HLA) alleles (including the typically protective HLA-B∗51) have also emerged from the region. Given that acquisition of HIV preadapted to host HLA negatively impacts clinical outcome, we hypothesized that HIV-host adaptation may be elevated in Saskatchewan.
Design:
Comparative analysis of population-level HIV sequence datasets from Saskatchewan and elsewhere in Canada/USA.
Methods:
We analyzed 1144 HIV subtype B Pol sequences collected in Saskatchewan between 2000 and 2016, comprising ∼65% of cumulative provincial HIV cases, for the presence of 70 HLA-associated Pol mutations. Sequences from British Columbia (N = 6525) and elsewhere in Canada/USA (N = 6517) were used for comparison. HIV adaptation levels to 34 HLA alleles were also computed. Putative HIV transmission clusters were identified, and the prevalence of HLA-associated adaptations within and outside these clusters was investigated.
Results:
Analyses confirmed significantly elevated and temporally increasing levels of HIV adaptation to commonly expressed HLA alleles, in particular B∗51. Notably, HLA-adapted HIV strains were significantly enriched among phylogenetic clusters in Saskatchewan.
Conclusion:
Extensive circulating HIV adaptation to HLA in Saskatchewan provides a plausible explanation for accelerated progression, while enrichment of adapted variants in phylogenetic clusters suggests they are being widely transmitted. Results highlight the utility of Pol sequences, routinely collected for drug resistance monitoring, for surveillance of HIV-host adaptation, and underscore the urgent need to expand HIV prevention and treatment programmes in Saskatchewan.
Introduction
Just as the transmission of drug-resistant HIV compromises therapy efficacy [1], transmission of HIV strains harbouring human leukocyte antigen (HLA) class I-restricted CD8+ cytotoxic T lymphocyte (CTL) escape mutations can compromise host antiviral immunity [2–4]. In fact, the extent to which an individual's transmitted/founder HIV sequence is ‘preadapted’ to its host's HLA allele profile explains a substantial portion of variation in clinical outcomes [2–4]; moreover, differential population-level spread of HIV immune escape variants [3,5–8] explains regional variation in clinical outcomes attributed to host HLA carriage [3]. The classic protective allele HLA-B∗57 for example is not associated with HIV control in Botswana due to the high prevalence of B∗57-associated escape mutations in circulation [7]; similarly, HLA-B∗51, typically associated with HIV control [5,9–12], has lost its protective status in Japan due to population-level accumulation of the B∗51-associated I135T escape mutation in reverse transcriptase [5,11,13]. Similar phenomena are likely occurring in epidemics elsewhere.
Since 2003 a concentrated HIV epidemic has been expanding in the Canadian province of Saskatchewan [14,15], where injection drug use represents the primary risk factor (60%) and where nearly 80% of infected persons self-identify as having Indigenous ancestry [16]. Overall HIV incidence in Saskatchewan (14.5 new diagnoses per 100 000 in 2016) is more than twice the national average [16] and among the highest in North America [17], but regional rates are up to fourfold higher (e.g., 67.8 per 100 000 in the former Prince Albert Parkland Health Region) [16].
The Saskatchewan epidemic may also have unique clinical characteristics. Anecdotal reports of more rapid than expected progression to HIV-induced immunodeficiency are emerging from the province [18], and the 2016 Saskatchewan HIV Prevention and Control report estimated that fewer than 50% of persons diagnosed since 2007 remained alive in 2016 [16]. Furthermore, studies in the neighbouring province of Manitoba [19,20] have reported accelerated HIV progression among individuals expressing certain HLA alleles, including the typically protective B∗51 [5,9–12]. This is notable, as a study investigating HLA diversity among Indigenous Saskatchewan residents identified B∗51 as the most common HLA-B allele in this group [21].
These observations suggest that Saskatchewan HIV strains possess properties that enhance their pathogenicity in an HLA-dependent manner. We hypothesized that circulating HIV adaptation to HLA could explain this, at least in part. Given the link between population-level spread of the B∗51-associated RT-I135X escape mutation and erosion of HIV control by this allele in other global regions [5,11,13], we further reasoned that HIV adaptation to B∗51 might be particularly elevated in Saskatchewan. We investigated this using comprehensive population-based HIV Pol sequence datasets, collected between 2000 and 2016, from Saskatchewan and other locations in Canada and the USA.
Methods
HIV Pol sequence datasets
The British Columbia Centre for Excellence in HIV/AIDS laboratory, which is accredited by the College of American Pathologists (CAP), the Diagnostic Accreditation Program of the College of Physicians and Surgeons of British Columbia (DAP) and the World Health Organization (WHO), has provided HIV drug resistance genotyping for virtually all Canadian provinces and territories since 1998. As described previously [22,23], genotyping involves extraction of total nucleic acids from blood plasma, bulk amplification of protease and partial reverse transcriptase sequences by nested RT-PCR using HIV-specific oligonucleotide primers, followed by direct Sanger sequencing [detailed standard operating procedures (SOPs) are available for sharing; please contact corresponding author]. All clinical laboratory staff undergo rigorous training and qualification tests, and many procedures, including chromatogram base calling [24], are nearly or fully automated to minimize error and interoperator variability. SOPs also feature daily and monthly Quality Control steps, which include comparison of all new HIV sequences to all previously collected sequences from that individual, as well as those collected from other individuals (where sequences are flagged for further investigation if they are too distant from other within-host sequences or too close to HIV sequences from other individuals).
HIV sequences from Saskatchewan and British Columbia analyzed in the present study were collected as part of comprehensive clinical monitoring systems for HIV drug resistance in these provinces, where baseline (pretherapy) genotyping is standard-of-care (in fact, genotyping has been performed automatically on all new HIV diagnoses in British Columbia since 2005). Between January 2000 and July 2016, a total of 2352 genotypes from 1360 unique HIV-infected Saskatchewan residents were performed (‘Saskatchewan dataset’). Similarly, a total of 6525 genotypes from unique HIV subtype B-infected British Columbians collected between 2000 and 2016 were used as a regional comparison group (’BC dataset’). For both the Saskatchewan and BC datasets, each individual was represented only once (where longitudinal genotypes were performed, analysis was restricted to the earliest one). Overall, the Saskatchewan dataset is estimated to comprise 65% of HIV cases reported in the province between 1985 and 2016 [25] while the BC dataset is estimated to represent more than 50% of persons living with HIV in the province [26].
In addition, all 6540 HIV subtype B Pol sequences annotated with Canada or USA as the country of origin and a date between 2000–2016 were retrieved from the Los Alamos (LANL) HIV sequence database in July 2017, after limiting the output to a single sequence per individual. Following a literature review and correspondence with relevant study authors [27], 23 sequences of possible Saskatchewan origin were subsequently removed from this dataset, yielding a final N = 6517 as a continental comparison group (’CA/US’ dataset).
Ethics statement
Ethical approval for this study was granted by the institutional review boards at Providence Healthcare/UBC and Simon Fraser University. As Saskatchewan and BC HIV sequences were collected for clinical purposes and irreversibly anonymized prior to study, the requirement for written informed consent was waived. No information other than collection year was available for study.
Phylogenetic inference and cluster analysis
Nucleic acid sequence datasets incorporating the HIV-1 subtype B reference strain HXB2 (GenBank K03455) were aligned using MAFFT [28] in a codon-aware manner and inspected using AliView [29]. Subtype classification was performed using the Recombination Identification Program (window size 400) [30]. Phylogenetic inference was performed by approximate Maximum Likelihood using FastTree2 under a general time reversible (GTR) model of nucleotide substitution [31] on alignments trimmed to Protease and the first 240 codons of reverse transcriptase; codons associated with HIV surveillance drug resistance mutations [32] were additionally removed prior to phylogenetic inference for clustering analyses to control for potential effects on tree topology. Trees were visualized using FigTree v1.4.3 (http://tree.bio.ed.ac.uk/software/figtree/). Clusters were recognized by identifying all pairs of sequences whose patristic (tip-to-tip) distances were less than 0.02 expected nucleotide substitutions/site; this cutoff corresponded to the 95% quantile of within-host and the 0.1% quantile of between-host Pol genetic distances in an independent analysis of 27 296 Pol sequences from 7747 unique individuals [33]. A cluster was defined as a set of five or more sequences where at least one member was less than 0.02 nucleotide substitutions per site away from at least one other (i.e. duos, triplets and quadruplets were not identified). This is done to mitigate the risk of identifying individuals as possible HIV transmission sources, as HIV nondisclosure remains criminally prosecutable in Canada. For this reason, HIV sequences were not deposited publicly and are instead available for sharing under IRB and institutionally approved processes (contact corresponding author).
Quantification of human leukocyte antigen-associated HIV adaptation
HLA-associated adaptations, which represent the inferred HLA-associated escape variant at a given viral codon, have been identified across HIV by statistical association [3,34–38]. We analyzed Saskatchewan, BC and CA/US HIV sequences for the presence of 70 HLA-associated adaptations, which represented all HLA-adapted associations identified in the studied Pol region at the stringent statistical threshold of q less than 0.05 (corresponding to 2.9e−92 < P < 1.4e−4) in an independent study of more than 1800 antiretroviral-naive individuals with chronic HIV subtype B infection [38]. The overall degree of adaptation of HIV sequences to specific HLA alleles was also determined using a published metric that uses a probabilistic model to compare what an HIV sequence would ‘look like’ if it were to evolve indefinitely in a host whose immune system either solely targeted HIV epitopes restricted by the HLA allele in question, versus what it would look like if it were to evolve indefinitely in the absence of immune pressure [3]. The resulting ratio of these two scenarios is scaled so that it ranges from −1 to 1, where the extremes denote zero and complete adaptation to the HLA allele in question, respectively.
Results
A total of 1360 HIV Pol sequences from unique Saskatchewan individuals collected between 2000 and 2016, estimated to represent 65% of HIV cases reported in the province between 1985 and 2016 [25], were available for study (Figure S1A). As HLA-associated adaptations are to some extent HIV subtype-specific [38–42], analyses were restricted to the 1144 (84.1%) subtype B sequences. Of these, 992 (86.7%) extended to reverse transcriptase codon 400 while 152 (13.3%) extended to codon 240. Broken down by year, 44 sequences (3.8%) were collected between 2000 and 2007, while 67 (5.9%), 35 (3.1%), 105 (9.2%), 128 (11.2%), 123 (10.8%), 169 (14.8%), 186 (16.3%), 203 (17.7%), and 84 (7.3%) were collected from 2008 through 2016, respectively (Figure S1B).
Phylogenies inferred from Saskatchewan Pol sequences, and those from elsewhere in Canada and the USA (‘CA/US’) revealed that, while total HIV subtype B diversity was broadly comparable between the two datasets (HXB2-rooted tree heights were 0.1377 and 0.1446 nucleotide substitutions/site respectively), tree topologies differed markedly (Fig. 1a, S2, S3). In particular, 77.6% (888 of 1144) Saskatchewan sequences resided within 26 clusters, where the three largest, containing 207, 182 and 176 sequences respectively, comprised 40.6% of the data (Fig. 1b, S2). In contrast, only 15% of CA/US sequences resided within clusters (117 total, the largest containing only 42 sequences) (Fig. 1b, S3), even though this dataset featured dense sampling of regional HIV epidemics including in Western Canada (excluding Saskatchewan) [27,43,44] and San Diego [45,46]. The high genetic divergence between major Saskatchewan sequence clusters is also readily apparent in a combined Saskatchewan/CA/US phylogeny (Figure S4). Further confirming Saskatchewan's unique HIV molecular epidemiology, the extent of clustering observed in 6525 HIV subtype B sequences from British Columbia collected between 2000 and 2016, estimated to represent more than 50% of persons living with HIV in the province [26], was also substantially lower than in Saskatchewan (198 clusters comprising 48% of sequences, where the three largest comprised less than 10% of the data) (Figures S5A, S5B, S6).
Fig. 1.
HIV Pol diversity and consensus amino acid comparison in Saskatchewan and elsewhere in Canada/USA.
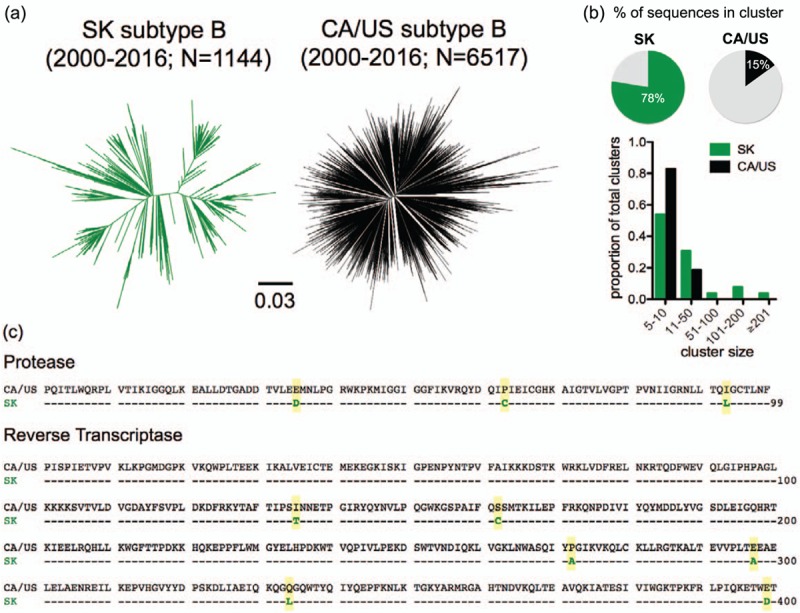
Panel a: Unrooted maximum-likelihood phylogenies inferred from 1144 and 6517 HIV subtype B sequences collected between 2000–2016 from Saskatchewan (‘SK’; green) and other areas of Canada and the USA (’CA/US’; black) respectively, depicted on the same distance scale. Panel b: Top: Percentage of sequences in Saskatchewan and CA/US datasets that fall within a cluster. Bottom: cluster size distributions in Saskatchewan and CA/US (expressed as a proportion of the total number of clusters in each dataset). Panel c: Consensus Pol amino acid sequences in CA/US versus Saskatchewan, with the 9 differences highlighted in yellow.
Strikingly, the Saskatchewan HIV consensus sequence differed from that in the rest of Canada/USA at 9 Pol codons: E35D, P63C and I93L in Protease, and I135T, S162C, P272A, E297A, Q334L, and E399D in reverse transcriptase (Fig. 1c), a remarkable observation given Pol's relative conservation and the long-term stability of the North American [8] and global (http://www.hiv.lanl.gov/) subtype B Pol consensus sequences. For all but RT-272 [where Proline and Alanine occur at comparable (47% vs. 45%) frequencies in subtype B globally], the magnitude of these differences was also striking. For example, whereas more than 60% of CA/US HIV subtype B sequences harbor Serine at RT-162, more than 80% of Saskatchewan sequences harbor Cysteine at this position. Moreover, eight of the nine Pol codons where Saskatchewan and CA/US consensus sequences differed represented HLA-associated sites (the polymorphic RT-272 being the sole exception), and for seven of these the Saskatchewan consensus represented the specific adapted form for one or more HLA alleles (Fig. 2a). E35D and I93L in Protease are adaptations to B∗44 : 02 and B∗15 : 01 respectively, while in RT, I135T is an adaptation to B∗51 : 01, S162C to B∗07 : 02, E297A to B∗35 : 03, Q334L to B∗15 : 01, and E399D to A∗32 : 01 [38]. Furthermore, most of these HLA alleles are common: B∗07 : 02, B∗15 : 01, B∗35 : 01, B∗44 : 02, and B∗51 : 01 rank among the six most prevalent alleles in CA/US HIV cohorts [38] while B∗51, B∗15 : 01 and B∗35 are the three most frequently-expressed HLA-B alleles among Indigenous persons in Saskatchewan [21] (Figure S7). This finding alone indicates that HIV adaptations to common HLA alleles dominate in Saskatchewan, and suggests that the skewed viral amino acid distributions in Saskatchewan are due to HIV's unique molecular epidemiology in this region.
Fig. 2.
Human leukocyte antigen (HLA)-associated adaptations in HIV Pol, and their frequencies in Saskatchewan and CA/US.
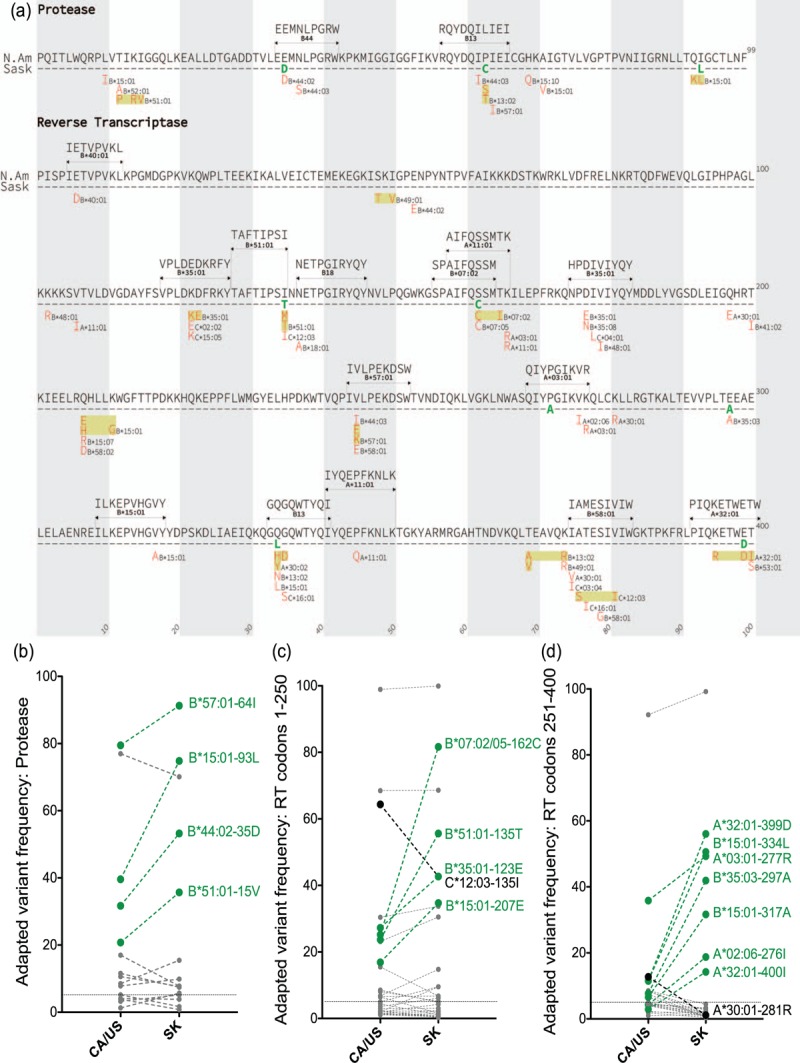
Panel a: The 70 HLA-associated adaptations in HIV subtype B Protease and the first 400 codons of RT, defined at q < 0.05 in an independent population-based study [38], that were investigated in the present analysis. Specific adaptations are shown in red, along with their restricting HLA, below the CA/US and Saskatchewan consensus Pol amino acid sequences. Nearby adaptations restricted by the same HLA allele are boxed together in yellow. For HLA-adapted variants that occur within an optimally-described HIV CTL epitope restricted by that allele [47], the published epitope sequence and HLA restriction are shown. In total, the 70 adaptations are restricted by 34 HLA class I alleles (6 HLA-A, 23 HLA-B and 5 HLA-C), occur at 52 Protease and RT codons, and include numerous experimentally verified immune escape mutations in optimally-described CTL epitopes (e.g., B∗51 : 01-RT- I135T that occurs at the C-terminus of the B∗51 : 01-restricted TI8 epitope and abrogates its ability to bind this allele [5,12,38]). The list includes HIV codons where the same adapted form is selected by distinct HLA alleles (e.g., both B∗07 : 02 and B∗07 : 05 select RT-S162C), codons where two or more adapted forms are described for the same allele at a given position (e.g., B∗51 : 01 normally selects RT-I135T but may also select I135 M) and codons where distinct alleles select opposing mutations (e.g., B∗51 : 01 selects I135T, while C∗12 : 03 induces pressure to maintain the global consensus I at this site). Panel b: Frequency comparison of HLA-associated adapted variants in HIV Protease in Saskatchewan versus CA/US, shown as linked pairs. Adapted variants with more than 10% higher frequency in Saskatchewan compared to CA/US are labeled in green; those with more than 10% lower frequency in Saskatchewan are labeled in black. Horizontal dotted line indicates the 5% frequency threshold used to define common HLA-adapted variants. Panels c, d: same as (b), for HLA-associated adapted variants in RT codons 1–250 and 251–400 respectively.
Comparison of the prevalence of all 70 published HLA-adapted variants (shown in Fig. 2a) in Saskatchewan versus CA/US revealed further striking differences (Fig. 2b–d). First, common HLA-adapted variants (those observed at ≥5% frequency in both regions) were significantly overrepresented in Saskatchewan compared to CA/US [median (IQR) 15% (5.5–50%) in Saskatchewan versus 11% (6.6–26%) in CA/US, Mann–Whitney P = 0.01]. Moreover, 17 HLA-adapted variants differed by more than 10% between datasets; of these, 15 (88.2%) were more prevalent in Saskatchewan compared to CA/US (P = 0.0024). The latter included the B∗07 : 02 (and B∗07 : 05)-associated RT-S162C adaptation (that occurs at position 7 of the optimally-described B∗07 : 02-restricted SM9 CTL epitope [47]; Fig. 2a), whose frequency was 81.6% in Saskatchewan compared to 25.2% in CA/US (Fig. 2c), the A∗32 : 01-associated RT-3499D adaptation at position 8 of the optimally described A∗32 : 01-restricted PW10 epitope, present at 56.0% frequency in Saskatchewan compared to 11.6% in CA/US (Fig. 2d) and the B∗51 : 01-associated RT-I135T escape mutation at the C-terminus of the optimally-described B∗51 : 01-restricted TI8 epitope [5,12,38], present at 55.6% frequency in Saskatchewan compared to 23.7% in CA/US (Fig. 2c). Only two adaptations exhibited more than 10% lower frequency in Saskatchewan than CA/US, one of which was the C∗12 : 03-associated RT-I135 (the opposing mutation to the B∗51 : 01-associated RT-I135T adaptation at the same site). Common HLA-adapted variants were similarly significantly overrepresented in Saskatchewan compared to BC (Mann-Whitney P = 0.01), with 12 occurring at more than 10% higher frequency in the former versus the latter province (Figures S5C-E).
We next examined temporal trends for the 15 HLA-adapted variants whose frequencies were more than 10% elevated in Saskatchewan compared to CA/US (Fig. 3). With the exception of the A∗03 : 01-associated RT-K277R and A∗32 : 01-associated RT-400I mutations whose prevalence declined (albeit not significantly) over the study period, all others increased over time, in seven of 15 cases (46.7%) significantly so. The latter included RT-K123E (adapted to B∗35 : 01), RT-I135T (B∗51 : 01), RT-S162C (B∗07 : 02/05), RT-Q207E (B∗15 : 01), RT-C297A (B∗35 : 03), RT-V317A (B∗15 : 01) and RT-E399D (A∗32 : 01) (all P ≤ 0.05). Of note, RT-I135T, S162C and V317A increased on average by 1.6%, 2.7% and 3.2% per year, respectively (Fig. 3). Nevertheless, prior to 2008, these 15 HLA-adapted variants were already more prevalent in Saskatchewan than in CA/US (Wilcoxon matched pairs test P < 0.0001; not shown), suggesting that founder effects, combined with frequent onward transmission, could explain their elevated frequency in Saskatchewan.
Fig. 3.
Rates of increase of select human leukocyte antigen (HLA)-adapted variants during the Saskatchewan epidemic.
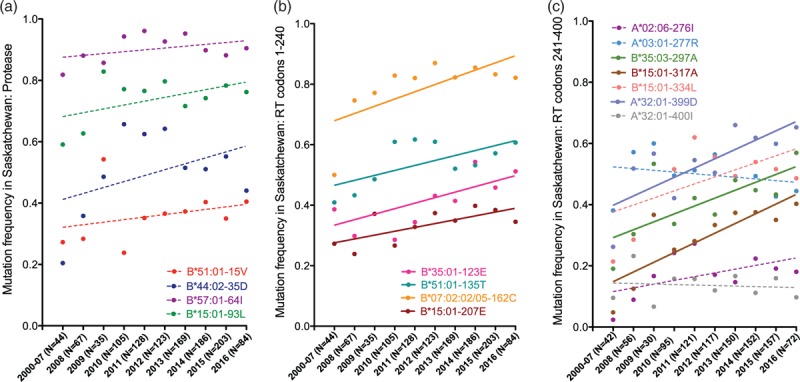
Annual frequencies (dots) and rates of change (regression lines) are depicted for the 15 HLA-associated adapted variants in Protease (Panel a), RT codons 1–250 (Panel b) and RT codons 251–400 (Panel c) whose frequencies were overall more than 10% higher frequency in Saskatchewan versus elsewhere in Canada and the USA. Sequences collected prior to 2008 are combined into a single group due to small N. The number of sequences analyzed per year is indicated on the x-axis. Variants whose annual rates of increase are statistically significant are shown as solid lines; others are shown as dotted lines.
Analyses to this point have focused on individual mutations, however this does not fully reflect HIV immune escape transmission dynamics because a given host will generally select, and thus transmit, multiple adaptations restricted by the same HLA allele (e.g., this Pol region includes eight B∗15 : 01- and four B∗51 : 01-associated sites; Fig. 2a). We thus quantified the overall level of adaptation of every Pol sequence to each of the 34 studied HLA alleles using a published metric [3]. Here, each HIV sequence is assigned a value from −1 to 1 reflecting its level of adaptation to each HLA allele of interest. Overall, HLA-specific adaptation levels were significantly higher in Saskatchewan compared to CA/US sequences (P = 0.002, Wilcoxon matched pairs test; Fig. 4a), and were particularly elevated for seven HLA alleles (A∗03 : 01, A∗32 : 01, B∗07 : 02/05, B∗15 : 01, B∗35 : 01, B∗44 : 02, B∗51 : 01), most of which are relatively common (Figure S7). Comparisons of HIV adaptation distributions for each of the 34 HLA alleles revealed that for 16 (47%) of them, adaptation was higher in Saskatchewan compared to CA/US (Fig. 4b; all P < 0.0001). In contrast, adaptation was lower in Saskatchewan for only 7 (21%) alleles (Fig. 4c; all P < 0.005), where the magnitudes of these differences were significantly smaller than those for which adaptation was significantly elevated in Saskatchewan (P = 0.0065, not shown). For the remaining 10 (29%) HLA alleles, adaptation was not significantly different between regions after multiple comparisons correction (Fig. 4d).
Fig. 4.
Comparison of human leukocyte antigen (HLA)-specific Pol adaptation scores in Saskatchewan vs. CA/US.
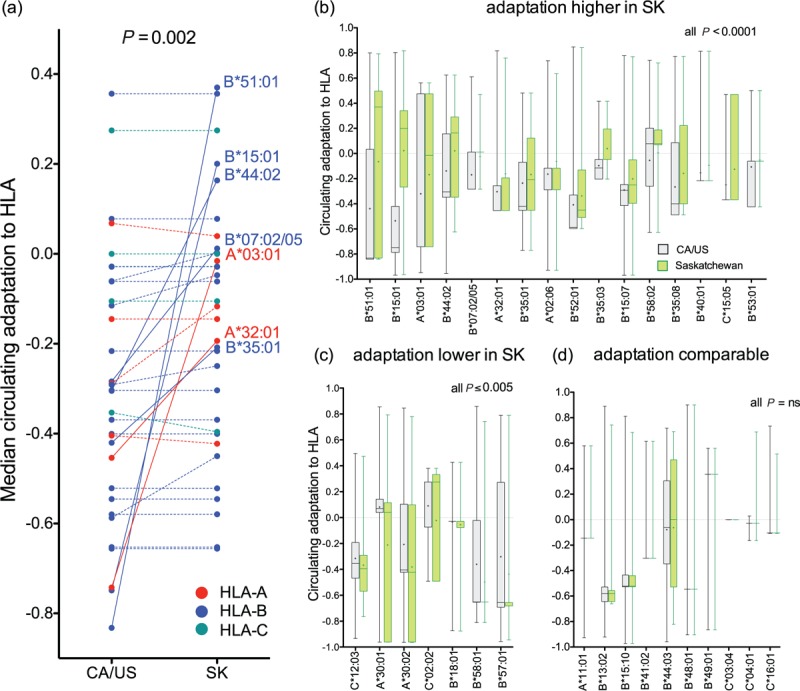
Panel a: Median HLA allele-specific adaptation scores of CA/US and Saskatchewan Pol sequences are shown as linked pairs, coloured according to the locus (HLA-A, B and C) to which the allele in question belongs. HLA alleles whose adaptation scores are more than 0.2 units higher in Saskatchewan versus CA/US are labeled and linked with solid lines; others are linked with dotted lines. Panel b: Paired histograms detailing adaptation distributions in Saskatchewan (green) and CA/US (gray) datasets for the 16 HLA alleles whose adaptation scores were significantly higher in Saskatchewan vs. CA/US after correction for multiple comparisons (all pairwise comparisons P < 0.0001 by Wilcoxon-rank sum test). Box horizontal line and edges represent median and interquartile range; cross represents mean, whiskers denote range. Panel c: Paired histograms depicting overall HLA allele-specific adaptation distributions for the 7 HLA alleles whose adaptation scores were significantly lower in Saskatchewan vs. CA/US after multiple comparisons correction. Panel d: Paired histograms depicting overall HLA allele-specific adaptation distributions for the 10 HLA alleles whose adaptation scores were not significantly higher in Saskatchewan vs. CA/US after multiple comparisons correction.
We next investigated the prevalence of HLA-associated adaptations within and outside Saskatchewan phylogenetic sequence clusters, reasoning that, if the elevated prevalence of HLA adaptations in Saskatchewan HIV sequences is due to their frequent and widespread transmission in the epidemic, then Saskatchewan clusters should be enriched in HIV sequences harbouring these adaptations. We had identified 26 Saskatchewan clusters that included 888 sequences (77.6% of the dataset) (Fig. 1b, S2). For each of the 70 HIV adaptations of interest we computed their odds ratios and P-values of being located in an HIV sequence within a cluster, using Fisher's exact test. For example, of the 888 Saskatchewan HIV sequences located in a cluster, 805 (90.7%) harboured RT-162C (Fig. 5a) whereas of the 256 sequences not within a cluster, only 129 (50.4%) harboured RT-162C. RT-162C was thus enriched in Saskatchewan clusters with an odds ratio of 9.6 (P < 0.0001). The B∗51 : 01-associated RT-I135T provides another example: 60% of HIV sequences within a Saskatchewan cluster harboured RT-I135T (Fig. 5b) compared to only 40% of those not within a cluster, an odds ratio of 2.2 (P < 0.0001).
Fig. 5.
HLA-associated adapted variants enriched in Saskatchewan are preferentially located within provincial sequence clusters.
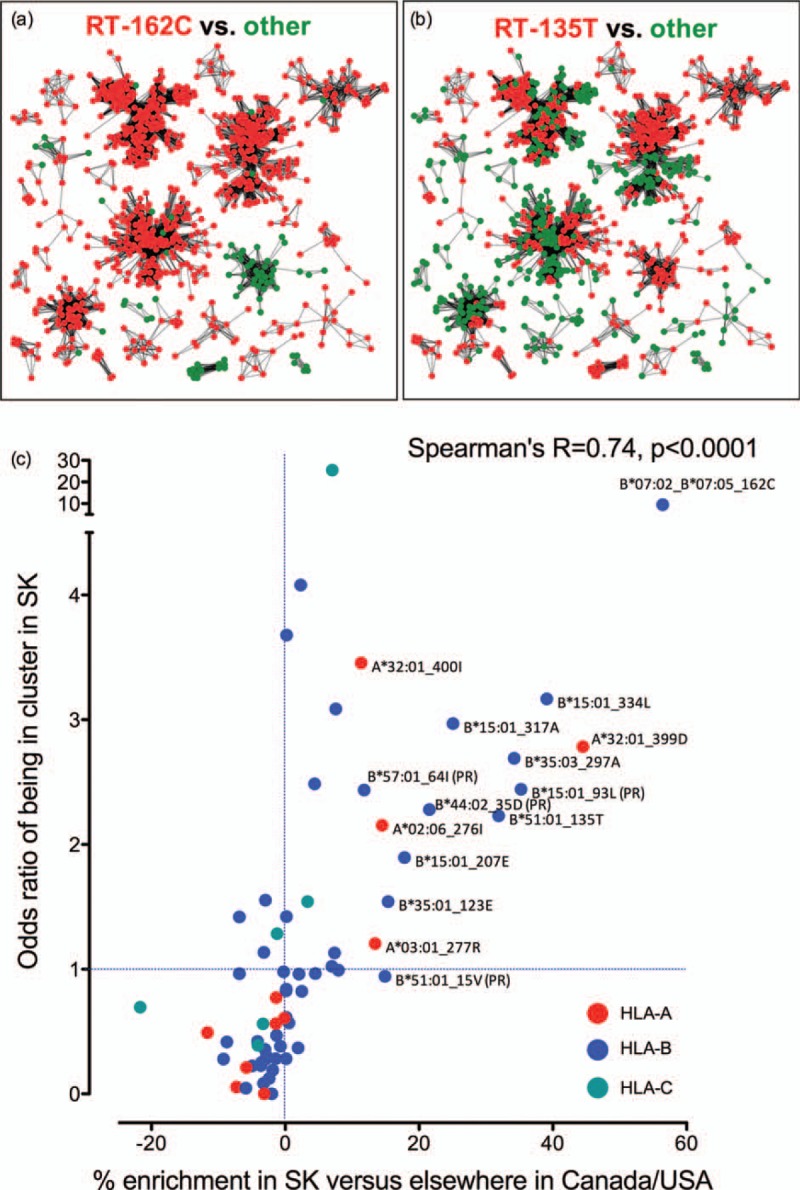
Panel a: Distribution of the B∗07 : 02/05-associated RT-S162C adaptation within major Saskatchewan sequence clusters. Sequences identified as being in a cluster of minimum size 5 were graphed such that each node represented an HIV sequence from a unique individual, and edges represent patristic (tip-to-tip) distances separating them. Sequences harbouring 162C are red, all others are green. Panel b: Distribution of the B∗51 : 01-associated RT-I135T adaptation within major Saskatchewan sequence clusters. Sequences harbouring 135T in red, all others are green. Panel c: Each dot represents one of the 70 HLA-associated adapted variants investigated in the present study, coloured by the HLA locus restricting it (HLA-A: red; HLA-B: blue and HLA-C: teal). For each mutation, its absolute % enrichment in Saskatchewan vs. CA/US (that is, the Δ of the linked values shown in Figures 2b–d) is plotted alongside its odds ratio of being located in an HIV sequence residing in a cluster in Saskatchewan, revealing a highly significant correlation between these factors. Mutations that are more than 10% enriched in Saskatchewan versus CA/US are labeled with their identity and HLA restriction. Results indicate that HLA-associated adapted variants enriched in Saskatchewan are preferentially located within sequence clusters.
We further reasoned that the extent of enrichment of a given HIV adaptation within Saskatchewan sequence clusters should correlate with the extent to which it is enriched in Saskatchewan compared to elsewhere on the continent. Thus, for each HLA-associated adaptation we plotted its % enrichment in Saskatchewan compared to CA/US (that is, the Δ of the linked frequencies shown in Fig. 2b–d) versus its odds ratio of being located within a Saskatchewan cluster (computed as above). We confirmed a highly significant positive correlation between these two parameters (Spearman's R = 0.74, P < 0.0001; Fig. 5c). The equivalent analysis performed on Saskatchewan versus BC datasets was similarly highly significant (Spearman's R = 0.69, P < 0.0001, not shown). Together, our observations support the notion that HIV sequences harbouring major HLA-adapted mutations are being transmitted in Saskatchewan more widely and frequently than those not harbouring such mutations, thus providing an explanation for why HIV adaptation to HLA is so elevated in this epidemic.
Discussion
Using comprehensive population-level datasets of HIV Pol sequences collected for drug resistance testing, we demonstrate that HIV strains circulating in Saskatchewan harbour markedly elevated levels of adaptation to numerous HLA alleles, and that these levels are generally increasing over time. Six common adaptations were especially elevated in Saskatchewan compared to BC and elsewhere in CA/US: B∗15 : 01-I93L in Protease and B∗51 : 01-I135T, B∗07 : 02/05-S162C, B∗35 : 03-E297A, B∗15 : 01-Q334L and A∗32 : 01- E399D in RT. Strikingly, more than 98% of Saskatchewan sequences collected in 2015/2016 harboured at least one of these mutations, more than 80% harboured at least three and 40% harboured five or more. Adaptation to the typically protective B∗51 [5,9–12] was particularly elevated: more than 75% of Saskatchewan sequences from 2015/2016 featured at least one B∗51-associated mutation, a level comparable to that typically observed within chronically-infected, antiretroviral-naive B∗51-expressing hosts (that is, under conditions where HIV had been evolving under direct B∗51-mediated selection for years [5,12,38]). This effectively means that a B∗51-expressing Saskatchewan resident has a more than 75% chance of being infected with an HIV strain that is likely to be inherently capable of evading their cellular immune responses to at least some extent. Similarly, 2015/2016 mutation levels indicate that persons expressing B∗07, B∗15 : 01 or B∗35 : 01 would have more than 80%, more than 85% and more than 75% chances, respectively, of acquiring an HIV strain preadapted to host cellular immunity.
Eighty percent of HIV-infected individuals in Saskatchewan have Indigenous ancestry [16]. Due to a strong population genetic bottleneck coincident with European colonization, genetic diversity in present-day North American Indigenous peoples is comparatively reduced [48,49]. Although HLA data were unavailable for the individuals presently studied, published HLA-A and HLA-B serotype distributions among Indigenous persons in Saskatchewan are indeed consistent with reduced HLA allelic diversity [21]. For example, the three most common HLA-B serotypes, observed in 30, 23, and 23% of Saskatchewan Indigenous persons respectively [21], were B51, B35, and B62 (where the latter includes B∗15 : 01) (Figure S7), such that Indigenous Saskatchewan residents have an estimated ∼70% probability of expressing at least one of these alleles. In contrast, no individual HLA-B allele was observed at more than 20% prevalence in a large HIV cohort in Canada/USA [38], where B∗51 : 01 and B∗35 : 01 frequencies were 10 and 13%, respectively (Figure S7).
More importantly, the HLA alleles to which the circulating HIV sequences in Saskatchewan harbor the highest level of adaptation, notably B∗51, B∗15 : 01, B∗35, B∗07 and B∗44, are those that are most commonly expressed in Indigenous persons (see Fig. 5 and S7). Though the lack of linked HLA and clinical information on studied individuals precludes our ability to directly link HIV adaptation to clinical outcomes, our observations nevertheless reveal plausible mechanisms to explain, at least in part, the extensive levels HIV preadaptation to HLA in Saskatchewan, as well as reports of rapid progression in the region [18]. Specifically, our findings support a scenario where limited host population immunogenetic diversity facilitates the initial selection of HIV immune escape mutations, which then stably persist upon transmission to hosts sharing these same HLA alleles, thereby undermining antiviral immunity which in turn accelerates progression [2–4] (in fact, our observations also potentially explain reports of B∗51-linked rapid HIV progression in neighbouring Manitoba [19,20], where 40% of HIV-infected persons self-identify as Indigenous [50] and where B∗51 prevalence is ∼20% [20]). Indeed, the abundance of short terminal branch lengths in the Saskatchewan phylogeny [overall median 1.57e−3 (interquartile range, IQR 5.50e−4 to 6.18e−3) nucleotide substitutions/site] compared to 1.62e–2 [IQR 4.50e–3 to 2.75e–3] in CA/USA, P < 0.0001 is consistent with widespread circulation of HIV strains preadapted to host HLA, where limited de-novo HIV evolution occurs following transmission.
In many ways the situation in Saskatchewan parallels that in Japan, where high population HLA-B∗51 prevalence has facilitated the spread of B∗51-adapted HIV variants, thereby eroding B∗51-associated protective effects in the region [5,7,11,13]. However, Saskatchewan's unique HIV molecular epidemiology, in particular the presence of large phylogenetic clusters that are significantly enriched in HLA-adapted HIV sequences, distinguishes it from other epidemics with documented HLA adaptation: specifically, our observations suggest that highly HLA-adapted HIV variants are being more widely and frequently transmitted than those with lower levels of adaptation. Though the lack of sociodemographic and ethnicity data precludes us from investigating this directly, it is tempting to speculate that the HIV strains that are being widely transmitted among Indigenous communities are the ones that harbor the highest levels of adaptation to HLA alleles expressed in these populations.
Some additional caveats and limitations merit mention. While the BC and Saskatchewan HIV sequences were collected as part of comprehensive clinical HIV drug resistance monitoring programmes (which include baseline genotyping, and in the case of BC, automatic genotyping upon HIV diagnosis, as standard of care) and are thus highly representative of HIV diversity in these provinces, the continental comparison (LANL) dataset simply represents all publicly-deposited HIV subtype B Pol sequences from Canada or the USA that had been annotated by collection year; and as such may not be representative of the full range of HIV diversity across these two countries. Nevertheless our observations that HIV adaptation to HLA in Saskatchewan is far higher than in either BC or elswhere in Canada/USA further supports our overall conclusions that the Saskatchewan epidemic is unique in terms of HIV molecular epidemiology and HLA adaptation. Although our analysis included 34 HLA subtypes (>95% of HIV-infected persons in Canada/USA are estimated to express at least one of these [38]), some HLA alleles (e.g., A∗02 : 01 and B∗08 : 01) were not assessed because they do not strongly select adaptations in the studied Pol region. Pol-based analyses may also underestimate HIV adaptation to HLA alleles whose main epitopes are located elsewhere, in particular Gag (e.g., B∗27 and B∗57) [51–54]. Nevertheless, Pol contains numerous conserved CTL epitopes critical to immune control [12,38,41,51,55] and Pol preadaptation is the strongest significant negative correlate of outcomes following infection [3], supporting the proposed link between widespread circulating Pol adaptation to common HLA alleles in the population (including in key epitopes targeted in early infection [56]) and poorer clinical outcomes following acquisition of such strains [3]. Finally, without linked sociodemographic, clinical and HLA information on our cohorts we cannot directly link infection with preadapted HIV to poorer clinical outcomes, nor can we reconstruct the dynamics of HLA-driven selection and reversion of these mutations in Saskatchewan. Nevertheless the population-level accumulation of HIV immune escape mutations in other global epidemics, and the demonstrated negative clinical consequences of preadapted HIV transmission, broadly support our inferences [3,5–8].
In conclusion, our observations highlight the utility of HIV Pol sequences, routinely collected for antiretroviral drug resistance testing, for regional surveillance of circulating HIV adaptation to host cellular immunity. Our results also identify Saskatchewan as the first documented example of a North American HIV epidemic characterized by significant circulating viral adaptation to HLA [6,8]. As no licensed preventive or therapeutic vaccine exists to boost anti-HIV immunity against strains harbouring transmitted (or de novo selected) immune escape mutations, our observation of extensive HIV adaptation to host HLA in Saskatchewan, combined with regional reports of rapid HIV progression [18], add even greater urgency to addressing this epidemic in which infection burden is already concentrated among the most marginalized persons [14,15]. Our findings underscore the critical need to partner with local communities to scale-up HIV diagnosis, treatment, prevention and harm reduction initiatives to reduce HIV-related morbidity and mortality and to curb onward HIV transmission in the province.
Acknowledgements
Z.L.B. designed the study, analyzed and interpreted data, and drafted the manuscript. N.N.K., S.S., A.W., E.M., K.D.C., P.S., P.N.L. and P.R.H. collected, contributed, analyzed and/or interpreted data. JBJ performed the phylogenetic clustering analyses, interpreted data and contributed to writing. All authors provided critical feedback on the manuscript.
We thank Roland Dyck for helpful discussions and permission to incorporate HLA frequencies, published in reference [21], into our study. We thank Jonathan M. Carlson for helpful discussions and the development of the adaptation algorithm, Richard Liang for database assistance, Mark A. Brockman for critical feedback and the laboratory staff at the BC Centre for Excellence in HIV/AIDS for technical support.
This work was supported by a Canadian Institutes for Health Research (CIHR) Project grant (PJT-148621) to Z.L.B. N.N.K. was supported by a Frederick Banting and Charles Best CIHR MSc award. Z.L.B. is supported by a Scholar Award from the Michael Smith Foundation for Health Research.
Conflicts of interest
There are no conflicts of interest.
Supplementary Material
References
- 1.Wainberg MA, Friedland G. Public health implications of antiretroviral therapy and HIV drug resistance. JAMA 1998; 279:1977–1983. [DOI] [PubMed] [Google Scholar]
- 2.Monaco DC, Dilernia DA, Fiore-Gartland A, Yu T, Prince JL, Dennis KK, et al. Balance between transmitted HLA preadapted and nonassociated polymorphisms is a major determinant of HIV-1 disease progression. J Exp Med 2016; 213:2049–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carlson JM, Du VY, Pfeifer N, Bansal A, Tan VY, Power K, et al. Impact of preadapted HIV transmission. Nat Med 2016; 22:606–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Katoh J, Kawana-Tachikawa A, Shimizu A, Zhu D, Han C, Nakamura H, et al. Rapid HIV-1 disease progression in individuals infected with a virus adapted to its host population. PLoS One 2016; 11:e0150397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawashima Y, Pfafferott K, Frater J, Matthews P, Payne R, Addo M, et al. Adaptation of HIV-1 to human leukocyte antigen class I. Nature 2009; 458:641–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cotton LA, Kuang XT, Le AQ, Carlson JM, Chan B, Chopera DR, et al. Genotypic and functional impact of HIV-1 adaptation to its host population during the North American epidemic. PLoS Genet 2014; 10:e1004295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Payne R, Muenchhoff M, Mann J, Roberts HE, Matthews P, Adland E, et al. Impact of HLA-driven HIV adaptation on virulence in populations of high HIV seroprevalence. Proc Natl Acad Sci U S A 2014; 111:E5393–E5400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kinloch NN, MacMillan DR, Le AQ, Cotton LA, Bangsberg DR, Buchbinder S, et al. Population-level immune-mediated adaptation in HIV-1 polymerase during the North American epidemic. J Virol 2015; 90:1244–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Brien SJ, Gao X, Carrington M. HLA and AIDS: a cautionary tale. Trends Mol Med 2001; 7:379–381. [DOI] [PubMed] [Google Scholar]
- 10.Carrington M, O’Brien SJ. The influence of HLA genotype on AIDS. Annu Rev Med 2003; 54:535–551. [DOI] [PubMed] [Google Scholar]
- 11.Kawashima Y, Kuse N, Gatanaga H, Naruto T, Fujiwara M, Dohki S, et al. Long-term control of HIV-1 in hemophiliacs carrying slow-progressing allele HLA-B∗5101. J Virol 2010; 84:7151–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Peng Y, Yan H, Xu K, Saito M, Wu H, et al. Multilayered defense in HLA-B51-associated HIV viral control. J Immunol 2011; 187:684–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koga M, Kawana-Tachikawa A, Heckerman D, Odawara T, Nakamura H, Koibuchi T, et al. Changes in impact of HLA class I allele expression on HIV-1 plasma virus loads at a population level over time. Microbiol Immunol 2010; 54:196–205. [DOI] [PubMed] [Google Scholar]
- 14.Vogel L. HIV in Saskatchewan merits urgent response. CMAJ 2015; 187:793–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vogel L. Saskatchewan won’t declare HIV emergency. CMAJ 2016; 188:1071–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Government_of_Saskatchewan. HIV prevention and control report for 2016. Regina, SK: Ministry of Health; 2017. [Google Scholar]
- 17.Centers_for_Disease_Control_and_Prevention. HIV Surveillance Report, 2016; vol. 28. Diagnoses of HIV infection in the United States and dependent areas. Atlanta, GA: Centers for Disease Control and Prevention; 2017. [Google Scholar]
- 18.Hunt K, Mondal P, Konrad S, Skinner S, Gartner K, Lim HJ. Identifying factors associated with changes in CD4(+) count in HIV-infected adults in Saskatoon, Saskatchewan. Can J Infect Dis Med Microbiol 2015; 26:207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keynan Y, Becker M, Rueda Z, Bresler K, Kasper K. Rapid human immunodeficiency virus disease progression is associated with human leukocyte antigen-B homozygocity and human leukocyte antigen-B51 in a cohort from Manitoba, Canada. Infect Dis (Lond) 2015; 47:447–452. [DOI] [PubMed] [Google Scholar]
- 20.Keynan Y, Rueda ZV, Bresler K, Becker M, Kasper K. HLA B51 is associated with faster AIDS progression among newly diagnosed HIV-infected individuals in Manitoba, Canada. Int J Immunogenet 2015; 42:336–340. [DOI] [PubMed] [Google Scholar]
- 21.Dyck R, Bohm C, Klomp H. Increased frequency of HLA A2/DR4 and A2/DR8 haplotypes in young saskatchewan aboriginal people with diabetic end-stage renal disease. Am J Nephrol 2003; 23:178–185. [DOI] [PubMed] [Google Scholar]
- 22.Galli RA, Sattha B, Wynhoven B, O'Shaughnessy MV, Harrigan PR. Sources and magnitude of intralaboratory variability in a sequence-based genotypic assay for human immunodeficiency virus type 1 drug resistance. J Clin Microbiol 2003; 41:2900–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gonzalez-Serna A, Min JE, Woods C, Chan D, Lima VD, Montaner JS, et al. Performance of HIV-1 drug resistance testing at low-level viremia and its ability to predict future virologic outcomes and viral evolution in treatment-naive individuals. Clin Infect Dis 2014; 58:1165–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woods CK, Brumme CJ, Liu TF, Chui CK, Chu AL, Wynhoven B, et al. Automating HIV drug resistance genotyping with RECall, a freely accessible sequence analysis tool. J Clin Microbiol 2012; 50:1936–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Government_of_Saskatchewan. Interim 2016 HIV/AIDS Key Information. Regina, SK: Ministry of Health; 2017. [Google Scholar]
- 26.BC_Centre_for_Disease_Control. British Columbia annual summary of reportable diseases. Vancouver, BC: 2017. [Google Scholar]
- 27.Ragonnet-Cronin M, Aris-Brosou S, Joanisse I, Merks H, Vallee D, Caminiti K, et al. Genetic diversity as a marker for timing infection in HIV-infected patients: evaluation of a 6-month window and comparison with BED. J Infect Dis 2012; 206:756–764. [DOI] [PubMed] [Google Scholar]
- 28.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 2002; 30:3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Larsson A. AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014; 30:3276–3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Siepel AC, Halpern AL, Macken C, Korber BT. A computer program designed to screen rapidly for HIV type 1 intersubtype recombinant sequences. AIDS Res Hum Retroviruses 1995; 11:1413–1416. [DOI] [PubMed] [Google Scholar]
- 31.Price MN, Dehal PS, Arkin AP. FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS One 2010; 5:e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shafer RW, Rhee SY, Pillay D, Miller V, Sandstrom P, Schapiro JM, et al. HIV-1 protease and reverse transcriptase mutations for drug resistance surveillance. AIDS 2007; 21:215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Poon AF, Joy JB, Woods CK, Shurgold S, Colley G, Brumme CJ, et al. The impact of clinical, demographic and risk factors on rates of HIV transmission: a population-based phylogenetic analysis in British Columbia, Canada. J Infect Dis 2015; 211:926–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moore CB, John M, James IR, Christiansen FT, Witt CS, Mallal SA. Evidence of HIV-1 adaptation to HLA-restricted immune responses at a population level. Science 2002; 296:1439–1443. [DOI] [PubMed] [Google Scholar]
- 35.Brumme ZL, Brumme CJ, Heckerman D, Korber BT, Daniels M, Carlson J, et al. Evidence of differential HLA class i-mediated viral evolution in functional and accessory/regulatory genes of HIV-1. PLoS Pathog 2007; 3:e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rousseau CM, Daniels MG, Carlson JM, Kadie C, Crawford H, Prendergast A, et al. HLA class-I driven evolution of human immunodeficiency virus type 1 subtype C proteome: immune escape and viral load. J Virol 2008; 82:6434–6446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brumme ZL, John M, Carlson JM, Brumme CJ, Chan D, Brockman MA, et al. HLA-associated immune escape pathways in HIV-1 subtype B Gag, Pol and Nef proteins. PLoS ONE 2009; 4:e6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carlson JM, Brumme CJ, Martin E, Listgarten J, Brockman MA, Le AQ, et al. Correlates of protective cellular immunity revealed by analysis of population-level immune escape pathways in HIV-1. J Virol 2012; 86:13202–13216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carlson JM, Le AQ, Shahid A, Brumme ZL. HIV-1 adaptation to HLA: a window into virus-host immune interactions. Trends Microbiol 2015. [DOI] [PubMed] [Google Scholar]
- 40.Carlson JM, Schaefer M, Monaco DC, Batorsky R, Claiborne DT, Prince J, et al. Selection bias at the heterosexual HIV-1 transmission bottleneck. Science 2014; 345:1254031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van Tran G, Chikata T, Carlson JM, Murakoshi H, Nguyen DH, Tamura Y, et al. A strong association of human leukocyte antigen-associated Pol and Gag mutations with clinical parameters in HIV-1 subtype A/E infection. AIDS 2016; 30:681–689. [DOI] [PubMed] [Google Scholar]
- 42.Kinloch NNGQ, Lee JM, Brumme CCJ, Byakwaga H, Muzoora C, et al. Subtype-specific constraints on HIV-1 adaptation to host cellular immunity revealed through statistical and functional analyses. 9th IAS conference on HIV Science Paris, France 2017. [Google Scholar]
- 43.Ragonnet-Cronin M, Ofner-Agostini M, Merks H, Pilon R, Rekart M, Archibald CP, et al. Longitudinal phylogenetic surveillance identifies distinct patterns of cluster dynamics. J Acquir Immune Defic Syndr 2010; 55:102–108. [DOI] [PubMed] [Google Scholar]
- 44.Ragonnet-Cronin M, Aris-Brosou S, Joanisse I, Merks H, Vallee D, Caminiti K, et al. Adaptive evolution of HIV at HLA epitopes is associated with ethnicity in Canada. PLoS ONE 2012; 7:e36933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hightower GK, May SJ, Perez-Santiago J, Pacold ME, Wagner GA, Little SJ, et al. HIV-1 clade B pol evolution following primary infection. PLoS One 2013; 8:e68188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Little SJ, Kosakovsky Pond SL, Anderson CM, Young JA, Wertheim JO, Mehta SR, et al. Using HIV networks to inform real time prevention interventions. PLoS One 2014; 9:e98443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Llano A, Williams A, Olvera A, Silva-Arrieta S, Brander C. Best-characterized HIV-1 CTL epitopes: the 2013 update. In: HIV Molecular Immunology 2013. In Yusim K, Korber B, Brander C. HIV molecular immunology 2013. Los Alamos: Theoretical Biology and Biophysics Group, Los Alamos National Laboratory, Los Alamos, NM. LA-UR 13-27758; 2013. pp. 3–19. [Google Scholar]
- 48.O’Fallon BD, Fehren-Schmitz L. Native Americans experienced a strong population bottleneck coincident with European contact. Proc Natl Acad Sci U S A 2011; 108:20444–20448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mulligan CJ, Hunley K, Cole S, Long JC. Population genetics, history, and health patterns in native americans. Annu Rev Genomics Hum Genet 2004; 5:295–315. [DOI] [PubMed] [Google Scholar]
- 50.2017. 2016 Manitoba HIV Program Update. [Google Scholar]
- 51.Kiepiela P, Ngumbela K, Thobakgale C, Ramduth D, Honeyborne I, Moodley E, et al. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat Med 2007; 13:46–53. [DOI] [PubMed] [Google Scholar]
- 52.Rolland M, Heckerman D, Deng W, Rousseau CM, Coovadia H, Bishop K, et al. Broad and Gag-biased HIV-1 epitope repertoires are associated with lower viral loads. PLoS ONE 2008; 3:e1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zuniga R, Lucchetti A, Galvan P, Sanchez S, Sanchez C, Hernandez A, et al. Relative dominance of Gag p24-specific cytotoxic T lymphocytes is associated with human immunodeficiency virus control. J Virol 2006; 80:3122–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Edwards BH, Bansal A, Sabbaj S, Bakari J, Mulligan MJ, Goepfert PA. Magnitude of functional CD8+ T-cell responses to the gag protein of human immunodeficiency virus type 1 correlates inversely with viral load in plasma. J Virol 2002; 76:2298–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Payne RP, Kloverpris H, Sacha JB, Brumme Z, Brumme C, Buus S, et al. Efficacious early antiviral activity of HIV Gag- and Pol-specific HLA-B 2705-restricted CD8+ T cells. J Virol 2010; 84:10543–10557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Altfeld M, Kalife ET, Qi Y, Streeck H, Lichterfeld M, Johnston MN, et al. HLA alleles associated with delayed progression to AIDS contribute strongly to the initial CD8(+) T Cell Response against HIV-1. PLoS Med 2006; 3:e403. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.