

Supplemental Digital Content is available in the text.
Keywords: angiogenesis; mice, transgenic; myocardial infarction; myocytes, cardiac; regeneration
Abstract
Background:
Although c-Kit+ adult progenitor cells were initially reported to produce new cardiomyocytes in the heart, recent genetic evidence suggests that such events are exceedingly rare. However, to determine if these rare events represent true de novo cardiomyocyte formation, we deleted the necessary cardiogenic transcription factors Gata4 and Gata6 from c-Kit–expressing cardiac progenitor cells.
Methods:
Kit allele–dependent lineage tracing and fusion analysis were performed in mice following simultaneous Gata4 and Gata6 cell type–specific deletion to examine rates of putative de novo cardiomyocyte formation from c-Kit+ cells. Bone marrow transplantation experiments were used to define the contribution of Kit allele–derived hematopoietic cells versus Kit lineage–dependent cells endogenous to the heart in contributing to apparent de novo lineage-traced cardiomyocytes. A Tie2CreERT2 transgene was also used to examine the global impact of Gata4 deletion on the mature cardiac endothelial cell network, which was further evaluated with select angiogenesis assays.
Results:
Deletion of Gata4 in Kit lineage–derived endothelial cells or in total endothelial cells using the Tie2CreERT2 transgene, but not from bone morrow cells, resulted in profound endothelial cell expansion, defective endothelial cell differentiation, leukocyte infiltration into the heart, and a dramatic increase in Kit allele–dependent lineage-traced cardiomyocytes. However, this increase in labeled cardiomyocytes was an artefact of greater leukocyte-cardiomyocyte cellular fusion because of defective endothelial cell differentiation in the absence of Gata4.
Conclusions:
Past identification of presumed de novo cardiomyocyte formation in the heart from c-Kit+ cells using Kit allele lineage tracing appears to be an artefact of labeled leukocyte fusion with cardiomyocytes. Deletion of Gata4 from c-Kit+ endothelial progenitor cells or adult endothelial cells negatively impacted angiogenesis and capillary network integrity.
Clinical Perspective.
What Is New?
This is the first study to genetically delete the necessary cardiogenic transcription factors Gata4/6 from c-Kit+ cardiac progenitor cells, which remarkably resulted in greater apparent cardiomyocyte derivation from these c-Kit+ cells.
Deletion of Gata4 from c-Kit–derived endothelial progenitors alters the integrity of the endothelial cell network in the heart, resulting in more c-Kit+–derived leukocytes entering the heart and fusing with cardiomyocytes.
We demonstrate a new role for Gata4 in endothelial differentiation, specifically showing for the first time that Gata4 is essential for vascular development via the c-Kit lineage.
This study shows that leukocyte-to-cardiomyocyte fusion is the primary basis for past lineage-tracing results incorrectly suggesting that c-Kit+ cardiac progenitor cells generated de novo cardiomyocytes in the heart.
What Are the Clinical Implications?
Our data demonstrate that c-Kit+ cardiac progenitor cells are much less likely to differentiate de novo into cardiomyocytes than previously reported, suggesting that such cells are not therapeutically meaningful as a source of new cardiomyocytes.
Our study highlights a capillary-driven mechanism of increased fusion of bone marrow–derived cells (leukocytes) with existing cardiomyocytes, which could have significant clinical implications in its own right.
Kit lineage and global endothelial cell deletion of Gata4 reveal apparent organ-specific regulation of microvascular differentiation, highlighting Gata4 as a potential target for angiogenic control in the human heart.
Large-scale cardiomyocyte loss from a cardiac ischemic event initially elicits a dramatic inflammatory response, followed by fibroblast activation with scar formation and fibrosis, and then ventricular remodeling and eventually heart failure.1 To combat this profile of progressive cardiac deterioration after ischemic injury, cell-specific approaches have emerged with emphasis on altering the hematopoietic response,2 ameliorating fibrotic remodeling,3–6 increasing collateral circulation,7,8 and preserving or replacing cardiomyocytes.9,10 Earlier reports that endogenous cardiac stem cells exist and might be efficacious in mediating cardiac regeneration generated a great deal of excitement in the field.11,12 c-Kit+ cardiac progenitor cells (CPCs), named for the presence of c-Kit tyrosine kinase receptor that marks hematopoietic stem cells,13 have been the focus of numerous cardiac regenerative studies.14–16 Select clinical trials evaluating the administration of bone marrow cells after myocardial infarction have shown minimal efficacy.17–19 However, expanded cardiac c-Kit+ cells were reported to potentially impart greater functional benefit with scar reduction when administered to patients post–myocardial infarction injury.20,21
Although injection of exogenously expanded CPCs may indeed positively impact the myocardial infarction–injured heart, several recent studies have definitively shown that the heart lacks an endogenous c-Kit+ CPC capable of producing new cardiomyocytes in vivo.22–24 For example, we determined that endothelial cells are the major fate of Kit lineage–traced cells in the heart and that only 1 in 17 000 cardiomyocytes might be produced de novo when an 80% fusion rate is taken into account.22 Sultana and colleagues23 confirmed these results, demonstrating that a large proportion of lineage-traced Kit allele–derived cells are endothelial, whereas Kit allele lineage–traced cardiomyocytes coexpressing cardiac troponin T in the adult mouse heart were exceptionally rare. Furthermore, a novel Cre/Dre dual recombinase mouse genetic system by He and colleagues,24 which no longer relies on the heterozygosity of the Kit allele, showed that c-Kit+ cells never produce de novo cardiomyocytes in the adult heart at baseline or with injury. However, the potential to genetically reprogram c-Kit–derived cells and other cardiac mesenchymal cells into cardiomyocytes remains attractive for future development.25,26
The goal of the current study was to determine the contribution of true versus apparent Kit allele lineage–derived cardiomyocytes by simultaneously deleting the cardiomyogenic transcription factors Gata4 and Gata6, which, when deleted from mesodermal progenitors during early development, results in acardia and absence of cardiomyocytes.27 We report here that deletion of Gata4 from c-Kit+ cells preferentially impacted a population of c-Kit–expressing endothelial cells and their differentiation state, resulting in defective cardiac capillary formation and immune cell infiltration, which secondarily resulted in a dramatically enhanced rate of fusion of c-Kit–derived immune cells with cardiomyocytes.
Methods
An expanded Methods section is available in the online-only Data Supplement.
The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedure by contacting the corresponding author. The RNA sequencing data were deposited with the GEO database group and given accession number GSE109661.
Animals
All experiments involving mice were approved by the Institutional Animal Care and Use Committee at Cincinnati Children’s Hospital Medical Center. Kit-Cre recombinase knock-in mice (KitCre) and Kit mice with a knock-in of the tamoxifen-inducible MerCreMer cDNA (KitMCM), and Rosa26 lineage-dependent reporter mice, as well, were each previously described.22 LoxP-targeted Gata4 (Gata4fl) and Gata6 (Gata6fl) mice were described previously.28,29 The endothelial specific Tie2CreERT2 transgenic mouse model was described elsewhere (Y. Zheng and colleagues, unpublished data).29a Detailed descriptions of tamoxifen-dosing procedures and euthanasia procedures are provided in the online-only Data Supplement.
Protein Analysis
Immunofluorescent stains were performed on cryoembedded tissues,22 and Western blots were performed on cells isolated by fluorescence-activated cell sorting and whole cardiac ventricle tissue as described previously.30 Descriptions of the antibodies used are provided in the online-only Data Supplement.
Vascular Endothelial Growth Factor-A Overexpression
An adeno-associated virus-9 vector expressing VEGF-A (vascular endothelial growth factor-A) was prepared and titered (online-only Data Supplement Methods). Adult C57Bl/6 mice were administered adeno-associated virus-9–VEGF-A by tail vein injection at concentration of 1×1012 viral particles in 200 µL of sterile phosphate-buffered saline.
Bone Marrow Transplantation
Bone marrow transplantations were performed by transplanting bone marrow cells from 6- to 8-week-old donor mice into 8- to 10-week-old recipients. Bone marrow cells (BMCs) were isolated by aseptically flushing femurs and tibiae with Hanks’ Balanced Salt Solution. Bone marrow transplant recipient mice first received lethal irradiation (12 Gy in a divided dose 4 hours apart31) and then immediately received a tail vein injection of BMCs at ≈50 million per mouse in 200 µL of Hanks’ Balanced Salt Solution.
Retinal Angiogenesis Assay
To observe the superficial vascular plexus to assess endothelial developmental defects, retinal flat mounts were prepared as described earlier.5 In brief, postnatal day (p) 0 mice were given intraperitoneal injections of 200 μg tamoxifen (Sigma T5648; dissolved in 5 μL ethanol and 95 μL peanut oil) for gene inactivation and Tie2 (Tek gene) lineage tracing. Neonatal mice were euthanized, their eyes were harvested at p8 and fixed, and the retinas were dissected and mounted for qualitative comparison by enhanced green fluorescent protein (eGFP) fluorescence.
Flow Cytometry and Cell Sorting
Cells were prepared for flow cytometry studies on cardiac interstitial cells and BMCs as described in detail before.4,22,32 BMCs and cardiac interstitial cells were stained with surface markers using allophycocyanin-conjugated antibodies, and optimum gating strategy was determined using singly labeled controls.
RNA Sequencing Analysis
Endothelial cells were collected as described above for RNA harvest and production of cDNA libraries for sequencing. Libraries were sequenced on the Illumina HiSeq2500 following the manufacturer’s protocol. Selected genes were verified by quantitative polymerase chain reaction.
Statistics
See Methods in the online-only Data Supplement for an in-depth description of statistics.
Results
Kit Lineage-Specific Gata4-, Gata6-, and Gata4/6-Deleted Mice
To examine the hypothesis that the deletion of Gata4 and Gata6 from c-Kit+ CPCs in the heart would block all de novo cardiomyocyte formation, we crossed KitMCM mice with mice containing loxP site (fl)–targeted alleles for Gata4 and Gata6 (Figure 1A). Again, loss of both transcription factors should lead to the inability of CPCs to activate the cardiac transcriptional program required for cardiogenesis.27,33,34 In addition, mice were bred to harbor either the Rosa26-loxP-stop-loxP-eGFP (R26eGFP) allele or the Rosa26-membrane-Tomato-loxP-Stop-loxP-membrane-eGFP allele35 (R26mT/mG) to trace total Kit allele–derived cells in the heart and to examine fusion-derived cardiomyocytes, respectively (Figure 1A). Fusion is scored when both the m-eGFP and m-Tomato signals are observed in the same cardiomyocyte, whereas a de novo transdifferentiation event from a c-Kit+ CPC would give cardiomyocytes that are only m-eGFP positive. After weaning, mice were put on tamoxifen food for continuous labeling of Kit lineage–derived cells (Figure 1B). Recombination of these loxP-targeted alleles was assessed by polymerase chain reaction on control and tamoxifen-treated mice, showing specific recombination (Figure 1C). Mice were euthanized after 1, 2, and 4 months of tamoxifen treatment for histological and flow cytometry analysis of heart and other tissues. Unexpectedly, total eGFP+ cardiomyocytes were increased in mice with deletion of Gata4/6 or Gata4, but not Gata6 alone with ≈2-fold, 4-fold, and 10-fold increases at 1, 2, and 4 months, respectively (Figure 1D through 1F). Dual reporter labeling in Gata4/6- and Gata4-deleted mice revealed significantly more fusion-derived cardiomyocytes (91%–93%) versus 80% to 83% in KitMCM R26mT/mG controls (Figure 1G and 1H). It is interesting to note that skeletal muscle showed the appearance of eGFP+-fused myofibers in Gata4/6- and Gata4-deleted Kit-lineage mice for the first time (Figure I in the online-only Data Supplement), whereas other known immune cell–based fusion-prone tissues such as the liver36,37 did not (data not shown). These results collectively show that deletion of Gata4 from Kit allele–derived cells dramatically enhances the relative rate of apparent cardiomyocyte de novo formation, which is potentially attributable to augmented fusion of Kit allele–derived immune cells with tissue parenchymal cells as previously shown.38,39
Figure 1.
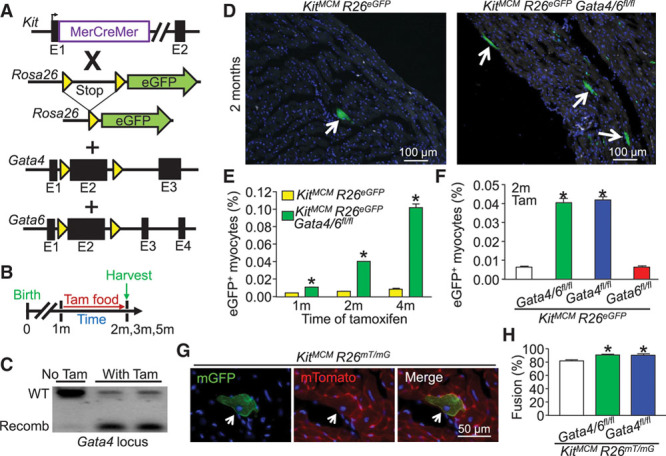
Kit allele-lineage deletion of Gata4/6 and Gata4 alone leads to increased lineage-traced cardiomyocytes attributable to fusion. A, Genetic models with the indicated alleles in mice that were crossed. B, Experimental design of the protocol used in this figure. C, PCR showing recombination (recomb) of the Gata4 locus with tamoxifen treatment to activate the MerCreMer protein (n=2) or untreated that did not show recombination (n=1) in KitMCM Gata4/6fl/fl R26eGFP mice. The analysis used Kit lineage tracing in which eGFP+ bone marrow cells were first collected. D, Immunofluorescent images of cardiac histological sections from Kit allele lineage–traced mice with and without Gata4/6 deletion. Arrows indicate eGFP+ cardiomyocytes. E, Time-course quantification of the percentage of eGFP+ cardiomyocytes within the entire heart in response to Gata4/6 deletion. Error bars are SEM, n=4 per group (10 sections per heart). *P<0.05 vs. KitMCM R26eGFP at each time point. F, Comparative quantitation of the percentage of eGFP+ cardiomyocytes within the entire mouse heart in response to Gata4, Gata6, or double Gata4/6 deletion in comparison with Kit lineage tracing controls. Error bars are SEM, n=4 per group (10 sections per heart). *P<0.05 vs. KitMCM R26eGFP for each genotype. G, Immunofluorescent cardiac histological section showing dual reporter evidence of a fusion-derived mTomato+ mGFP+ cardiomyocyte. H, Percentage of fusion-derived Kit allele lineage–traced cardiomyocytes (mTomato+mGFP+) vs. presumed de novo cardiomyocytes (only mGFP+). Error bars are SEM, n=4 per group (10 sections per heart). *P<0.05 vs. KitMCM R26mT/mG for each genotype. eGFP indicates enhanced green fluorescent protein; PCR, polymerase chain reaction; Tam, tamoxifen; and WT, wild type.
Immunofluorescence staining of Gata4 was frequently positive in total aggregates of BMCs, which is also a known site of c-Kit expression as shown by lineage tracing from the Kit allele to produce eGFP expression (Figure 2A). It is interesting to note that CD133+ endothelial progenitor cells40 from the bone marrow were dramatically enhanced in Kit allele-lineage Gata4/6–deleted mice in comparison with controls, possibly suggesting a functional role for Gata4 in endothelial cells (Figure 2B). Indeed, previous lineage tracing with the KitCre mouse model demonstrated that cardiac Kit-derived cell populations in the heart comprised 77% endothelial cells (CD31+) and 18% leukocytes (CD45+).22 Western blotting of these populations for Gata4 protein demonstrated detectable expression in CD31+ endothelial cells but not in CD45+ leukocytes or total CD31− CD45− cardiac interstitial cells, or when Gata4 was deleted from CD31+ cells using a Tie2CreERT2 transgene (Figure 2C). Flow cytometry analysis for CD45+ cell content in the hearts of Kit-lineage Gata4/6- and Gata4-deleted mice after 4 months of tamoxifen showed a ≈50% increase in leukocytes (Figure 2D and 2F) and a ≈60% increase in CD31+ endothelial cells in comparison with controls (Figure 2E and 2G). Taken together, these results show a clear alteration of Kit allele–derived populations in the heart in the absence of Gata4, such as a dramatic increase in endothelial and CD45 cell content.
Figure 2.
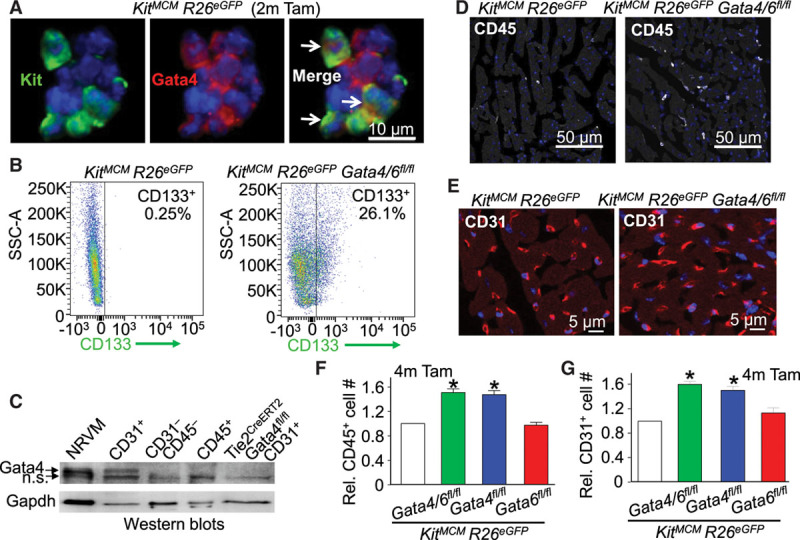
Cardiac leukocytes and endothelial cells are increased in Kit lineage Gata4-deleted mice. A, Immunofluorescent staining of Gata4 (red) and DAPI (blue) in bone marrow cells from KitMCM R26eGFP mice (2 months tamoxifen as control) showing some overlap of Kit lineage–traced eGFP+ cells with Gata4. B, Flow cytometry plots showing CD133+ endothelial progenitor cells in Kit allele–derived bone marrow cells. C, Western blot showing relative Gata4 levels in c-Kit lineages such as CD45+ leukocytes, CD31+ endothelial cells, and CD31− CD45− interstitial cells, or CD31+ cells that were deleted for Gata4 with the Tie2CreERT2 transgene. The positive control was neonatal rat ventricular myocytes (NRVM) loaded at a reduced amount to show the Gata4 band accurately. Although Gapdh was used as a processing and loading control, this protein is more highly expressed in cardiomyocytes and select other cell types. D, Immunofluorescent staining of CD45+ (white) and DAPI (blue) in cardiac histological sections. E, Immunofluorescent staining of CD31+ (red) and DAPI (blue) in cardiac histological sections. F, Relative quantification of cardiac CD45+ cells based on flow cytometry of the interstitial cell fraction from dissociated hearts. Error bars are SEM, n=3 per group. *P<0.05 vs. KitMCM R26eGFP for each genotype. G, Relative quantification of cardiac CD31+ cells based on flow cytometry from the interstitial cell fraction in dissociated hearts. Error bars are SEM, n=3 per group. *P<0.05 vs. KitMCM R26eGFP for each genotype. DAPI indicates 4′,6-diamidino-2-phenylindole; n.s., nonspecific; SSC-A, side scatter area; and Tam, tamoxifen.
Bone Marrow Transplant to Identify Cardiovascular Impact of Kit-Lineage Gata4/6 Loss
We hypothesized that the increase in relative Kit allele lineage–traced eGFP+ cardiomyocytes with Gata4/6 deletion was attributable to greater leukocyte activity or altered endothelial cell function. To test this hypothesis, we performed a bone marrow transplant that would isolate the impact of Gata4/6 effects from the hematopoietic compartment. Here, the donating bone marrow was from KitMCM Gata4/6fl/fl R26eGFP mice (or controls not deleted for Gata4/6) that had already undergone full recombination with 6 weeks of prior tamoxifen treatment, which would produce eGFP+ circulating immune cells and even endothelial progenitors in recipients (Figure 3A). The recipient mice were in the R26mT genetic background so endogenous cells could be parsed from eGFP+ cells. Recipient mice were irradiated, then given the bone marrow transplant and allowed to reconstitute for 8 weeks (Figure 3A). Full bone marrow niche reconstitution was confirmed with Kit-lineage R26eGFP recombination (Figure 3B) and a general lack of native mTomato+ bone marrow (Figure 3C). eGFP+ cardiomyocytes were observed in the hearts of both control and Gata4/6-deleted bone marrow recipients at the same level, which was attributable to fusion because 100% of the cells were both eGFP+ and mTomato+ (Figure 3D and 3E). Furthermore, flow cytometry analysis of cardiac interstitial cells revealed no difference in total CD31+ cells (Figure 3F), demonstrating that Gata4/6-deleted bone marrow alone did not recapitulate the phenotype of endothelial expansion or the large relative increase in eGFP+ cardiomyocytes observed in the standard Kit lineage–traced Gata4/6-deleted mice at baseline.
Figure 3.
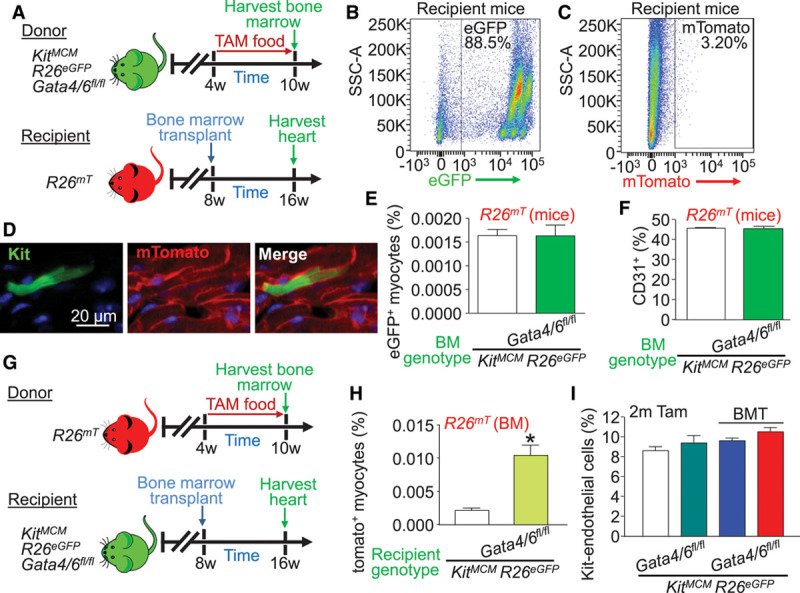
Increased cardiomyocyte cell fusion events occur with Kit lineage loss of Gata4/6 within heart cells during bone marrow transplant. A, Experimental design for bone marrow transplant experiments examining the impact of Gata4/6 loss in Kit allele–derived bone marrow on cardiac phenotyping. B, Representative FACS plot showing recombination of eGFP+ bone marrow from a Kit allele lineage–traced donor mouse in a bone marrow transplant recipient. C, Representative FACS plot showing effective irradiation and loss of native mTomato+ bone marrow in a recipient mouse. D, Representative eGFP+ mTomato+ fusion-derived cardiomyocyte from a cardiac histological section following bone marrow transplant. E, Percentage of Kit allele–dependent bone marrow–derived eGFP+ cardiomyocytes from the entire heart of the indicated mice. Error bars are SEM, n=6 per group (10 sections per heart). F, Quantification of cardiac CD31+ cells based on flow cytometry from the interstitial fraction in bone marrow transplant recipient hearts from the indicated mice. Error bars are SEM, n=3 per group, P=n.s. G, Experimental design for bone marrow transplantation to examine the impact of endogenous cardiac loss of Gata4/6 in Kit allele–dependent lineages on the apparent rate of new cardiomyocytes and endothelial cells. H, Percentage of wild-type bone marrow–derived mTomato+ cardiomyocytes within the entire heart following bone marrow transplant in the indicated mice. Error bars are SEM, n=6 per group (10 sections per heart), *P<0.05 vs. KitMCM R26eGFP recipient. I, Quantification from cardiac sections of endogenous endothelial cell number from the Kit lineage with 2 months of tamoxifen induction, with and without bone marrow transplantation, in the indicated mice. Error bars are SEM, n=3 per group, P=n.s. for all pairwise comparisons. BM indicates bone marrow; BMT, bone marrow transplantation; eGFP, enhanced green fluorescent protein; FACS, fluorescence-activated cell sorting; n.s., nonspecific; SSC-A, side scatter area; and Tam, tamoxifen.
Next, we performed a reverse bone marrow transplant strategy. Recipient KitMCM mice with or without Gata4/6 deletion were lethally irradiated at 8 weeks of age, whereafter they received control bone marrow from global R26mT donors (Figure 3G). After 2 months of tamoxifen treatment, mice were euthanized, and cardiac sections were analyzed for mTomato+-traced cardiomyocytes within Gata4/6 Kit lineage–deleted hearts. The data show an ≈4-fold increase in bone marrow–dependent mTomato+ cardiomyocytes with endogenous Kit allele-dependent Gata4/6 deletion in comparison with Gata4/6 wild-type controls, suggesting that it is the deletion of Gata4/6 within the existing heart cells that leads to greater apparent cardiomyocyte fusion/labeling (Figure 3H). It is important to note that Kit-lineage endothelial cell recombination in bone marrow recipient mice was not significantly changed from the ≈8% to 10% of total cardiac endothelium observed in both control and Gata4/6-deleted hearts, with and without bone marrow transplant (Figure 3I). Together, these experiments demonstrate that a Kit lineage–derived cell source endogenous to the heart is the mechanistic basis for the increased ability of wild-type bone marrow progenitor–derived cells (leukocytes) to fuse with cardiomyocytes in vivo.
Endothelial cells are the predominant Kit lineage–derived cell type present in the heart,22 suggesting the hypothesis that loss of Gata4/6 within endogenous cardiac endothelial cells might be mechanistically responsible for greater immune cell infiltration and fusion with cardiomyocytes. To test this hypothesis, we first attempted to alter the differentiation and permeability characteristics of endogenous cardiac endothelial cells by using VEGF-A. Previous work has shown that VEGF-A overexpression leads to weakening of endothelial junctions and increased permeability as these cells partially dedifferentiate or proliferate.41,42 Here, we used a VEGF-A adeno-associated virus-9 to overexpress this factor in the heart of Kit lineage–traced mice by tail vein injection (Figure IIA in the online-only Data Supplement). VEGF-A overexpression in the heart (Figure IIB in the online-only Data Supplement) produced a significant increase in eGFP+ cardiomyocytes and leukocyte infiltration (Figure IIC through IIE in the online-only Data Supplement), reminiscent of the increases observed in Gata4/6-deleted mice.
Global Endothelial Deletion of Gata4
Despite the modest proportion of Kit allele–derived endothelium (≈10%), the loss of Gata4 in these cells of the heart appeared to cause a global cardiac phenotype of endothelial cell expansion, leukocyte infiltration, and heterotypic cardiomyocyte fusion, suggesting that Kit allele–derived endothelial cells were critical in adult vascular maintenance. To further implicate endothelial cells as the causative cell type, we used a tamoxifen-inducible Tie2 Cre-expressing transgenic mouse (Tie2CreERT2) to achieve a global and adult endothelial cell–specific deletion of Gata4. Tie2 lineage–traced mice were used as controls versus lineage-traced Gata4-deleted mice with a continuous tamoxifen regimen, again beginning after weaning (Figure 4A and 4B). It is remarkable that after 2 months of tamoxifen, Gata4-deleted endothelial cells showed a ≈2-fold increase in Tie2-eGFP+ in the heart (Figure 4C and 4D) with a significant shift from CD31 high-expressing to CD31 low-expressing cells by flow cytometry (Figure 4G and 4H), suggesting a less differentiated state. Adult endothelial cell–specific deletion of Gata4 also resulted in the greater presence of CD45+ cells in the heart (Figure 4E and 4F). In addition, 5-ethynyl-2′-deoxyuridine staining of cardiac sections showed a 3-fold increase in endothelial cell–specific proliferation (Figure 4I). Skeletal muscle showed similar results with a shift toward CD31 low-expressing cells and increased 5-ethynyl-2′-deoxyuridine staining (Figure IIIA and IIIB in the online-only Data Supplement). Together, these data suggest that loss of Gata4 in adult endothelial cells alters their differentiated state and produces greater proliferation and leukocyte diapedesis.
Figure 4.
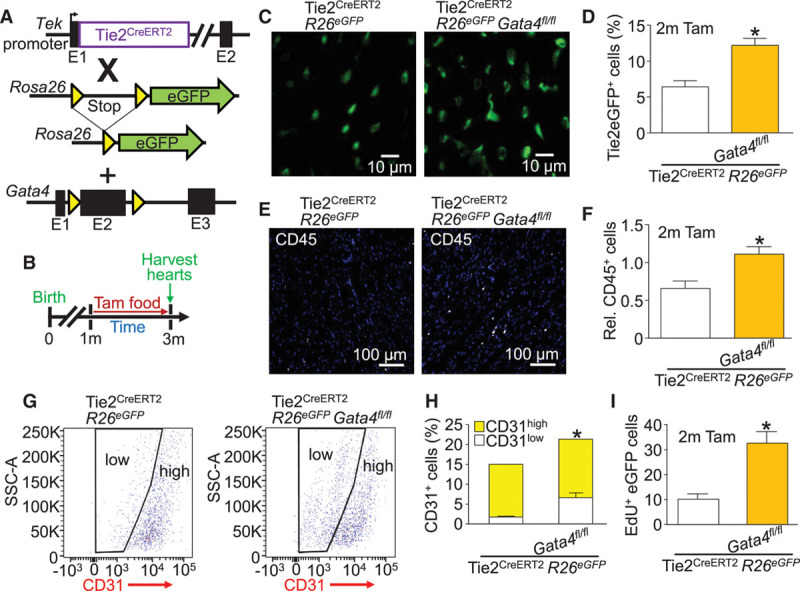
Global Tie2-lineage deletion of Gata4 expands the immature endothelial cell population and increases diapedesis. A, Genetic models with the indicated alleles or transgenes in mice that were crossed. B, Experimental design of the protocol used in this figure. The Tie2 promoter from the Tek gene was used to drive the Cre-ERT2 cDNA. C, Representative cardiac histological sections showing Tie2 lineage–traced eGFP+ cells (green) from the indicated genotypes of mice. D, Quantification of Tie2 lineage–traced eGFP+ endothelial cells by flow cytometry from the interstitial fraction in hearts of the indicated lines of mice. Error bars are SEM, n=3 per group, *P<0.05 vs. Tie2CreERT2 R26eGFP. E, Representative cardiac histological sections showing CD45+ cells (white) and DAPI (blue) from the indicated genotypes of mice. F, Quantification of CD45+ cells by flow cytometry of the interstitial fraction in the hearts of the indicated lines of mice. Error bars are SEM, n=3 per group, *P<0.05 vs. Tie2CreERT2 R26eGFP. G, Flow cytometry plots showing cardiac CD31+ cells with gating to display low- and high-expressing cells from the indicated genotypes of mice. H, Quantification of CD31low, CD31high, and total CD31+ cells from flow cytometry of the interstitial fraction of hearts of the indicated genotypes of mice. Error bars are SEM, n=3 per group, P=0.0257 for CD31 low comparison, P=0.1799 for CD31 high comparison, and P<0.0001 for total CD31+ comparison vs. Tie2CreERT2 R26eGFP. I, Quantification of EdU+ nuclei in Tie2 lineage-traced eGFP+ cardiac endothelial cells from cardiac histological sections from the indicated genotypes of mice. Error bars are SEM, n=3 per group (6 EdU-stained sections per heart). *P<0.05 vs. Tie2CreERT2 R26eGFP. DAPI indicates 4′,6-diamidino-2-phenylindole; EdU, 5-ethynyl-2′-deoxyuridine; eGFP, enhanced green fluorescent protein; SSC-A, side scatter area; and Tam, tamoxifen.
To probe even further into the hypothesis that deletion of Gata4 from Kit allele lineage–derived endothelial cells is the primary reason for enhanced bone marrow–derived leukocyte fusion with endogenous cardiomyocytes, we used the KitMCM allele together with the Tie2CreERT2 transgene in Gata4fl/fl R26eGFP mice given continuous tamoxifen (Figure 5A and 5B). Unfortunately, the Tie2CreERT2 transgene is not active in bone marrow; hence, the KitMCM allele is needed to label fusigenic immune cells, even though both Cre alleles would inactivate Gata4 within endothelial cells of the heart (Figure 5C). The data again showed significantly greater rates of eGFP+-fused cardiomyocytes with KitMCM allele–mediated deletion of Gata4, and that enhanced deletion of Gata4 from essentially all endogenous cardiac endothelial cells with the Tie2CreERT2 transgene doubled the number of fused cardiomyocytes (Figure 5D and 5E). To show this even more conclusively, bone marrow transplantation was performed between a R26mT donor mouse and a Tie2CreERT2 Gata4fl/fl R26eGFP recipient mouse. In this manner, the recipient mice only have Tie2CreERT2 causing Gata4 deletion in adult endothelial cells without deletion of Gata4 from bone marrow–derived immune cells or endothelial progenitors. The data again show an enhanced rate of mTomato+-fused cardiomyocytes, indicating that the effect is exclusively attributable to defects within endogenous endothelial cells of the heart when Gata4 is deleted (Figure 5G and 5H).
Figure 5.
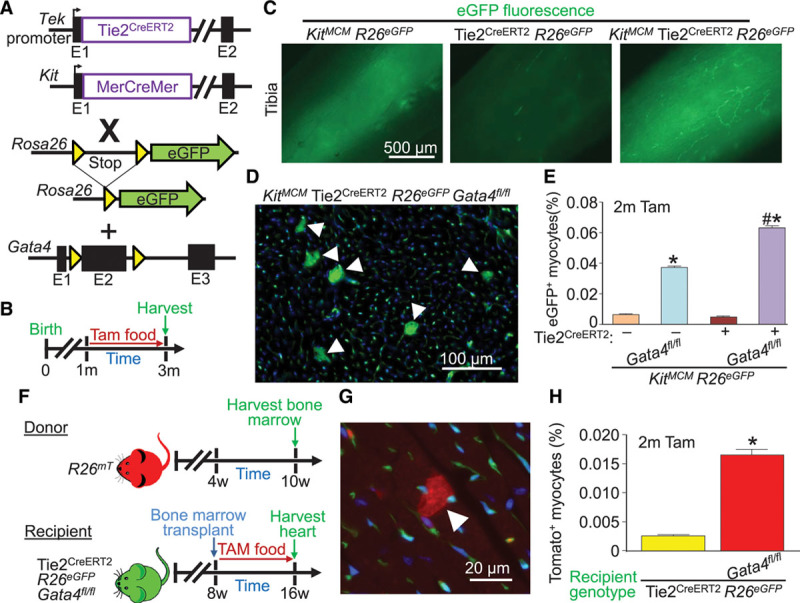
Global Tie2-lineage deletion of Gata4 augments appearance of Kit lineage– and bone marrow–labeled cardiomyocytes. A, Genetic models with the indicated alleles or transgenes in mice that were crossed. B, Experimental design of the protocol used in this figure. The Tie2 promoter from the Tek gene was used to drive the Cre-ERT2 cDNA. C, Whole-mount imaging of tibias from mice of the indicated genotypes of mice, highlighting labeled bone marrow cells (green) when the Kit lineage–tracing allele was used. D, Representative image showing accumulation of Kit allele–derived eGFP+ cardiomyocytes (arrowheads) in cardiac histological sections from the indicated mouse genotype 2 months after Gata4 deletion. E, Quantification of eGFP+ cardiomyocytes from KitMCM lineage tracing and KitMCM Tie2CreERT2 dual lineage tracing, with or without Gata4 deletion. Error bars are SEM, n=4 per group. *P<0.001 vs. KitMCM R26eGFP controls by 2-way ANOVA. #P<0.001 vs. dual KitMCM Tie2CreERT2 R26eGFP by 2-way ANOVA. F, Experimental design for bone marrow transplant of wild-type mTomato+ bone marrow into Tie2CreERT2 recipient mice with or without Gata4 deletion. G, Representative cardiac histological image of an mTomato+ cardiomyocyte (arrowhead) derived from bone marrow following bone marrow transplant from the mice shown in F. H, Quantification of mTomato+ cardiomyocytes from cardiac histological sections of recipient mice of the indicated genotypes following bone marrow transplant. Error bars are SEM, n=6 per group (10 sections per heart). *P<0.05 vs. Tie2CreERT2 R26eGFP recipient mice. eGFP indicates enhanced green fluorescent protein; and Tam, tamoxifen.
Ex Vivo and Developmental Analysis of Gata4-Deleted Endothelial Cells
Isolation and culturing of endothelial cells from these genetically modified mouse hearts grossly showed that Gata4-replete cells form a typical cobblestone monolayer, whereas Gata4-deleted cells are less adherent and show a more torturous morphology (Figure 6A). Analysis of cell culture supernatant showed an increase in secretion of angiopoietin-2 and decreased VEGF-A from Gata4-deleted endothelial cells, which suggests a vascular-regressive state (Figure 6B and 6C).43,44 Endothelial cells mixed with Matrigel from these mice were subcutaneously injected into wild-type recipient mice to evaluate vascular tube formation with or without Gata4 deletion (Figure 6D). After 2 weeks, Matrigel plugs were collected and analyzed by confocal microscopy, which showed a limited ability of Gata4-deleted endothelial cells to form differentiated vascular networks in comparison with Gata4-replete cells using z-stacked confocal microscopy (Figure 6E).
Figure 6.
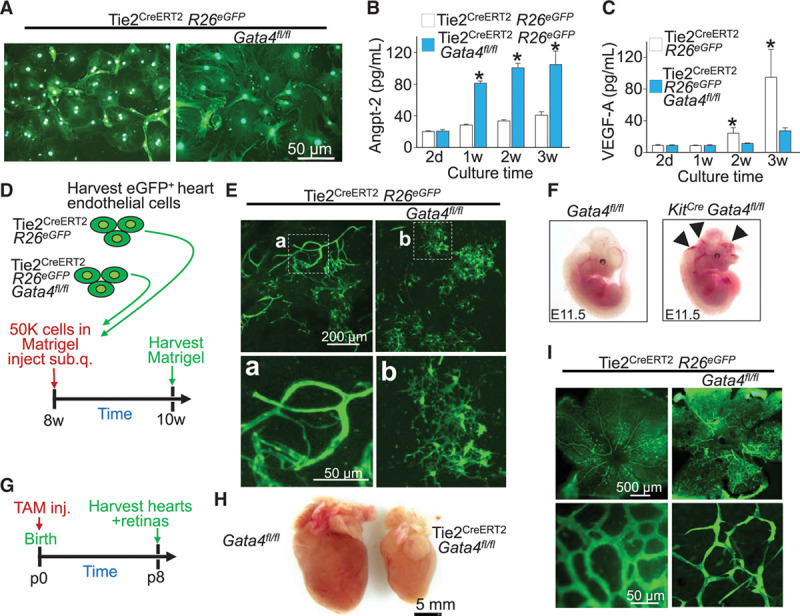
Gata4-deleted endothelial cells demonstrate poor differentiation and impaired tube formation. A, eGFP-based imaging of cardiac endothelial cells grown in culture after isolation by fluorescence-activated cell sorting of Tie2-eGFP+ CD31+ cells from the indicated genotypes of mice. n=3 per group (12 wells of 10 000 cells per heart). Angiopoeitin-2 (B) and VEGF-A protein (C) quantification from the collected supernatants of cardiac endothelial cell cultures as described in A. Error bars are SEM, n=3 per group (3 replicates each). *P<0.05 vs. Tie2CreERT2 R26eGFP at each time point. D, Experimental design for ex vivo Matrigel angiogenesis assay from the indicated genotypes of mice over the 2-week time period. E, Representative images of tube formation from harvested Matrigel plugs (images presented as flattened z-stack) from the indicated genotypes of mice. The bottom 2 panels are enlarged versions of the smaller windows shown in the top 2 panels. n=4 per group. F, Pictures of whole-mount embryos from constitutive KitCre expressing mice demonstrating hemorrhagic phenotype with Gata4 deletion, but not in control embryos at embryonic day (E) 11.5 of development. n=4. The arrowheads show areas of hemorrhage. G, Experimental design for the retinal angiogenesis assay from neonatal times of postnatal day 0 through postnatal day 8. H, Representative whole-mount images of postnatal day 8 hearts from control and Tie2CreERT2 Gata4-deleted hearts. I, Representative whole-mount images of neonatal retinas with eGFP imaging based on Tie2CreERT2 lineage tracing. The genotypes of mice used are shown, and the bottom 2 panels are higher magnification images of the upper 2 panels. n=6 per group. Angpt-2 indicates angiopoeitin-2; eGFP, enhanced green fluorescent protein; TAM, tamoxifen; and VEGF-A, vascular endothelial growth factor-A.
Previously, embryonic endothelial-specific deletion of Gata4 with a constitutive Tie2-Cre transgene demonstrated late embryonic or perinatal lethality because of defective epithelial-to-mesenchymal transition and improper heart valve formation.45 Here, we also observed embryonic lethality with a constitutive KitCre allele to delete Gata4. More specifically, constitutive KitCre Gata4fl/fl mice were found to be hemorrhagic at embryonic day 11.5 with noticeable areas of blood pooling and defective vasculogenesis, leading to embryonic lethality (Figure 6F). Constitutive Cdh5Cre (endothelial cell–specific cadherin 5 gene promoter)–mediated deletion of Gata4 also produced embryonic lethality at the same time (data not shown). Taken together, these results suggest that Kit allele–derived cells critically underlie early hemangioblast progenitor cell activity during development and that Gata4 is essential in these cells for proper vascular development.
Development of the superficial vascular plexus in the neonatal mouse eye begins at birth, with radial outgrowth of endothelial vessels reaching the retinal edge by p8.46 To test the role of Gata4 in retinal vascularization, Tie2-dependent lineage-traced mice were injected with tamoxifen at p0 and harvested at p8 for cardiac phenotyping and retinal angiogenesis (Figure 6G). Tie2CreERT2 Gata4fl/fl mice were underdeveloped in comparison with Gata4fl/fl controls with significantly reduced heart size (Figure 6H) and improper patterning of both micro- and macrovasculature in the retina in comparison with Tie2CreERT2 R26eGFP controls (Figure 6I). Together, these findings suggest that, in addition to being required embryonically for proper organogenesis, Gata4 is required for continued maturation and maintenance of the adult microvasculature.
Gene Expression Alterations in Adult Gata4-Deficient Endothelial Cells
To elucidate the molecular basis for Gata4-dependent regulation of endothelial cell differentiation, global RNA sequencing was performed from cardiac endothelial cells isolated from Tie2CreERT2 R26eGFP Gata4fl/fl mice versus Tie2CreERT2 R26eGFP controls (Figure 7). Cells double-positive for endogenous Tie2 lineage tracing (eGFP+) and fluorescently labeled for CD31 were obtained by fluorescence-activated cell sorting at 8 weeks of age after 4 weeks of tamoxifen induction. Bioinformatics analysis of the data showed a gene expression signature in which many of the top categories are related to previously known partners and targets of Gata4, and genes related to the function and maintenance of the vasculature (Figure 7). Although signature endothelial-defining genes47 (Cdh5, Edn1, Kdr, Pecam1, S1pr1, Vwf) were largely unchanged, well-established regulatory partners of Gata4, including Fog2 (Zfpm2),48,49 Hand2,50 Tbx20,51 Dkk3,52 Mlc2a (Mly7),53 Sox9,54 and Klf15,55 were significantly downregulated. A variety of putatively Gata4-regulated genes were also identified using the Harmonizome database,56 many of which have evidence for vascular regulation. For example, Figf (Vegfd), a predicted target of Gata4, was downregulated in this study and has a known role in endothelial cell regulation.57 A host of genes related in angiogenesis and extracellular matrix formation were impacted with Gata4 deletion including downregulation of Itgb4, Mmp2, Timp2, Vcam1, and Vcan and upregulation of Apold1 and Has3 (Figure 7). In addition, a number of genes related to the cell cycle in endothelial cells were upregulated, such as E2f7, Egr1/2/3, Esm1, and Plk3. Verification of mRNA expression differences was performed for most of these gene changes by quantitative reverse transcription polymerase chain reaction, showing consistency with the patterns found by RNA sequencing analysis (Figure IV in the online-only Data Supplement). Together, these gene expression data suggest a global impact of Gata4 deletion on the endothelial cell differentiation gene program, helping to validate the observed differences in proliferation, permeability, and maturation.
Figure 7.
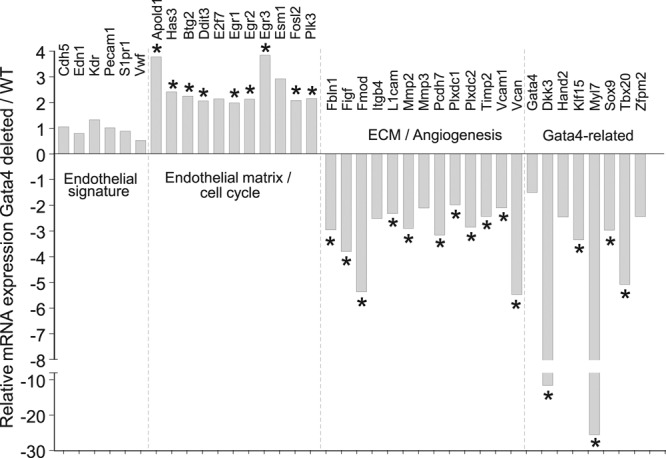
Gata4-deleted adult cardiac endothelial cells have dysregulation of cell cycle and angiogenesis-related genes. RNA sequencing analysis of selected genes from sorted Tie2 lineage–traced eGFP+ CD31+ cells from hearts of adult mice with or without Gata4 deletion mediated by the Tie2CreERT2 transgene (4 weeks of tamoxifen). Data show fold change in mRNA between Gata4-deleted and wild-type endothelial cells, and are organized to highlight genes relevant to previously defined endothelial gene sets or those known to interact with the transcription factor Gata4. n=2 per group (2 combined digested hearts each). See Methods in the online-only Data Supplement for description of bioinformatics and by DESeq2 analysis to show genes differentially expressed. *P<0.05 vs. WT endothelial cells. Eight of the mRNAs shown were not statistically significant by DESeq2 but are included because they were significantly changed by RT-PCR (see Figure IV in the online-only Data Supplement). ECM indicates extracellular matrix; eGFP, enhanced green fluorescent protein; RT-PCR, reverse transcriptase polymerase chain reaction; and WT, wild type.
Discussion
Here, we used a genetic approach to determine if the very low level of presumed new cardiomyocyte generation in the heart from the Kit lineage really occurs, especially because it was recently reported that adult cardiac Kit-dependent CPCs never truly transdifferentiate into cardiomyocytes.24 Indeed, deletion of Gata4 and Gata6 simultaneously from Kit lineage cells did not reduce the apparent rate of cardiomyocyte labeling in the heart by genetic lineage tracing, and in fact caused as much as a 10-fold increase. Because Gata4/6 are lacking, it is even more unlikely that new cardiomyocytes could be created from a progenitor cell source, even embryonic stem cells.27 This consideration, along with the very dramatic increase in cell fusion we observed in the current study, suggest that most past results reporting a minor contribution of cardiomyocytes from the Kit lineage are likely because of cellular fusion between lineage-traced immune cells and host endogenous cardiomyocytes, or from ectopic induction of the KitMCM allele in cardiomyocytes.24 Indeed, our past results, which were subtracted for cellular fusion effects, still showed apparent de novo cardiomyocyte production at ≈1 in 17 000, but, as suggested in the recent literature, this likely reflects ectopic activation of the KitCre allele within rare cardiomyocytes.24,58 Hence, it could be argued that Kit lineage–traced cells from any source, whether bone marrow or endogenous to the heart, essentially lack all cardiomyocyte transdifferentiation ability.
In attempting to understand how Gata4 deletion from the Kit lineage produced an apparent 10-fold increase in eGFP+ cardiomyocytes, we uncovered an essential role for this transcription factor in regulating the endothelial cell gene program. Although endothelial cell–specific deletion of Gata4 was shown to cause developmental lethality because of alterations in endothelial cell activity in the cardiac cushions and newly developing heart valves,45 a role for Gata4 in adult cardiac vascular maintenance had not been previously explored. Gata4 was also recently identified as a master regulator of liver sinusoidal endothelial cells, determining their organ specificity and requirement in liver development.59 In addition, in this transition from sinusoidal to capillary formed vascular networks in the liver, the authors observed a distinct shift in CD31low cells to CD31high cells.59 Conversely, we observed a shift toward more permeable vasculature and greater CD31low cells (less differentiated) in the heart and skeletal muscle with deletion of Gata4, but this effect was not observed in the liver or lung of our study using Tie2CreERT2 (data not shown).
We observed that Gata6 deletion in both Kit allele and endothelial lineages did not overtly alter the angiogenic and vascular processes as shown with Gata4 deletion. Indeed, we observed that constitutive KitCre-expressing Gata6fl/fl-deleted mice were not lethal and, when lineage-traced through development to 5 months of age, did not show increases in eGFP+ cardiomyocytes. Endothelial cell–specific deletion of Gata6 with the inducible Tie2-specific Cre mice also did not demonstrate an increase in CD31+ cells or increased vascular permeability, suggesting that only Gata4 plays a specific role in regulating the expression of genes involved in endothelial cell biology. We previously observed a similar paradigm at the level of the cardiomyocyte in which angiogenic genes in this cell type were preferentially regulated by Gata4 over Gata6.29
Currently, we speculate that Gata4 is important in endothelial cell maturation/differentiation largely because of the observed ex vivo defective tube formation and the appearance of CD31low endothelial cells in Tie2CreERT2 transgene-mediated Gata4-deleted mice. The overall increase in CD31+ and Tie2 lineage–traced eGFP+ cells could be a compensatory response of the cardiac vasculature because of the inability of endothelial cells to maintain secure junctions when Gata4-regulated gene expression is compromised. Thus, new endothelial cells are formed as an attempt to bolster the vasculature, yet leukocytes are still able to infiltrate more easily, giving greater fusion events with endogenous cardiomyocytes. Alternatively, poorly differentiated endothelial cells resulting from the deletion of Gata4 generate a tissue injury–like signal in these cells that recruits leukocytes and makes them more active. Overall, our results suggest that Gata4 could be used as a therapeutic leverage point in affecting endothelial cell biology for selective therapeutic approaches in humans.
Acknowledgments
Author contributions: B.D.M. conducted experiments and acquired the data along with assistance from O.K., J.K., H.K., X.F., J.G.B., and V.P. Y.Z. provided Tie2-Cre transgenic mice used in this study. J.D.M. and B.D.M. designed the experiments, analyzed the data, and wrote the manuscript. J.D.M. directed and supervised the study.
Sources of Funding
This work was supported by grants from the National Institutes of Health to Dr Molkentin and by the Howard Hughes Medical Institute to Dr Molkentin. B.D. Maliken was supported by the National Institutes of Health, National Heart, Lung, and Blood Institute F30 grant HL137239-01, American Heart Association predoctoral grant 17PRE33410368, and National Institutes of Health grant T32 HL007752-23S1.
Disclosures
Dr Molkentin received significant research support from National Institutes of Health grant R37HL60562; however, no direct conflicts of interest related to this study are declared.
Supplementary Material
Footnotes
Sources of Funding, see page 1022
The online-only Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/CIRCULATIONAHA.118.033703.
References
- 1.Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ. Heart disease and stroke statistics–2012 update: a report from the American Heart Association. Circulation. 2011;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. doi:10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Toldo S, Abbate A. The NLRP3 inflammasome in acute myocardial infarction. Nat Rev Cardiol. 2018;15:203–214. doi: 10.1038/nrcardio.2017.161. doi: 10.1038/nrcardio.2017.161. [DOI] [PubMed] [Google Scholar]
- 3.Talman V, Ruskoaho H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res. 2016;365:563–581. doi: 10.1007/s00441-016-2431-9. doi: 10.1007/s00441-016-2431-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, J Lin SC, Aronow BJ, Tallquist MD, Molkentin JD. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun. 2016;7:12260. doi: 10.1038/ncomms12260. doi: 10.1038/ncomms12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, Vagnozzi RJ, Liu R, Huynh T, Lee SJ, Karch J, Molkentin JD. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest. 2017;127:3770–3783. doi: 10.1172/JCI94753. doi: 10.1172/JCI94753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saxena A, Bujak M, Frunza O, Dobaczewski M, Gonzalez-Quesada C, Lu B, Gerard C, Frangogiannis NG. CXCR3-independent actions of the CXC chemokine CXCL10 in the infarcted myocardium and in isolated cardiac fibroblasts are mediated through proteoglycans. Cardiovasc Res. 2014;103:217–227. doi: 10.1093/cvr/cvu138. doi: 10.1093/cvr/cvu138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berry C, Balachandran KP, L’Allier PL, Lespérance J, Bonan R, Oldroyd KG. Importance of collateral circulation in coronary heart disease. Eur Heart J. 2007;28:278–291. doi: 10.1093/eurheartj/ehl446. doi: 10.1093/eurheartj/ehl446. [DOI] [PubMed] [Google Scholar]
- 8.Zimarino M, D’Andreamatteo M, Waksman R, Epstein SE, De Caterina R. The dynamics of the coronary collateral circulation. Nat Rev Cardiol. 2014;11:191–197. doi: 10.1038/nrcardio.2013.207. doi: 10.1038/nrcardio.2013.207. [DOI] [PubMed] [Google Scholar]
- 9.Senyo SE, Lee RT, Kühn B. Cardiac regeneration based on mechanisms of cardiomyocyte proliferation and differentiation. Stem Cell Res. 2014;13(3 p)(t B):532–541. doi: 10.1016/j.scr.2014.09.003. doi: 10.1016/j.scr.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kawaguchi N, Nakanishi T. Cardiomyocyte regeneration. Cells. 2013;2:67–82. doi: 10.3390/cells2010067. doi: 10.3390/cells2010067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu L, Li F, Lu J. Identification of functional tissue-resident cardiac stem/progenitor cells in adult mouse. Cell Biol Int Rep (2010) 2012;19:e00016. doi: 10.1042/CBR20120001. doi: 10.1042/CBR20120001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bollini S, Smart N, Riley PR. Resident cardiac progenitor cells: at the heart of regeneration. J Mol Cell Cardiol. 2011;50:296–303. doi: 10.1016/j.yjmcc.2010.07.006. doi: 10.1016/j.yjmcc.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 13.Fazel S, Cimini M, Chen L, Li S, Angoulvant D, Fedak P, Verma S, Weisel RD, Keating A, Li RK. Cardioprotective c-kit+ cells are from the bone marrow and regulate the myocardial balance of angiogenic cytokines. J Clin Invest. 2006;116:1865–1877. doi: 10.1172/JCI27019. doi: 10.1172/JCI27019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hosoda T. C-kit-positive cardiac stem cells and myocardial regeneration. Am J Cardiovasc Dis. 2012;2:58–67. [PMC free article] [PubMed] [Google Scholar]
- 15.Tallini YN, Greene KS, Craven M, Spealman A, Breitbach M, Smith J, Fisher PJ, Steffey M, Hesse M, Doran RM, Woods A, Singh B, Yen A, Fleischmann BK, Kotlikoff MI. c-kit expression identifies cardiovascular precursors in the neonatal heart. Proc Natl Acad Sci U S A. 2009;106:1808–1813. doi: 10.1073/pnas.0808920106. doi: 10.1073/pnas.0808920106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, Leri A, Kajstura J, Nadal-Ginard B, Anversa P. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003;114:763–776. doi: 10.1016/s0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- 17.Nowbar AN, Mielewczik M, Karavassilis M, Dehbi HM, Shun-Shin MJ, Jones S, Howard JP, Cole GD, Francis DP DAMASCENE writing group. Discrepancies in autologous bone marrow stem cell trials and enhancement of ejection fraction (DAMASCENE): weighted regression and meta-analysis. BMJ. 2014;348:g2688. doi: 10.1136/bmj.g2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jeevanantham V, Butler M, Saad A, Abdel-Latif A, Zuba-Surma EK, Dawn B. Adult bone marrow cell therapy improves survival and induces long-term improvement in cardiac parameters: a systematic review and meta-analysis. Circulation. 2012;126:551–568. doi: 10.1161/CIRCULATIONAHA.111.086074. doi: 10.1161/CIRCULATIONAHA.111.086074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kudo M, Wang Y, Wani MA, Xu M, Ayub A, Ashraf M. Implantation of bone marrow stem cells reduces the infarction and fibrosis in ischemic mouse heart. J Mol Cell Cardiol. 2003;35:1113–1119. doi: 10.1016/s0022-2828(03)00211-6. [DOI] [PubMed] [Google Scholar]
- 20.Bolli R, Chugh AR, D’Amario D, Loughran JH, Stoddard MF, Ikram S, Beache GM, Wagner SG, Leri A, Hosoda T, Sanada F, Elmore JB, Goichberg P, Cappetta D, Solankhi NK, Fahsah I, Rokosh DG, Slaughter MS, Kajstura J, Anversa P. Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO): initial results of a randomised phase 1 trial. Lancet. 2011;378:1847–1857. doi: 10.1016/S0140-6736(11)61590-0. doi: 10.1016/S0140-6736(11)61590-0. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Chugh AR, Beache GM, Loughran JH, Mewton N, Elmore JB, Kajstura J, Pappas P, Tatooles A, Stoddard MF, Lima JA, Slaughter MS, Anversa P, Bolli R. Administration of cardiac stem cells in patients with ischemic cardiomyopathy: the SCIPIO trial: surgical aspects and interim analysis of myocardial function and viability by magnetic resonance. Circulation. 2012;126(11)(suppl 1):S54–S64. doi: 10.1161/CIRCULATIONAHA.112.092627. doi: 10.1161/CIRCULATIONAHA.112.092627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Berlo JH, Kanisicak O, Maillet M, Vagnozzi RJ, Karch J, Lin SC, Middleton RC, Marbán E, Molkentin JD. c-kit+ cells minimally contribute cardiomyocytes to the heart. Nature. 2014;509:337–341. doi: 10.1038/nature13309. doi: 10.1038/nature13309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sultana N, Zhang L, Yan J, Chen J, Cai W, Razzaque S, Jeong D, Sheng W, Bu L, Xu M, Huang GY, Hajjar RJ, Zhou B, Moon A, Cai CL. Resident c-kit(+) cells in the heart are not cardiac stem cells. Nat Commun. 2015;6:8701. doi: 10.1038/ncomms9701. doi: 10.1038/ncomms9701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He L, Li Y, Li Y, Pu W, Huang X, Tian X, Wang Y, Zhang H, Liu Q, Zhang L, Zhao H, Tang J, Ji H, Cai D, Han Z, Han Z, Nie Y, Hu S, Wang QD, Sun R, Fei J, Wang F, Chen T, Yan Y, Huang H, Pu WT, Zhou B. Enhancing the precision of genetic lineage tracing using dual recombinases. Nat Med. 2017;23:1488–1498. doi: 10.1038/nm.4437. doi: 10.1038/nm.4437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Z, Zhu W, Bender I, Gong W, Kwak IY, Yellamilli A, Hodges TJ, Nemoto N, Zhang J, Garry DJ, van Berlo JH. Pathologic stimulus determines lineage commitment of cardiac c-kit+ cells. Circulation. 2017;136:2359–2372. doi: 10.1161/CIRCULATIONAHA.117.030137. doi: 10.1161/CIRCULATIONAHA.117.030137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kannappan R, Matsuda A, Ferreira-Martins J, Zhang E, Palano G, Czarna A, Cabral-Da-Silva MC, Bastos-Carvalho A, Sanada F, Ide N, Rota M, Blasco MA, Serrano M, Anversa P, Leri A. p53 modulates the fate of cardiac progenitor cells ex vivo and in the diabetic heart in vivo. EBioMedicine. 2017;16:224–237. doi: 10.1016/j.ebiom.2017.01.028. doi: 10.1016/j.ebiom.2017.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao R, Watt AJ, Battle MA, Li J, Bondow BJ, Duncan SA. Loss of both GATA4 and GATA6 blocks cardiac myocyte differentiation and results in acardia in mice. Dev Biol. 2008;317:614–619. doi: 10.1016/j.ydbio.2008.03.013. doi: 10.1016/j.ydbio.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oka T, Maillet M, Watt AJ, Schwartz RJ, Aronow BJ, Duncan SA, Molkentin JD. Cardiac-specific deletion of Gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ Res. 2006;98:837–845. doi: 10.1161/01.RES.0000215985.18538.c4. doi: 10.1161/01.RES.0000215985.18538.c4. [DOI] [PubMed] [Google Scholar]
- 29.van Berlo JH, Aronow BJ, Molkentin JD. Parsing the roles of the transcription factors GATA-4 and GATA-6 in the adult cardiac hypertrophic response. PLoS One. 2013;8:e84591. doi: 10.1371/journal.pone.0084591. doi: 10.1371/journal.pone.0084591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29a.Feng Y, Liu M, Guo F, Liu W, Sampson L, You L-R, Kuan C-Y, Zheng Y. Novel method to study mouse bone marrow endothelial cells in vivo and in vitro. Blood. 2012;120:617. [Google Scholar]
- 30.Karch J, Schips TG, Maliken BD, Brody MJ, Sargent MA, Kanisicak O, Molkentin JD. Autophagic cell death is dependent on lysosomal membrane permeability through Bax and Bak. eLife. 2017;6 doi: 10.7554/eLife.30543. doi: 10.7554/eLife.30543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cui YZ, Hisha H, Yang GX, Fan TX, Jin T, Li Q, Lian Z, Ikehara S. Optimal protocol for total body irradiation for allogeneic bone marrow transplantation in mice. Bone Marrow Transplant. 2002;30:843–849. doi: 10.1038/sj.bmt.1703766. doi: 10.1038/sj.bmt.1703766. [DOI] [PubMed] [Google Scholar]
- 32.Accornero F, Schips TG, Petrosino JM, Gu SQ, Kanisicak O, van Berlo JH, Molkentin JD. BEX1 is an RNA-dependent mediator of cardiomyopathy. Nat Commun. 2017;8:1875. doi: 10.1038/s41467-017-02005-1. doi: 10.1038/s41467-017-02005-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maitra M, Schluterman MK, Nichols HA, Richardson JA, Lo CW, Srivastava D, Garg V. Interaction of Gata4 and Gata6 with Tbx5 is critical for normal cardiac development. Dev Biol. 2009;326:368–377. doi: 10.1016/j.ydbio.2008.11.004. doi: 10.1016/j.ydbio.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xin M, Davis CA, Molkentin JD, Lien CL, Duncan SA, Richardson JA, Olson EN. A threshold of GATA4 and GATA6 expression is required for cardiovascular development. Proc Natl Acad Sci U S A. 2006;103:11189–11194. doi: 10.1073/pnas.0604604103. doi: 10.1073/pnas.0604604103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- 36.Vassilopoulos G, Wang PR, Russell DW. Transplanted bone marrow regenerates liver by cell fusion. Nature. 2003;422:901–904. doi: 10.1038/nature01539. doi: 10.1038/nature01539. [DOI] [PubMed] [Google Scholar]
- 37.Camargo FD, Finegold M, Goodell MA. Hematopoietic myelomonocytic cells are the major source of hepatocyte fusion partners. J Clin Invest. 2004;113:1266–1270. doi: 10.1172/JCI21301. doi: 10.1172/JCI21301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alvarez-Dolado M, Pardal R, Garcia-Verdugo JM, Fike JR, Lee HO, Pfeffer K, Lois C, Morrison SJ, Alvarez-Buylla A. Fusion of bone-marrow-derived cells with Purkinje neurons, cardiomyocytes and hepatocytes. Nature. 2003;425:968–973. doi: 10.1038/nature02069. doi: 10.1038/nature02069. [DOI] [PubMed] [Google Scholar]
- 39.Nygren JM, Jovinge S, Breitbach M, Säwén P, Röll W, Hescheler J, Taneera J, Fleischmann BK, Jacobsen SE. Bone marrow-derived hematopoietic cells generate cardiomyocytes at a low frequency through cell fusion, but not transdifferentiation. Nat Med. 2004;10:494–501. doi: 10.1038/nm1040. doi: 10.1038/nm1040. [DOI] [PubMed] [Google Scholar]
- 40.Yin AH, Miraglia S, Zanjani ED, Almeida-Porada G, Ogawa M, Leary AG, Olweus J, Kearney J, Buck DW. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood. 1997;90:5002–5012. [PubMed] [Google Scholar]
- 41.Gavard J, Gutkind JS. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol. 2006;8:1223–1234. doi: 10.1038/ncb1486. doi: 10.1038/ncb1486. [DOI] [PubMed] [Google Scholar]
- 42.Bates DO. Vascular endothelial growth factors and vascular permeability. Cardiovasc Res. 2010;87:262–271. doi: 10.1093/cvr/cvq105. doi: 10.1093/cvr/cvq105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, Compton D, McClain J, Aldrich TH, Papadopoulos N, Daly TJ, Davis S, Sato TN, Yancopoulos GD. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997;277:55–60. doi: 10.1126/science.277.5322.55. [DOI] [PubMed] [Google Scholar]
- 44.Hakanpaa L, Sipila T, Leppanen VM, Gautam P, Nurmi H, Jacquemet G, Eklund L, Ivaska J, Alitalo K, Saharinen P. Endothelial destabilization by angiopoietin-2 via integrin β1 activation. Nat Commun. 2015;6:5962. doi: 10.1038/ncomms6962. doi: 10.1038/ncomms6962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rivera-Feliciano J, Lee KH, Kong SW, Rajagopal S, Ma Q, Springer Z, Izumo S, Tabin CJ, Pu WT. Development of heart valves requires Gata4 expression in endothelial-derived cells. Development. 2006;133:3607–3618. doi: 10.1242/dev.02519. doi: 10.1242/dev.02519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stahl A, Connor KM, Sapieha P, Chen J, Dennison RJ, Krah NM, Seaward MR, Willett KL, Aderman CM, Guerin KI, Hua J, Löfqvist C, Hellström A, Smith LE. The mouse retina as an angiogenesis model. Invest Ophthalmol Vis Sci. 2010;51:2813–2826. doi: 10.1167/iovs.10-5176. doi: 10.1167/iovs.10-5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ho M, Yang E, Matcuk G, Deng D, Sampas N, Tsalenko A, Tabibiazar R, Zhang Y, Chen M, Talbi S, Ho YD, Wang J, Tsao PS, Ben-Dor A, Yakhini Z, Bruhn L, Quertermous T. Identification of endothelial cell genes by combined database mining and microarray analysis. Physiol Genomics. 2003;13:249–262. doi: 10.1152/physiolgenomics.00186.2002. doi: 10.1152/physiolgenomics.00186.2002. [DOI] [PubMed] [Google Scholar]
- 48.Manuylov NL, Tevosian SG. Cardiac expression of Tnnt1 requires the GATA4-FOG2 transcription complex. ScientificWorldJournal. 2009;9:575–587. doi: 10.1100/tsw.2009.75. doi: 10.1100/tsw.2009.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou B, Ma Q, Kong SW, Hu Y, Campbell PH, McGowan FX, Ackerman KG, Wu B, Zhou B, Tevosian SG, Pu WT. Fog2 is critical for cardiac function and maintenance of coronary vasculature in the adult mouse heart. J Clin Invest. 2009;119:1462–1476. doi: 10.1172/JCI38723. doi: 10.1172/JCI38723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dai YS, Cserjesi P, Markham BE, Molkentin JD. The transcription factors GATA4 and dHAND physically interact to synergistically activate cardiac gene expression through a p300-dependent mechanism. J Biol Chem. 2002;277:24390–24398. doi: 10.1074/jbc.M202490200. doi: 10.1074/jbc.M202490200. [DOI] [PubMed] [Google Scholar]
- 51.Stennard FA, Costa MW, Elliott DA, Rankin S, Haast SJ, Lai D, McDonald LP, Niederreither K, Dolle P, Bruneau BG, Zorn AM, Harvey RP. Cardiac T-box factor Tbx20 directly interacts with Nkx2-5, GATA4, and GATA5 in regulation of gene expression in the developing heart. Dev Biol. 2003;262:206–224. doi: 10.1016/s0012-1606(03)00385-3. [DOI] [PubMed] [Google Scholar]
- 52.Pei Y, Yao Q, Yuan S, Xie B, Liu Y, Ye C, Zhuo H. GATA4 promotes hepatoblastoma cell proliferation by altering expression of miR125b and DKK3. Oncotarget. 2016;7:77890–77901. doi: 10.18632/oncotarget.12839. doi: 10.18632/oncotarget.12839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holtzinger A, Rosenfeld GE, Evans T. Gata4 directs development of cardiac-inducing endoderm from ES cells. Dev Biol. 2010;337:63–73. doi: 10.1016/j.ydbio.2009.10.003. doi: 10.1016/j.ydbio.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Manuylov NL, Fujiwara Y, Adameyko II, Poulat F, Tevosian SG. The regulation of Sox9 gene expression by the GATA4/FOG2 transcriptional complex in dominant XX sex reversal mouse models. Dev Biol. 2007;307:356–367. doi: 10.1016/j.ydbio.2007.04.040. doi: 10.1016/j.ydbio.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fisch S, Gray S, Heymans S, Haldar SM, Wang B, Pfister O, Cui L, Kumar A, Lin Z, Sen-Banerjee S, Das H, Petersen CA, Mende U, Burleigh BA, Zhu Y, Pinto YM, Pinto Y, Liao R, Jain MK. Kruppel-like factor 15 is a regulator of cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A. 2007;104:7074–7079. doi: 10.1073/pnas.0701981104. doi: 10.1073/pnas.0701981104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rouillard AD, Gundersen GW, Fernandez NF, Wang Z, Monteiro CD, McDermott MG, Ma’ayan A. The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database. 2016 doi: 10.1093/database/baw100. doi: 10.1093/database/baw100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Karkkainen MJ, Petrova TV. Vascular endothelial growth factor receptors in the regulation of angiogenesis and lymphangiogenesis. Oncogene. 2000;19:5598–5605. doi: 10.1038/sj.onc.1203855. doi: 10.1038/sj.onc.1203855. [DOI] [PubMed] [Google Scholar]
- 58.Liu Q, Yang R, Huang X, Zhang H, He L, Zhang L, Tian X, Nie Y, Hu S, Yan Y, Zhang L, Qiao Z, Wang QD, Lui KO, Zhou B. Genetic lineage tracing identifies in situ Kit-expressing cardiomyocytes. Cell Res. 2016;26:119–130. doi: 10.1038/cr.2015.143. doi: 10.1038/cr.2015.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Géraud C, Koch PS, Zierow J, Klapproth K, Busch K, Olsavszky V, Leibing T, Demory A, Ulbrich F, Diett M, Singh S, Sticht C, Breitkopf-Heinlein K, Richter K, Karppinen SM, Pihlajaniemi T, Arnold B, Rodewald HR, Augustin HG, Schledzewski K, Goerdt S. GATA4-dependent organ-specific endothelial differentiation controls liver development and embryonic hematopoiesis. J Clin Invest. 2017;127:1099–1114. doi: 10.1172/JCI90086. doi: 10.1172/JCI90086. [DOI] [PMC free article] [PubMed] [Google Scholar]