

Abstract
Alcohol withdrawal symptoms contribute to excessive alcohol drinking and relapse in alcohol-dependent individuals. Among these symptoms, alcohol withdrawal promotes hyperalgesia, but the neurological underpinnings of this phenomenon are not known. Chronic alcohol exposure alters cell signaling in the central nucleus of the amygdala (CeA), and the CeA is implicated in mediating alcohol dependence-related behaviors. The CeA projects to the periaqueductal gray (PAG), a region critical for descending pain modulation, and may have a role in alcohol withdrawal hyperalgesia. Here, we tested the roles of (1) CeA projections to PAG, (2) CeA melanocortin signaling, and (3) PAG μ-opioid receptor signaling in mediating thermal nociception and alcohol withdrawal hyperalgesia in male Wistar rats. Our results demonstrate that alcohol dependence reduces GABAergic signaling from CeA terminals onto PAG neurons and alters the CeA melanocortin system, that CeA-PAG projections and CeA melanocortin signaling mediate alcohol withdrawal hyperalgesia, and that μ-opioid receptors in PAG filter CeA effects on thermal nociception.
SIGNIFICANCE STATEMENT Hyperalgesia is commonly seen in individuals with alcohol use disorder during periods of withdrawal, but the neurological underpinnings behind this phenomenon are not completely understood. Here, we tested whether alcohol dependence exerts its influence on pain modulation via effects on the limbic system. Using behavioral, optogenetic, electrophysiological, and molecular biological approaches, we demonstrate that central nucleus of the amygdala (CeA) projections to periaqueductal gray mediate thermal hyperalgesia in alcohol-dependent and alcohol-naive rats. Using pharmacological approaches, we show that melanocortin receptor-4 signaling in CeA alters alcohol withdrawal hyperalgesia, but this effect is not mediated directly at synaptic inputs onto periaqueductal gray-projecting CeA neurons. Overall, our findings support a role for limbic influence over the descending pain pathway and identify a potential therapeutic target for treating hyperalgesia in individuals with alcohol use disorder .
Keywords: pain, descending inhibition, mu opioid receptors, optogenetics, electrophysiology
Introduction
Alcohol use disorder (AUD) affects >15 million Americans (Substance Abuse and Mental Health Services Administration, 2015) and is closely tied to pain in humans. Individuals with AUD are often diagnosed with comorbid pain disorders (Egli et al., 2012), with 30% of Americans reporting chronic, recurrent, or long-lasting pain (Johannes et al., 2010). Epidemiological studies estimate that 20%–50% of U.S. adults with chronic pain use alcohol to self-medicate (Brennan et al., 2005; Riley and King, 2009). In the laboratory, acute alcohol increases pain thresholds in humans (Wolff et al., 1942). Humans develop tolerance to the analgesic effects of alcohol following chronic use, and withdrawal from chronic alcohol use can produce paradoxical hyperalgesia, much like what is seen with chronic opioid use (Angst and Clark, 2006). Blunted alcohol analgesia and sensitized withdrawal hyperalgesia may combine to drive alcohol abuse in humans, eventually leading to or perpetuating alcohol dependence (Koob, 2008).
The central nucleus of the amygdala (CeA) is a GABAergic brain region (Duvarci and Pare, 2014) that undergoes major neuroadaptations in animals and humans during alcohol dependence and periods of alcohol withdrawal (Gilpin et al., 2015). Important for this study, the CeA integrates nociceptive information and sends projections to regions that gate descending pathways that modulate nociceptive afferent activity, including the periaqueductal gray (PAG) (Rizvi et al., 1991). The descending inhibitory pain pathway from the PAG to the rostroventral medulla (RVM) to the spinal cord ends with gating of incoming sensory information in the dorsal horn of the spinal cord (Baliki and Apkarian, 2015). Previous work has demonstrated analgesic effects of CeA stimulation that is contingent on μ-opioid receptor (MOR) activity in the PAG (Oliveira and Prado, 2001; Xu et al., 2003), as well as hyperalgesic effects of CeA silencing (Itoga et al., 2016). The present study uses circuit-based techniques to demonstrate a role for the CeA and CeA-PAG projections in thermal nociception and alcohol withdrawal-induced hyperalgesia in rats.
Brain melanocortin-4 receptor (MC4R) activity is pronociceptive in rodents, and MC4R blockade attenuates (1) tolerance to the analgesic efficacy of opioids, and (2) the emergence of paradoxical hyperalgesia during morphine withdrawal (Kalange et al., 2007). Constitutively active (Tao, 2014) MC4Rs are widely distributed in both ascending and descending pain pathways (Caruso et al., 2014), including the lateral and medial subdivisions of the CeA and the PAG (Kishi et al., 2003), as are the endogenous MC4R ligand α-melanocyte-stimulating hormone (αMSH) and the endogenous antagonist/inverse agonist agouti-related protein. We previously reported that intraventricular and intranasal administration of MC4R antagonists reduce thermal hyperalgesia in alcohol-dependent rats tested during withdrawal (Roltsch Hellard et al., 2017). Here, we demonstrate that alcohol-dependent rats exhibit hyperalgesia that is mediated by MC4R signaling in the CeA and MOR signaling in the PAG. Collectively, our data reveal molecular targets and circuits that may be leveraged to reduce hyperalgesia in alcohol-dependent individuals.
Materials and Methods
Animals.
All procedures were approved by the Institutional Animal Care and Use Committee of the Louisiana State University Health Sciences Center and were in accordance with the National Institutes of Health guidelines. Adult male Wistar rats (Charles River; RRID:RGD_13508588) weighing ∼300 g at start of experiments were pair-housed in a humidity- and temperature-controlled (22°C) vivarium on a 12 h light/dark cycle (lights off at 8:00 A.M.), with ad libitum access to food and water. Rats were acclimated for 1 week before the start of experiments. Behavioral tests occurred during the dark period.
Drugs.
HS014, CNQX, dl-AP-5, and CGP 55845A were purchased from R&D Systems. DAMGO, αMSH, kynurenic acid, and picrotoxin (PTX) were purchased from Sigma-Aldrich.
Stereotaxic surgeries.
Rats were anesthetized with isoflurane and mounted into a stereotaxic frame (Kopf Instruments). For optogenetic experiments, stereotaxic injections of virus were delivered to the CeA, and optical fibers were implanted in either the CeA or ventrolateral periaqueductal gray (vlPAG). The CeA was targeted using the following coordinates from bregma: −2.5 mm posterior, ±4.0 mm lateral, and −8.4 mm ventral. Rats received 0.5 μl bilateral injections of halorhodopsin virus (AAV5-eNpHR3.0-mCherry, University of North Carolina Vector Core, Chapel Hill, NC), channelrhodopsin virus (AAV5-hSyn-hChR2-mCherry), or control virus (AAV5-hSyn-mCherry). Viral titers in all experiments were between 5.7 and 5.8 × 1012 viral molecules/ml virus.
Bilateral intracranial optical fiber and/or cannula implantation in the CeA or vlPAG was performed similarly. The CeA was targeted using the above coordinates; the vlPAG was targeted using the following coordinates from bregma: −7.6 mm posterior, ±0.8 mm lateral, −6.0 mm ventral. Fibers were secured to the skull with cranioplastic cement and stainless-steel anchor screws. For retrograde tracing experiments, 0.5 μl retrobeads (Lumafluor) were injected bilaterally into the vlPAG. After all surgical procedures, rats were monitored to ensure full recovery from anesthesia before being singly housed for one night. Rats were treated with the analgesic flunixin (2.5 mg/kg, s.c.) once immediately before surgery and once every 12 for 24 h postoperatively. Rats were treated with the antibiotic cefazolin (20 mg/kg, i.m.) once just before surgery and once 24 h postoperatively to prevent infection. Weight and appearance were also monitored for a period of 7 d following surgery to detect any signs of distress or postoperative complications. Rats used for behavioral experiments were allowed 1 week to recover from any surgical procedure before the resumption of behavioral testing.
Hargreaves Test for thermal nociception.
Rats were given five or six 30 min habituation sessions before baseline testing began. On all subsequent test days, rats were allowed 10 min to acclimate to the testing environment, then were placed in a 4 × 8 × 5 inch clear Plexiglas enclosure on top of a glass pane suspended 8 inches above the tabletop. After a 5 min habituation period, each hindpaw was stimulated by a halogen light heat source from an IITC model 309 Hargreaves apparatus (IITC Life Sciences). Latency to withdraw the hindpaw was measured twice for each hindpaw in an alternating order, with at least 1 min between measurements. A 20 s cutoff was used to prevent tissue damage in nonresponsive subjects. The average of these latency measurements was the thermal nociception score for each rat. Rats were excluded from analysis based on the following preestablished criteria: lost head caps, death during surgical procedure, inability to verify fiber placement, or off-target injections (total of 12 rats excluded).
Rats were tested for thermal nociception at least 3–5 times using the Hargreaves Test before any other manipulations. Following stable baseline hindpaw withdrawal values, rats underwent virus injection surgery. The mean baseline paw withdrawal latency was 10.92 ± 0.23 s for all rats; rats were assigned to their respective experimental group in a counterbalanced fashion. For chronic intermittent exposure (CIE) rats, testing was performed during the withdrawal period (6–8 h after vapor off) twice weekly to monitor the development of withdrawal hyperalgesia. Once withdrawal hyperalgesia was observed, rats underwent fiber implant surgery, then were subsequently rebaselined before the start of sham testing days (where cables were inserted, but no light delivered; 2–3 d/rat) and testing days.
Optical fibers for implantation were constructed by stripping and cleaving a 300 μm core multimodal fiber optic fiber (Thorlabs) with a Miller FOC-SS fiber optic cleaver (Precision Fiber Products). The fiber was inserted into a metal ferrule and held in place with a two-part heat cured epoxy (Precision Fiber Products). Each fiber was polished using 5, 3, 1, and 0.3 μm polishing pads, then visually inspected and tested for light transmission after construction. Only fibers with transmission values of >70% were implanted into experimental animals (average transmission value was 76%). During behavioral testing, the polished end of the optic fiber implant was connected via patch cable to the 100 mW laser light source at 473 nm for ChR2 experiments and 593.5 nm for eNpHR experiments (OEM Laser systems). Illumination was controlled by a Master-9 pulse stimulator (A.M.P.I.) and adjusted based on transmission values to allow for 10 mW light output at the fiber tip as measured by an optical power meter.
Trains of 5 ms light pulses were delivered at 20 Hz for up to 2 min for all ChR2 experiments, while illumination was constant for up to 2 min for all eNpHR experiments. In ChR2 experiments, rats were attached to the fiber optic cable, placed in the Hargreaves Test apparatus, and given 2 min to habituate to the apparatus on each test day. Behavioral testing days were divided into light on/off versus light off/on test days. Test days were alternated and repeated, and the order was counterbalanced between groups (4 yellow light test sessions per rat). The fifth test session used blue light, either light on/off or light off/on, with the order counterbalanced between groups. Lasers were on for at least 30 s preceding the first light-on data point, and all light-on data were collected within a 2 min period. Each test day consisted of 4 data points per rat: withdrawal times for each hindpaw per light condition, with a 2 min rest period between light conditions.
Drug infusions.
Drug infusions were delivered through guide cannulae implanted bilaterally above the CeA or vlPAG. Drugs were administered through 33-gauge injectors over 2.5 min at a rate of 0.2 μl/min, and 1 additional minute was allowed for diffusion before injector removal. Animals underwent sham infusions on days preceding drug infusion to acclimate them to the infusion procedure.
CIE to alcohol vapor.
A CIE to alcohol vapor protocol was used to induce dependence in rats. Rats were pair-housed in sealed chambers (La Jolla Alcohol Research) and exposed to alcohol vapor for 14 h/d. Blood alcohol levels (BALs) were measured 1–2 times weekly and analyzed using an Analox AM1 analyzer (Analox Instruments). Rats were maintained within a BAL range of 150–250 mg/dl over the experimental time course (minimum 4 weeks; mean BAL = 170 ± 9.5 mg/dl across experiments). For behavioral experiments, rats were exposed to 4–9 weeks of CIE (number of weeks varied between cohorts to allow emergence of stable withdrawal hyperalgesia). For electrophysiological experiments, rats were exposed to 4–15 weeks CIE, with longer CIE duration for virus-injected rats; controls were exposed to ambient air for equal amounts of time. All alcohol-dependent rats were given 1 d without vapor exposure presurgical and postsurgical procedures to minimize potential interactions between alcohol and the anesthetic isoflurane.
In vitro electrophysiology.
CIE rats were sacrificed between 6 and 8 h after cessation of vapor exposure, when BALs were near zero. Under isoflurane anesthesia, rats were transcardially perfused with 80 ml room temperature (∼25°C) NMDG aCSF containing the following (in mm): 92 NMDG, 2.5 KCl, 1.25 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 2 thiourea, 0.5 CaCl2, 10 MgSO4·7 H2O, 5 Na-ascorbate, and 3 Na-pyruvate. The 300-μm-thick coronal sections containing the CeA or PAG were collected using a vibratome (VT1200S, Leica Microsystems). Sections were incubated in NMDG aCSF at 37°C for 12 min, then transferred to a room temperature holding aCSF solution containing the following (in mm): 92 NaCl, 2.5 KCl, 1.25 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 2 thiourea, 2 CaCl2, 2 MgSO4·7 H2O, 5 Na-ascorbate, and 3 Na-pyruvate. Slices were allowed to recover for 1 h before recording. Slices were visualized with oblique infrared light illumination, a w60 water-immersion objective (LUMPLFLN60X/W, Olympus) and a CCD camera (Retiga 2000r, QImaging). Data were sampled at 10 kHz and Bessel filtered at 4 kHz using an acquisition control software package Ephus (Suter et al., 2010).
CeA recordings were performed in the medial subdivision of the CeA. Sections were transferred to a recording aCSF solution containing the following (in mm): 130 NaCl, 3.5 KCl, 2 CaCl2, 1.25 NaH2PO4, 1.5 MgSO4·7 H2O, 24 NaHCO3, and 10 glucose. Recording aCSF was maintained at 32°C–34°C using an in-line heater (Warner Instruments). Retrobead-containing (vlPAG-projecting) neurons were identified by their fluorescence using a filter set (Chroma). Resting membrane potential and rheobase values were recorded in current clamp using an internal recording solution containing the following (in mm): 140 K-gluconate, 5 KCl, 0.2 EGTA, 10 HEPES, 2 MgCl2·6 H2O, 4 Mg-ATP, 0.3 Na2-GTP, and 10 Na2-phosphocreatine, pH 7.2–7.3 (285–295 mOsm). sIPSC recordings were performed using a KCl internal solution containing the following (in mm): 145.0 KCl, 5 EGTA, 5 MgCl2, 10 HEPES, 2 Na-ATP, and 0.2 Na-GTP, pH 7.2–7.3 (285–295 mOsm), with cells clamped at −60 mV. To isolate sIPSCs, kynurenic acid (2 mm) and CGP 55845A (1 μm) were included in the recording aCSF. sEPSC recordings were performed using an internal solution containing the following (in mm): 145.0 K-gluconate, 0.5 EGTA, 2 MgCl2, 10 HEPES, 2 Na-ATP, and 0.2 Na-GTP, pH 7.2–7.3 (285–295 mOsm), with cells clamped at −60 mV. To isolate sEPSCs, PTX (25 μm) was included in the recording aCSF. Liquid junction potentials were not corrected during recordings. Experiments with a series resistance >30 mΩ or a >20% change in series resistance were excluded from analysis (total of 3 cells from 3 rats).
PAG-containing sections were prepared as described above, and recordings were performed in the vlPAG. Recordings were performed with an internal recording solution containing the following (in mm): 120 K-gluconate, 4 KCl, 2 K-EGTA, 10 HEPES, 4 Mg-ATP, 0.3 Na2-GTP, and 10 Na2-phosphocreatine, pH 7.2–7.3 (285–295 mOsm). For optical stimulation experiments, voltage-clamp recordings were performed with cells clamped at −50 mV. Light pulses (∼13 mW/mm2 power density, 0.5 ms pulse duration, 5 Hz pulse frequency) were delivered. In a separate series of experiments, recordings were performed in current-clamp mode in the presence of CNQX (10 μm) and AP-5 (50 μm). Current was injected to maintain the cell at an average resting membrane potential of −60 mV. Electrical stimulation was delivered from a bipolar chromium wire electrode.
Western blotting.
Under light isoflurane anesthesia, rats (n = 8 CIE and n = 8 naive) were decapitated, and brains were snap-frozen in isopentane. Animals were sacrificed 8 h after cessation of vapor exposure, when BAL = 0. Tissue punches of the CeA were taken using a 17-gauge punch from 500-μm-thick coronal sections. Samples were homogenized by sonication in lysis buffer containing the following (in mm): 320 sucrose, 5 HEPES, 1 EGTA, 1 EDTA, and 1% SDS, with protease inhibitor mixture and phosphatase inhibitor cocktails II and III (diluted 1:100; Sigma-Aldrich), heated at 100°C for 5 min, and stored at −80°C until the determination of protein concentration by a detergent-compatible Lowry method (Bio-Rad); 15 μg protein was subjected to SDS-PAGE on 8% acrylamide gels using a Tris/glycine/SDS buffer system (Bio-Rad), followed by electrophoretic transfer to PVDF membranes (GE Healthcare). Membranes were blocked overnight in 5% nonfat milk at 4°C, then incubated in primary antibody (rabbit anti-MC4R [1:1000; Alomone Labs; catalog #AMR-024; RRID:AB_2039980] and mouse anti-β-tubulin [1:500,000; Santa Cruz Biotechnology; catalog #sc-53140; RRID:AB_793543]). Membranes were washed and labeled with species-specific peroxidase-conjugated secondary antibody (1:10,000; Bio-Rad) for 1 h at 25°C. Following chemiluminescence detection (SuperSignal West Pico, Thermo Fisher Scientific), blots were stripped (Restore, Thermo Fisher Scientific) and reprobed for β-tubulin levels. Immunoreactivity was quantified by densitometry (ImageJ 1.45S) under linear exposure conditions. Densitized values were expressed as a percentage of the mean of control values for each gel to normalize data across blots. Individual MC4R levels were normalized to individual total β-tubulin protein levels.
Immunohistochemistry.
Under isoflurane anesthesia, rats (n = 10 CIE and n = 9 naive) were transcardially perfused with saline followed by 150 ml 4% PFA. Animals were sacrificed 8 h after cessation of vapor exposure, when BAL = 0. After perfusion, brains were postfixed for 24 h, transferred to 30% sucrose solution for 24–48 h, and then snap-frozen in isopentane. The 40-μm-thick coronal sections were collected and stored in a 0.1% sodium azide in 1× PBS until processing.
Sections were incubated in 1% H2O2 for 20 min, followed by three 5 min washes PBS. Slices were incubated in Vectastain ABC Elite blocking reagent (Vector Laboratories) for 2 h before a 3 h incubation in 1:10,000 rabbit anti-αMSH (Phoenix Pharmaceuticals; catalog #H-043-01; RRID:AB_10013604). Following three 5 min rinses in PBS, slices were incubated in Vectastain ABC Elite biotinylated anti-rabbit secondary antibody for 1 h, washed 3× in PBS, and then incubated in Vectastain ABC Reagent A and Reagent B solution for 30 min. After three 5 min washes in PBS, slices were processed with DAB HRP substrate kit (Vector Laboratories), then rinsed and mounted on microscope slides with Permount.
To quantify αMSH immunoreactivity, 10× images of CeA, paraventricular nucleus of the hypothalamus (PVN), and BNST were analyzed using ImageJ. For each image, immunoreactivity density of the area of interest and a representative background area were quantified. Immunoreactivity is represented as the density of the area of interest divided by the density of the background area, averaged over three rostral-caudal depths. All values were normalized to immunoreactivity in brain sections from alcohol-naive controls.
Imaging.
Images of AAV5/hSyn-mCherry-containing neurons in CeA and terminals in vlPAG (see Fig. 1A) were obtained with a confocal microscope (DMi8, Leica Microsystems). Images of retrobead-injected brains (see Fig. 2A) and αMSH-stained tissue sections (see Fig. 4A) were obtained with a fluorescent/brightfield microscope (Olympus).
Figure 1.
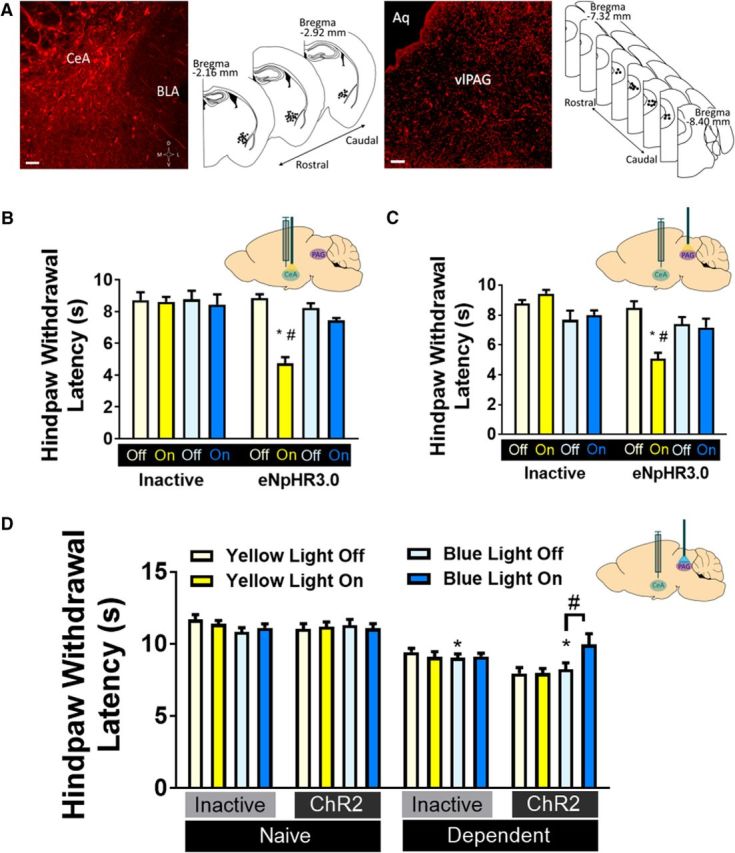
CeA-PAG projections mediate thermal nociception in rats. A, Virus expression. Left, Representative 20× image of hSyn-mCherry+ neurons in CeA, and map of virus placements. Scale bar, 100 μm. Right, representative 20× image of hSyn-mCherry+ terminals in PAG, and map of cannulae placements. Scale bar, 50 μm. BLA, Basolateral amygdala; Aq, aqueduct. B, Thermal hyperalgesia is induced by yellow light inactivation of CeA neurons (inset) transfected with eNpHR3.0 (n = 7 or 8/group). *p < 0.05 versus inactive virus. #p < 0.05 versus yellow light off. C, Thermal hyperalgesia is induced by yellow light inactivation of CeA terminals in PAG (inset) transfected with eNpHR3.0 (n = 7 or 8/group). *p < 0.05 versus inactive virus. #p < 0.05 versus yellow light off. D, Alcohol dependence-induced thermal hyperalgesia was reversed by blue light activation of CeA terminals in PAG (inset) transfected with ChR2 (n = 7–11/group). *p < 0.05 versus inactive virus. #p < 0.05 versus blue light off. B–D, Data are mean ± SEM. Hindpaw withdrawal latencies (s) in Hargreaves Test in response to light stimulation. Yellow bars represent yellow light trials. Blue bars represent blue light trials.
Figure 2.

CeA-PAG circuit activity alterations following alcohol dependence. A, Retrograde labeling of PAG neurons. Left, 4× image of retrobeads (red) injected in PAG, with map of retrobead injections in naive (black) and alcohol-dependent (red) rats. Aq, Aqueduct. Scale bar, 500 μm. Right, 40× image of retrobead-containing (PAG-projecting) neurons in CeA. Nuclei are stained with DAPI and appear blue. White arrow indicates position of a representative retrobead+ neuron. Scale bar, 50 μm. B, Map indicating approximate recording positions of PAG-projecting CeA neurons from naive (black) and alcohol-dependent (red) rats; recordings were performed in the medial subdivision of the CeA. C, Average resting membrane potential of PAG-projecting CeA neurons does not significantly differ between naive and dependent rats. D, Average rheobase values do not significantly differ between populations. E, Representative sIPSC traces recorded in PAG-projecting CeA neurons of naive (black) and alcohol-dependent (red) rats. There were no significant differences in baseline sIPSC frequency (F) but a tendency toward an increase in sIPSC amplitude (p = 0.067) (G) of PAG-projecting CeA neurons from naive or alcohol-dependent rats. H, Representative sEPSC traces recorded in PAG-projecting CeA neurons of naive (black) and alcohol-dependent (red) rats. There were no significant differences in baseline sEPSC frequency (I) or amplitude (J) of PAG-projecting CeA neurons from naive or alcohol-dependent rats. C, D, F, G, I, J, Data are mean ± SEM.
Figure 4.

Alcohol dependence reduces MC4R expression in CeA. A, 10× image of αMSH expression in CeA of naive (left) and alcohol-dependent (right) rat. Approximate positions of CeA and BLA indicated. Scale bar, 200 μm. CeM, Medial subdivision of CeA; CeL, lateral subdivision of CeA. B, Average expression of αMSH in CeA, BNST, and PVN of alcohol-dependent (red) and naive (white) rats. C, CeA MC4R expression is significantly decreased in CeA of alcohol-dependent rats (red) compared with naive controls (white). *p < 0.05. Inset, Representative image of MC4R and tubulin levels from naive (N) and alcohol-dependent (D) rats. B, C, Data are mean ± SEM.
Experimental design and statistical analysis.
Statistical analysis was performed in Prism version 7.03 (GraphPad Software). Data were analyzed using two-tailed t test and one-way and two-way ANOVAs, except where otherwise indicated. In the experiment shown in Figure 3B, Fisher's exact test was used to compare the number of vlPAG neurons from alcohol-dependent and naive rats in which evoked IPSCs were detected. In the experiment shown in Figure 5A, a linear trend analysis was used to test for dose–response drug effect. In the case of significant interaction and/or main effects, pairwise comparisons were probed with Tukey's post hoc analysis. A p value of <0.05 was considered significant. All tests and variables are identified in Results.
Figure 3.

CeA inputs to vlPAG are weaker in alcohol-dependent rats. A, Top, Map indicating approximate positions of PAG neurons recorded from naive (black) and alcohol-dependent (red) rats. ChR2-containing CeA terminals were stimulated in PAG-containing brain slices (inset). Closed circles represent neurons in which an optically evoked postsynaptic current was observed. Open circles represent neurons in which optical stimulation evoked no events. Bottom, Sample recordings of optically evoked postsynaptic current of PAG neuron from naive (black) and alcohol-dependent (red) rats. B, No significant differences exist in the proportion of neurons in which evoked events were observed between groups. C, Average amplitude of evoked postsynaptic current was significantly lower in PAG neurons from alcohol-dependent rats compared with naive controls. *p < 0.05. D, Representative trace showing optically evoked current of PAG neuron in the absence (black) and presence (orange) of PTX. E, Superfusion of PTX significantly decreased the relative amplitude of evoked postsynaptic currents of PAG neurons. C, E, Data are mean ± SEM.
Figure 5.
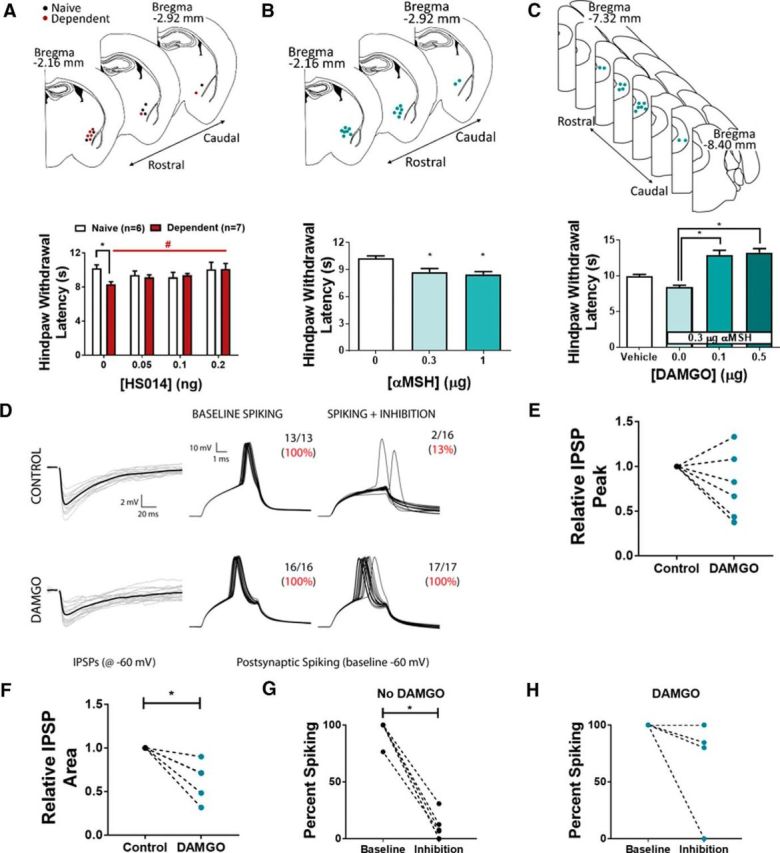
MC4R in CeA and MOR in vlPAG modulate thermal nociception. A, Hindpaw withdrawal latencies of naive (white) and alcohol-dependent (red) rats following intra-CeA infusion of HS014. *p < 0.05 versus naive. #p < 0.05, effect of HS014. B, Hindpaw withdrawal latency of naive rats is significantly reduced following intra-CeA infusion of αMSH. *p < 0.05 versus control. C, Hindpaw withdrawal latencies of naive rats following intra-PAG infusion of DAMGO and intra-CeA infusion of 0.3 μg αMSH. Intra-PAG administration of DAMGO before αMSH prevents thermal hyperalgesia. *p < 0.05 versus 0 μg DAMGO. A–C, Top, Cannulae placement. D, IPSPs are evoked in putative PAG pyramidal neurons by nearby electrical stimulation (left). Superthreshold current injection evoked action potentials (APs) under baseline conditions (middle), but APs are blocked by concurrent stimulation of local inhibition (right). Bath application of DAMGO reduces IPSPs in the same neuron shown in top. In the presence of DAMGO, stimulation of local inhibition fails to block AP generation while depolarizing neurons (middle, right). E, DAMGO reduces IPSP amplitude in 4 of 6 neurons recorded. F, DAMGO reduces integrated IPSP area in 6 of 6 neurons. *p < 0.05. G, Under control conditions, inhibition blocks AP generation. *p < 0.05. H, In the presence of DAMGO, inhibition fails to significantly block AP generation; 1 of 6 neurons was still blocked by inhibition. A–C, Data are mean ± SEM.
Results
PAG-projecting CeA neurons mediate thermal nociception and hyperalgesia in rats
The CeA is the major output nucleus of the amygdala and projects to brain regions that mediate behavioral and physiological responses to emotionally relevant events (Hopkins and Holstege, 1978; Pitkänen, 2000; Pape and Pare, 2010). The PAG coordinates descending inhibitory responses to fearful and painful stimuli (LeDoux et al., 1988; Millan, 2002). Dense CeA projections to PAG (Rizvi et al., 1991) promote antinociception that is blocked by MOR antagonists in PAG (Oliveira and Prado, 2001; Xu et al., 2003). Therefore, we used site-specific optical stimulation to test the effects of CeA neuronal activity and CeA-to-PAG projection activity on thermal nociception. We previously demonstrated that intra-CeA TTX produces thermal hyperalgesia in otherwise experimentally naive rats (Itoga et al., 2016). We began by replicating this observation using optogenetics (Fig. 1). In adult male rats, neurons were transfected with optically active halorhodopsin virus (AAV5/hSyn-eNpHR3.0-mCherry) or optically inactive control virus (AAV5/hSyn-mCherry) injected into the CeA (Fig. 1A). Silencing all CeA neurons via yellow light delivery into the CeA produced thermal hyperalgesia in otherwise experimentally naive rats, as indicated by significantly shorter hindpaw withdrawal latency in the Hargreaves Test during light ON trials (Fig. 1B), confirming our previous findings using site-specific TTX infusion into CeA (Itoga et al., 2016). Two-way ANOVA revealed that yellow light significantly reduced hindpaw withdrawal latencies (F(1,26) = 29.94; p < 0.0001) and that animals with active virus exhibited significantly shorter hindpaw withdrawal latencies (F(1,26) = 23.76; p < 0.0001), with a significant light × virus interaction effect (F(1,26) = 27.17; p < 0.0001). Tukey's multiple-comparisons test revealed that active virus rats exhibited significantly shorter hindpaw withdrawal latencies during yellow light ON trials relative to control virus rats during yellow light ON trials (p < 0.05), and also relative to light OFF trials in the active virus group (p < 0.05). To rule out nonspecific light effects, we performed additional trials with blue light delivery. A separate two-way ANOVA revealed no effect of virus (F(1,26) = 2.597; p = 0.119) or light (F(1,26) = 1.367; p = 0.253) on hindpaw withdrawal latencies during blue light trials.
To test whether this nociceptive effect was due specifically to CeA projections to vlPAG, we performed similar experiments with halorhodopsin or control virus injected in the CeA and optical stimulation in the vlPAG. Silencing of CeA terminals in vlPAG via yellow light delivery produced thermal hyperalgesia in otherwise experimentally naive rats, as indicated by significantly shorter hindpaw withdrawal latency in the Hargreaves Test during light ON trials (Fig. 1C). Two-way ANOVA revealed that yellow light significantly reduced hindpaw withdrawal latencies (F(1,26) = 13.3; p = 0.0012) and that animals with active virus exhibited significantly shorter hindpaw withdrawal latencies (F(1,26) = 39.17; p < 0.0001), with a significant light × virus interaction effect (F(1,26) = 29.71; p < 0.0001). Tukey's multiple-comparisons test revealed that active virus rats exhibited significantly shorter hindpaw withdrawal latencies during yellow light ON trials relative to control virus rats during yellow light ON trials (p < 0.05), and also relative to light OFF trials in the active virus group (p < 0.05). A separate two-way ANOVA revealed no effect of virus (F(1,26) = 1.146; p = 0.294) or light (F(1,26) = 0.013; p = 0.910) on hindpaw withdrawal latencies during blue light trials. The results of these experiments confirm that inactivation of CeA-PAG projection neurons produce thermal hyperalgesia and suggest that basal tonic activity of CeA-PAG projections may have antinociceptive effects.
Humans living with AUD commonly report higher pain sensitivity during periods of withdrawal (Jochum et al., 2010), and we previously demonstrated thermal hyperalgesia in alcohol-dependent rats during alcohol withdrawal (Roltsch Hellard et al., 2017). To test the role of CeA-PAG projection neurons in thermal hyperalgesia during withdrawal in alcohol-dependent rats, we combined optogenetics with CIE to alcohol vapor. Adult male rats were tested for baseline thermal nociception, then injected with AAV5/hSyn-Channelrhodopsin-mCherry (ChR2) or AAV5/hSyn-mCherry in the CeA before CIE. Following 4–9 weeks of CIE (number of weeks varied between cohorts to allow emergence of stable withdrawal hyperalgesia), rats demonstrated significantly lower paw withdrawal latencies relative to their own baseline and also relative to alcohol-naive controls. Blue light stimulation of ChR2-containing CeA terminals in the vlPAG attenuated this withdrawal hyperalgesia (Fig. 1D). Analysis of blue light OFF data revealed that alcohol-dependent rats exhibited significantly shorter hindpaw withdrawal latencies than alcohol-naive controls (F(1,30) = 46.26; p < 0.0001, two-way ANOVA), with no significant effect of virus (F(1,30) = 0.241; p = 0.627) or alcohol × virus interaction effect (F(1,30) = 3.073; p = 0.090). Two-way ANOVA analysis of data from alcohol-dependent and alcohol-naive rats injected with ChR2 revealed a significant vapor × light interaction effect (F(1,28) = 5.132; p = 0.0314), and Tukey's multiple-comparisons test showed that blue light stimulation increased hindpaw withdrawal latencies only in alcohol-dependent rats (p < 0.05). The lack of a blue light effect on nociception in alcohol-naive rats injected with ChR2 virus suggests that activation of CeA-PAG projection neurons may be occluded by normal basal function of this circuit. These data show that activation of CeA-PAG projection neurons partially rescues alcohol dependence-induced hyperalgesia, suggesting that alcohol withdrawal impairs CeA-PAG circuit function in alcohol-dependent rats. We did not observe any gross behavioral changes, aside from alterations in hindpaw withdrawal latency, during light stimulation of eNpHR- or ChR2-injected animals. Recent work from other laboratories has suggested a role for circuitry connecting the CeA and vlPAG in certain defensive behaviors, such as avoidance (LeDoux et al., 2017), freezing (Tovote et al., 2016), and flight (Fadok et al., 2017), suggesting that optogenetic manipulation of these brain regions could elicit overt behavioral responses under our experimental conditions. Our failure to observe these effects here may be due to one or more of these factors: (1) our rats were habituated to the test apparatus over days and before each test session and between test trials; (2) Hargreaves testing occurs in a confined space that may eliminate avoidance and flight from the behavioral repertoire; (3) species differences; and (4) subtle differences in virus injections and/or light stimulation protocols.
Alcohol withdrawal alters CeA-PAG circuit function in alcohol-dependent rats
We observed that silencing CeA terminals in the vlPAG produces thermal hyperalgesia in otherwise experimentally naive rats, that alcohol-dependent rats exhibit thermal hyperalgesia during periods of withdrawal, and that activating CeA terminals in the vlPAG attenuates withdrawal-induced hyperalgesia. We next sought to test the effect of alcohol withdrawal on CeA-PAG circuit function in the brains of alcohol-dependent rats. We used retrograde tracing and/or optogenetics in combination with in vitro electrophysiology to measure electrophysiological parameters in CeA and vlPAG neurons of alcohol-dependent rats relative to alcohol-naive controls.
To identify vlPAG-projecting neurons in CeA, we stereotaxically injected retrobeads into the vlPAG of adult male rats (Fig. 2A). Whole-cell recordings were performed in retrobead-containing (vlPAG-projecting) neurons in the medial subdivision of the CeA (Fig. 2B). We observed no difference in resting membrane potential of vlPAG-projecting CeA neurons of alcohol-dependent rats (−57.47 ± 2.18 mV, n = 15 cells from 5 rats) and alcohol-naive controls (−57.77 ± 2.55 mV, n = 13 cells from 5 rats; t(26) = 0.0908, p = 0.928, two-tailed t test; Fig. 2C). Similarly, we observed no difference in rheobase values of vlPAG-projecting CeA neurons of alcohol-dependent rats (26.25 ± 3.21 pA, n = 12 cells from 5 rats) and alcohol-naive controls (29.58 ± 5.42 pA, n = 12 cells from 5 rats; t(22) = 0.529, p = 0.602, two-tailed t test; Fig. 2D). We next assessed whether CeA synaptic transmission was altered in alcohol-dependent rats relative to alcohol-naive controls. sIPSCs were recorded in vlPAG-projecting CeA neurons (Fig. 2E). We observed no significant differences in baseline sIPSC frequency of vlPAG-projecting CeA neurons of alcohol-dependent rats (8.01 ± 1.19 Hz, n = 9 cells from 6 rats) and alcohol-naive controls (10.08 ± 2.27 Hz, n = 8 cells from 4 rats; t(15) = 0.836, p = 0.416, two-tailed t test; Fig. 2F). The baseline sIPSC frequency values are higher than those typically reported (e.g., Varodayan et al., 2016). This difference may be due to our slice preparation procedure, which is protective for GABA neurons (Ting et al., 2014; Pan et al., 2015) and/or differences in recording temperature across studies (32°C in current study vs room temperature). We observed a nonsignificant trend toward higher baseline sIPSC amplitude of vlPAG-projecting CeA neurons of alcohol-dependent rats (65.29 ± 4.95 pA, n = 9 cells from 6 rats) relative to alcohol-naive controls (50.07 ± 5.97 pA, n = 8 cells from 4 rats; t(15) = 1.978, p = 0.067, two-tailed t test; Fig. 2G). This trend toward a sIPSC amplitude effect is similar to what has been previously reported in unlabeled CeA cells (Roberto et al., 2004).
We next recorded sEPSCs in vlPAG-projecting CeA neurons (Fig. 2H). We observed no significant differences in baseline sEPSC frequency of vlPAG-projecting CeA neurons of alcohol-dependent rats (3.74 ± 1.26 Hz, n = 8 cells from 5 rats) and alcohol-naive controls (1.89 ± 0.61 Hz, n = 7 cells from 5 rats; t(13) = 1.28, p = 0.223, two-tailed t test; Fig. 2I). We observed no significant difference in the baseline sEPSC amplitude of vlPAG-projecting CeA neurons of alcohol-dependent rats (17.4 ± 2.97 pA, n = 8 cells from 5 rats) and alcohol-naive controls (11.29 ± 0.79 pA, n = 7 cells from 5 rats; t(13) = 1.868, p = 0.085, two-tailed t test; Fig. 2J). Collectively, these data indicate that basal excitability, whether intrinsic or modulated by inhibitory or excitatory transmission, of vlPAG-projecting CeA neurons is not significantly altered during alcohol withdrawal, although there may be a trend toward stronger synaptic inhibition of these projection neurons in alcohol-dependent rats.
Because we observed no effect of alcohol withdrawal on excitability of vlPAG-projecting neurons in the CeA, we next used optogenetics with in vitro electrophysiology to test the strength of CeA synaptic inputs to the vlPAG in alcohol-dependent and alcohol-naive rats. AAV5/hSyn-ChR2-mCherry was injected into the CeA of adult male rats; then rats were exposed to 8–15 weeks of CIE, during which time virus anterogradely transported to the vlPAG. Whole-cell recordings of vlPAG neurons near the presence of ChR2-containing terminals were performed by delivering blue light to the slice to evoke postsynaptic currents in vlPAG neurons (Fig. 3A). Because neurons were clamped at −50 mV, encouraging outward current deflections, and because evoked postsynaptic currents were blocked by application of PTX (Fig. 3D), we conclude that the recorded events are putative eIPSCs. We observed no difference in the number of neurons in which eIPSCs were observed (37.3% of neurons from alcohol-naive rats vs 27.3% of neurons from alcohol-dependent rats demonstrated a response to light stimulation; p = 0.366, Fisher's exact test; Fig. 3B). The relationship between observed response amplitude and variability, and short-term synaptic dynamics (i.e., facilitation or depression) is consistent with a model where heterogeneities of observed response amplitudes are determined by presynaptic probability of release (Faber and Korn, 1991); there were no differences between alcohol-dependent and alcohol-naive rats in this respect. Photostimulation produced significantly smaller-amplitude eIPSCs in vlPAG neurons from alcohol-dependent rats (25.15 ± 8.52 pA, n = 9 cells from 4 rats) relative to alcohol-naive controls (132.7 ± 30.02 pA, n = 22 cells from 8 rats; t(29) = 2.255, p = 0.0319, two-tailed t test; Fig. 3C). We observed a greater range of eIPSC amplitudes in neurons from alcohol-naive compared with alcohol-dependent rats; the distribution of CeA-to-PAG synaptic inhibition in each dataset passed the test for log-normality. GABAergic projection neurons in CeA coexpress and likely corelease various neuromodulators, for example, dynorphin (Reyes et al., 2008; Jüngling et al., 2015), corticotropin-releasing factor (Pomrenze et al., 2015), and neurotensin (Batten et al., 2002). To confirm that synaptic effects of photostimulation of ChR2-positive CeA terminals in the vlPAG were attributable to GABA release, we repeated these experiments in the presence of the GABA blocker PTX (100 μm; Fig. 3D). Application of PTX significantly decreased the amplitude of eIPSCs in vlPAG neurons from naive rats by 92.5 ± 2.4% (n = 3 cells from 2 rats; t(4) = 38.86, p < 0.0001, two-tailed t test; Fig. 3E). Collectively, these data suggest that alcohol withdrawal reduces the strength of CeA GABAergic inhibition onto vlPAG neurons.
Alcohol withdrawal alters melanocortin system protein expression in the CeA of alcohol-dependent rats
Brain MC4R signaling modulates nociception as well as morphine analgesic tolerance and morphine withdrawal hyperalgesia in rodents (Kalange et al., 2007), and our laboratory previously reported that intraventricular (and intranasal) administration of endogenous and synthetic MC4R antagonists attenuates alcohol withdrawal hyperalgesia (Roltsch Hellard et al., 2017). Therefore, we next tested (1) the effect of alcohol withdrawal on melanocortin system protein expression in CeA, and (2) the role of MC4R signaling in CeA in alcohol withdrawal hyperalgesia.
We used immunohistochemistry to measure αMSH (endogenous melanocortin receptor ligand) expression in the CeA, PVN, and BNST of alcohol-dependent rats sacrificed during withdrawal relative to alcohol-naive controls. Each of these regions is densely innervated by αMSH projections from the arcuate hypothalamus (Eskay et al., 1979). Alcohol-dependent rats sacrificed during withdrawal did not exhibit significant changes in αMSH immunoreactivity in medial (t(17) = 1.475, p = 0.158) or lateral (t(17) = 1.603, p = 0.127, two-tailed t test) subdivisions of the CeA (Fig. 4A,B), or in PVN or BNST (Fig. 4B). We next used Western blot analysis to measure MC4R expression; alcohol-dependent rats sacrificed during withdrawal exhibited lower MC4R expression in the CeA compared with alcohol-naive controls (t(14) = 2.595, p = 0.0212, two-tailed t test; Fig. 4C).
MC4Rs in CeA modulate nociception, and this effect is filtered by MORs in vlPAG
To test the hypothesis that MC4R antagonism in CeA attenuates alcohol withdrawal hyperalgesia, we infused multiple doses of the synthetic MC4R antagonist HS014 into the CeA of alcohol-dependent and alcohol-naive rats and measured thermal nociception. As in previous experiments (e.g., Fig. 1D), we observed shorter hindpaw withdrawal latencies in alcohol-dependent rats tested during withdrawal relative to alcohol-naive controls (t(5) = 6.025, p = 0.0018, two-tailed t test; Fig. 5A). A linear trend analysis indicated that intra-CeA HS014 dose-dependently increased hindpaw withdrawal latencies (i.e., rescues thermal hyperalgesia) in alcohol-dependent rats (F(1,18) = 10.04, p = 0.0053) but did not affect hindpaw withdrawal latencies in alcohol-naive controls.
Next, we tested the hypothesis that infusion of αMSH into CeA is sufficient to produce thermal hyperalgesia in otherwise experimentally naive rats. Intra-CeA infusions of αMSH produced reductions in hindpaw withdrawal latency of alcohol-naive male rats (F(2,15) = 9.048; p = 0.0026, one-way ANOVA; Fig. 5B). Tukey's multiple-comparison test revealed that the middle (0.3 μg) and high (1.0 μg) αMSH doses reduced hindpaw withdrawal latencies relative to vehicle infusion (p < 0.05 in both cases).
It was previously reported that electrical stimulation of CeA has antinociceptive effects that are blocked by lidocaine infusion or MOR blockade in PAG (Oliveira and Prado, 2001; Xu et al., 2003). In earlier experiments (Fig. 1), we showed that CeA-vlPAG projections are necessary and sufficient for producing thermal hyperalgesia in rats. Therefore, we used dual-site pharmacology to test the effects of intra-vlPAG infusion of the MOR agonist DAMGO on thermal hyperalgesia induced by intra-CeA infusion of αMSH (middle dose from prior experiment; 0.3 μg). As in the previous experiment, intra-CeA infusion of 0.3 μg αMSH produced thermal hyperalgesia when preceded by intra-vlPAG infusion of vehicle (Fig. 5C). Pretreatment with intra-vlPAG infusion of the MOR agonist DAMGO blocked intra-CeA αMSH effects on hindpaw withdrawal latencies in the Hargreaves Test (F(3,44) = 25.16, p < 0.0001; one-way ANOVA; Fig. 5C). Tukey's multiple-comparisons test revealed that intra-vlPAG infusion of each DAMGO dose (0.1 and 0.5 μg) in combination with intra-CeA αMSH infusion elicited significantly higher hindpaw withdrawal latencies relative to intra-CeA αMSH infusion alone (p < 0.05). These data suggest that MORs in vlPAG filter or override the effects of CeA MC4Rs on thermal nociception.
To test the role of MORs in the vlPAG in gating local synaptic inhibition, we used in vitro electrophysiology to record from putative pyramidal neurons in vlPAG; neurons were targeted for recordings based on their cell body morphology (Mantyh, 1982). It is important to note that ∼30%-45% of vlPAG pyramidal neurons projecting to RVM (i.e., the descending pain inhibitory pathway) express mRNA for MOR (Commons et al., 2000; Wang and Wessendorf, 2002). Individual recorded vlPAG neurons were clamped at a current just below the threshold required for spontaneous firing, then injected with current to elicit action potentials in the presence or absence of local electrically evoked IPSPs (Fig. 5D). These experiments were performed in the absence of drug and also during superfusion of DAMGO (2 μm). Supra-threshold current injection evoked action potentials in vlPAG neurons, and this effect was blocked by concurrent stimulation of local inhibition (Fig. 5D). In the absence of drug, locally evoked inhibition blocked action potential generation in vlPAG neurons (t(10) = 14.19, p < 0.0001; two-tailed t test; Fig. 5G). Bath application of DAMGO reduced the amplitude of local electrically evoked IPSPs in 4 of 6 neurons recorded (Fig. 5E) and reduced the integrated IPSP area in 6 of 6 neurons (t(10) = 4.277, p = 0.0016; two-tailed t test; Fig. 5F). Furthermore, in the presence of DAMGO, locally evoked inhibition failed to block action potential generation in vlPAG neurons (t(10) = 1.419, p = 0.186; two-tailed t test; Fig. 5H). These data support a role for MORs in filtering local inhibition onto putative pyramidal neurons in vlPAG.
Because CeA projections to vlPAG are GABAergic and inhibit vlPAG interneurons (Fig. 3), a normally functioning CeA-vlPAG circuit would be expected to disinhibit vlPAG pyramidal neuron activity in vivo. CeA inactivation (Fig. 1) or intra-CeA pharmacological manipulations (Fig. 5) (Xu et al., 2003) promote nociception, likely in part by reducing inhibition (i.e., increasing activity) of vlPAG interneurons, thereby reducing activity of vlPAG projection neurons (Fig. 6). Our data suggest that MOR activation in vlPAG may block CeA-related thermal hyperalgesia by dampening the ability of local inhibition to reduce the activity of vlPAG pyramidal neurons (e.g., to the medulla for descending pain inhibition).
Figure 6.

Adaptations in CeA-PAG circuitry contribute to hyperalgesia following alcohol dependence. Left, Schematic depicting CeA-PAG circuitry under baseline conditions. CeA GABAergic neurons project to PAG, ultimately resulting in disinhibition of MOR-containing PAG neurons, initiating the descending pain inhibition pathway. Right, Following repeated cycles of alcohol exposure and withdrawal, MC4R expression is decreased in the CeA. There is a decrease in the strength of CeA projections to PAG, resulting in increased inhibition of PAG output neurons to RVM and/or forebrain regions (e.g., TH+ projections to BNST) and/or midbrain regions (e.g., VTA/SN). Ultimately, these adaptations contribute to dependence-induced hyperalgesia. Illustration generated by modifying images purchased in the Illustration Toolkit-Neuroscience from Motifolio.
MC4R drugs in CeA do not alter synaptic transmission onto CeA-PAG projection neurons
Finally, we used in vitro electrophysiology to test the effect of MC4R antagonist on CeA synaptic transmission in alcohol-dependent rats relative to alcohol-naive controls. Superfusion of the MC4R antagonist HS014 (0.5 μm) did not alter sIPSC or sEPSC characteristics of vlPAG-projecting CeA neurons of either alcohol-dependent or naive rats (data not shown). This suggests that the antihyperalgesic effects of MC4R antagonist infusion into the CeA of alcohol-dependent rats may not be mediated by inhibitory or excitatory transmission onto vlPAG-projecting neurons, and further suggests that perhaps intra-CeA effects of MC4R antagonist on behavior may be mediated at the level of CeA interneurons.
Discussion
Humans living with AUD exhibit heightened nociception during periods of alcohol withdrawal (Jochum et al., 2010). Here, we demonstrate that CeA projections to vlPAG modulate thermal nociception in naive rats and mediate alcohol withdrawal-induced hyperalgesia in alcohol-dependent rats. Our data suggest that hypofunctional CeA projections to vlPAG mediate alcohol withdrawal hyperalgesia in alcohol-dependent rats. In support of this hypothesis, alcohol-dependent rats killed during withdrawal exhibited reduced inhibition of vlPAG neurons evoked from CeA inputs, relative to alcohol-naive controls. In the vlPAG, we speculate that we recorded primarily from interneurons, based on cell body morphology (Mantyh, 1982). Tonic inhibition of vlPAG output neurons by local GABAergic neurons has been demonstrated to influence nociception (Depaulis et al., 1987) in rats, with antagonism of GABA receptors producing increases in the antinociceptive effect of opiates. A reduction in the synaptic strength of inhibitory CeA inputs onto vlPAG interneurons (e.g., in alcohol-dependent rats) would result in a net decrease in the activity of vlPAG projection neurons, likely leading to less descending pain inhibition (i.e., hyperalgesia). These data are supported by a recent report demonstrating selective inhibition of vlPAG glutamatergic neurons, or activation of vlPAG GABAergic neurons, facilitates nociception in rats (Samineni et al., 2017).
Melanocortin receptors modulate nociception, opioid analgesia, and the development of tolerance to this effect, and paradoxical opioid-induced hyperalgesia (Kalange et al., 2007). We previously demonstrated that intraventricular and intranasal administration of MC4R antagonists reduces alcohol withdrawal hyperalgesia (Roltsch Hellard et al., 2017). Here, we report that intra-CeA MC4R antagonism attenuates alcohol withdrawal hyperalgesia. We did not observe MC4R antagonist effects on synaptic transmission onto vlPAG-projecting CeA neurons using in vitro electrophysiology. Therefore, we speculate that MC4Rs may modulate nociception via direct action on CeA interneurons and subsequent downstream modulation of CeA projector neuron activity.
MORs in vlPAG mediate the antinociceptive effects of MOR activation in the amygdala (Pavlovic et al., 1996; Tershner and Helmstetter, 2000), suggesting that CeA modulation of nociception is filtered by MOR activity in vlPAG. This opens the possibility that CeA MC4R effects on nociception may also converge on MORs in vlPAG. We report that intra-CeA αMSH administration produces hyperalgesia in alcohol-naive rats, and this effect is blocked by intra-vlPAG administration of an MOR agonist. Furthermore, our in vitro electrophysiology data demonstrate that MORs on putative pyramidal neurons filter the ability of local vlPAG inhibition to reduce the activity of vlPAG projection neurons. Collectively, these data suggest that MORs on vlPAG pyramidal neurons (e.g., those projecting to RVM) filter the hyperalgesic effects of upstream events that terminate in altered activity of the inhibitory interneuron milieu in vlPAG (e.g., higher local inhibition of pyramidals in alcohol-dependent rats with weaker GABAergic inputs from CeA). We did not label the projection target of putative pyramidal neurons in our vlPAG electrophysiology experiments; therefore, it is possible these vlPAG neurons project to the forebrain (e.g., BNST) (Li et al., 2016) or midbrain (Omelchenko and Sesack, 2010; Suckow et al., 2013) (e.g., ventral tegmental area and substantia nigra). That said, we performed most of our recordings in the caudal vlPAG (Fig. 3), where there are dense RVM projectors that do not contain catecholamines (Suckow et al., 2013).
Collectively, these data support a model in which CeA projections to vlPAG tonically suppress nociception via descending pain inhibition pathways (summarized in Fig. 6). Under physiological conditions, we expect that CeA inputs disinhibit vlPAG pyramidal neurons via effects on local inhibitory interneurons in vlPAG. Alcohol-dependent animals tested during withdrawal exhibit thermal hyperalgesia, as well as weaker CeA inputs to vlPAG neurons. Alcohol withdrawal hyperalgesia is rescued by optogenetic activation of CeA projections to vlPAG and is mimicked by optogenetic silencing of CeA projections to vlPAG in alcohol-naive animals. In alcohol-dependent rats, weaker CeA inputs to vlPAG interneurons are accompanied by a reduction in CeA MC4R expression. MC4R blockade in CeA rescues alcohol withdrawal hyperalgesia, whereas MC4R activation in the CeA promotes thermal nociception, and this latter effect is blocked by MOR activation in vlPAG, which is likely a final point of convergence for CeA modulatory effects on nociception. These data provide new insight into the circuitry underlying thermal nociception and alcohol withdrawal hyperalgesia, and highlights MC4Rs as potential therapeutic targets for reducing pain in individuals living with AUD.
Footnotes
This work was supported by National Institute of Health Grants R01 AA023305 to N.W.G., Grant T32 AA007577 to E.M.A., C.A.I., and M.M.W., Grant F32 AA025831 to E.M.A., and Grant R00 AA020839 to S.E., and Veterans Administration Grant I01 BX003451 to N.W.G.
N.W.G. owns shares in Glauser Life Sciences, Inc., a start-up company with interest in development of therapeutics for treatment of mental illness. The remaining authors declare no competing financial interests.
References
- Angst MS, Clark JD (2006) Opioid-induced hyperalgesia: a qualitative systematic review. Anesthesiology 104:570–587. 10.1097/00000542-200603000-00025 [DOI] [PubMed] [Google Scholar]
- Baliki MN, Apkarian AV (2015) Nociception, pain, negative moods, and behavior selection. Neuron 87:474–491. 10.1016/j.neuron.2015.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batten TF, Gamboa-Esteves FO, Saha S (2002) Evidence for peptide co-transmission in retrograde- and anterograde-labelled central nucleus of amygdala neurones projecting to NTS. Auton Neurosci 98:28–32. 10.1016/S1566-0702(02)00026-7 [DOI] [PubMed] [Google Scholar]
- Brennan PL, Schutte KK, Moos RH (2005) Pain and use of alcohol to manage pain: prevalence and 3-year outcomes among older problem and non-problem drinkers. Addiction 100:777–786. 10.1111/j.1360-0443.2005.01074.x [DOI] [PubMed] [Google Scholar]
- Caruso V, Lagerström MC, Olszewski PK, Fredriksson R, Schiöth HB (2014) Synaptic changes induced by melanocortin signalling. Nat Rev Neurosci 15:98–110. 10.1038/nrn3657 [DOI] [PubMed] [Google Scholar]
- Commons KG, Aicher SA, Kow LM, Pfaff DW (2000) Presynaptic and postsynaptic relations of mu-opioid receptors to gamma-aminobutyric acid-immunoreactive and medullary-projecting periaqueductal gray neurons. J Comp Neurol 419:532–542. 10.1002/(SICI)1096-9861(20000417)419:4%3C532::AID-CNE8%3E3.0.CO;2-6 [DOI] [PubMed] [Google Scholar]
- Depaulis A, Morgan MM, Liebeskind JC (1987) GABAergic modulation of the analgesic effects of morphine microinjected in the ventral periaqueductal gray matter of the rat. Brain Res 436:223–228. 10.1016/0006-8993(87)91665-9 [DOI] [PubMed] [Google Scholar]
- Duvarci S, Pare D (2014) Amygdala microcircuits controlling learned fear. Neuron 82:966–980. 10.1016/j.neuron.2014.04.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egli M, Koob GF, Edwards S (2012) Alcohol dependence as a chronic pain disorder. Neurosci Biobehav Rev 36:2179–2192. 10.1016/j.neubiorev.2012.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskay RL, Giraud P, Oliver C, Brown-Stein MJ (1979) Distribution of alpha-melanocyte-stimulating hormone in the rat brain: evidence that alpha-MSH-containing cells in the arcuate region send projections to extrahypothalamic areas. Brain Res 178:55–67. 10.1016/0006-8993(79)90087-8 [DOI] [PubMed] [Google Scholar]
- Faber DS, Korn H (1991) Applicability of the coefficient of variation method for analyzing synaptic plasticity. Biophys J 60:1288–1294. 10.1016/S0006-3495(91)82162-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok JP, Krabbe S, Markovic M, Courtin J, Xu C, Massi L, Botta P, Bylund K, Müller C, Kovacevic A, Tovote P, Lüthi A (2017) A competitive inhibitory circuit for selection of active and passive fear responses. Nature 542:96–100. 10.1038/nature21047 [DOI] [PubMed] [Google Scholar]
- Gilpin NW, Herman MA, Roberto M (2015) The central amygdala as an integrative hub for anxiety and alcohol use disorders. Biol Psychiatry 77:859–869. 10.1016/j.biopsych.2014.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins DA, Holstege G (1978) Amygdaloid projections to the mesencephalon, pons, and medulla oblongata in the cat. Exp Brain Res 32:529–547. [DOI] [PubMed] [Google Scholar]
- Itoga CA, Roltsch Hellard EA, Whitaker AM, Lu YL, Schreiber AL, Baynes BB, Baiamonte BA, Richardson HN, Gilpin NW (2016) Traumatic stress promotes hyperalgesia via corticotropin-releasing factor-1 receptor (CRFR1) signaling in central amygdala. Neuropsychopharmacology 41:2463–2472. 10.1038/npp.2016.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jochum T, Boettger MK, Burkhardt C, Juckel G, Bär KJ (2010) Increased pain sensitivity in alcohol withdrawal syndrome. Eur J Pain 14:713–718. 10.1016/j.ejpain.2009.11.008 [DOI] [PubMed] [Google Scholar]
- Johannes CB, Le TK, Zhou X, Johnston JA, Dworkin RH (2010) The prevalence of chronic pain in United States adults: results of an internet-based survey. J Pain 11:1230–1239. 10.1016/j.jpain.2010.07.002 [DOI] [PubMed] [Google Scholar]
- Jüngling K, Lange MD, Szkudlarek HJ, Lesting J, Erdmann FS, Doengi M, Kügler S, Pape HC (2015) Increased GABAergic efficacy of central amygdala projections to neuropeptide S neurons in the brainstem during fear memory retrieval. Neuropsychopharmacology 40:2753–2763. 10.1038/npp.2015.125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalange AS, Kokare DM, Singru PS, Upadhya MA, Chopde CT, Subhedar NK (2007) Central administration of selective melanocortin 4 receptor antagonist HS014 prevents morphine tolerance and withdrawal hyperalgesia. Brain Res 1181:10–20. 10.1016/j.brainres.2007.08.054 [DOI] [PubMed] [Google Scholar]
- Kishi T, Aschkenasi CJ, Lee CE, Mountjoy KG, Saper CB, Elmquist JK (2003) Expression of melanocortin 4 receptor mRNA in the central nervous system of the rat. J Comp Neurol 457:213–235. 10.1002/cne.10454 [DOI] [PubMed] [Google Scholar]
- Koob GF. (2008) A role for brain stress systems in addiction. Neuron 59:11–34. 10.1016/j.neuron.2008.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux JE, Iwata J, Cicchetti P, Reis DJ (1988) Different projections of the central amygdaloid nucleus mediate autonomic and behavioral correlates of conditioned fear. J Neurosci 8:2517–2529. 10.1523/JNEUROSCI.08-07-02517.1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux JE, Moscarello J, Sears R, Campese V (2017) The birth, death and resurrection of avoidance: a reconceptualization of a troubled paradigm. Mol Psychiatry 22:24–36. 10.1038/mp.2016.166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Sugam JA, Lowery-Gionta EG, McElligott ZA, McCall NM, Lopez AJ, McKlveen JM, Pleil KE, Kash TL (2016) Mu opioid receptor modulation of dopamine neurons in the periaqueductal gray/dorsal raphe: a role in regulation of pain. Neuropsychopharmacology 41:2122–2132. 10.1038/npp.2016.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantyh PW. (1982) The midbrain periaqueductal gray in the rat, cat, and monkey: a nissl, Weil, and Golgi analysis. J Comp Neurol 204:349–363. 10.1002/cne.902040406 [DOI] [PubMed] [Google Scholar]
- Millan MJ. (2002) Descending control of pain. Prog Neurobiol 66:355–474. 10.1016/S0301-0082(02)00009-6 [DOI] [PubMed] [Google Scholar]
- Oliveira MA, Prado WA (2001) Role of PAG in the antinociception evoked from the medial or central amygdala in rats. Brain Res Bull 54:55–63. 10.1016/S0361-9230(00)00420-2 [DOI] [PubMed] [Google Scholar]
- Omelchenko N, Sesack SR (2010) Periaqueductal gray afferents synapse onto dopamine and GABA neurons in the rat ventral tegmental area. J Neurosci Res 88:981–991. 10.1002/jnr.22265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan G, Li Y, Geng HY, Yang JM, Li KX, Li XM (2015) Preserving GABAergic interneurons in acute brain slices of mice using the N-methyl-D-glucamine-based artificial cerebrospinal fluid method. Neurosci Bull 31:265–270. 10.1007/s12264-014-1497-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape HC, Pare D (2010) Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol Rev 90:419–463. 10.1152/physrev.00037.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlovic ZW, Cooper ML, Bodnar RJ (1996) Opioid antagonists in the periaqueductal gray inhibit morphine and β-endorphin analgesia elicited from the amygdala of rats. Brain Res 741:13–26. 10.1016/S0006-8993(96)00880-3 [DOI] [PubMed] [Google Scholar]
- Pitkänen A. (2000) Connectivity of the rat amygdaloid complex. In: The amygdala (Aggleton JP, ed). Oxford: Oxford UP. [Google Scholar]
- Pomrenze MB, Millan EZ, Hopf FW, Keiflin R, Maiya R, Blasio A, Dadgar J, Kharazia V, De Guglielmo G, Crawford E, Janak PH, George O, Rice KC, Messing RO (2015) A transgenic rat for investigating the anatomy and function of corticotrophin releasing factor circuits. Front Neurosci 9:487. 10.3389/fnins.2015.00487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes BA, Drolet G, Van Bockstaele EJ (2008) Dynorphin and stress-related peptides in rat locus coeruleus: contribution of amygdalar efferents. J Comp Neurol 508:663–675. 10.1002/cne.21683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley JL 3rd, King C (2009) Self-report of alcohol use for pain in a multi-ethnic community sample. J Pain 10:944–952. 10.1016/j.jpain.2009.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi TA, Ennis M, Behbehani MM, Shipley MT (1991) Connections between the central nucleus of the amygdala and the midbrain periaqueductal grey: topography and reciprocity. J Comp Neurol 303:121–131. 10.1002/cne.903030111 [DOI] [PubMed] [Google Scholar]
- Roberto M, Madamba SG, Stouffer DG, Parsons LH, Siggins GR (2004) Increased GABA release in the central amygdala of ethanol-dependent rats. J Neurosci 24:10159–10166. 10.1523/JNEUROSCI.3004-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roltsch Hellard EA, Impastato RA, Gilpin NW (2017) Intra-cerebral and intra-nasal melanocortin-4 receptor antagonist blocks withdrawal hyperalgesia in alcohol-dependent rats. Addict Biol 22:692–701. 10.1111/adb.12360 [DOI] [PubMed] [Google Scholar]
- Samineni VK, Grajales-Reyes JG, Copits BA, O'Brien DE, Trigg SL, Gomez AM, Bruchas MR, Gereau RW 4th (2017) Divergent modulation of nociception by glutamatergic and GABAergic neuronal subpopulations in the periaqueductal gray. eNeuro 4:ENEURO.0129–0116.2017. 10.1523/ENEURO.0129-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Substance Abuse and Mental Health Services Administration (2015) 2015 National Survey on Drug Use and Health. Table 5.6A—substance use disorder in past year among persons aged 18 or older, by demographic characteristics: numbers in thousands, 2014 and 2015. https://www.samhsa.gov/data/sites/default/files/NSDUH-DetTabs-2015/NSDUH-DetTabs-2015/NSDUH-DetTabs-2015.htm#tab5–6a. [PubMed]
- Suckow SK, Deichsel EL, Ingram SL, Morgan MM, Aicher SA (2013) Columnar distribution of catecholaminergic neurons in the ventrolateral periaqueductal gray (vlPAG) and their relationship to efferent pathways. Synapse 67:94–108. 10.1002/syn.21624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter BA, O'Connor T, Iyer V, Petreanu LT, Hooks BM, Kiritani T, Svoboda K, Shepherd GM (2010) Ephus: multipurpose data acquisition software for neuroscience experiments. Front Neural Circuits 4:100. 10.3389/fncir.2010.00100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao YX. (2014) Constitutive activity in melanocortin-4 receptor: biased signaling of inverse agonists. Adv Pharmacol 70:135–154. 10.1016/B978-0-12-417197-8.00005-5 [DOI] [PubMed] [Google Scholar]
- Tershner SA, Helmstetter FJ (2000) Antinociception produced by mu opioid receptor activation in the amygdala is partly dependent on activation of mu opioid and neurotensin receptors in the ventral periaqueductal gray. Brain Res 865:17–26. 10.1016/S0006-8993(00)02179-X [DOI] [PubMed] [Google Scholar]
- Ting JT, Daigle TL, Chen Q, Feng G (2014) Acute brain slice methods for adult and aging animals: application of targeted patch clamp analysis and optogenetics. Methods Mol Biol 1183:221–242. 10.1007/978-1-4939-1096-0_14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovote P, Esposito MS, Botta P, Chaudun F, Fadok JP, Markovic M, Wolff SB, Ramakrishnan C, Fenno L, Deisseroth K, Herry C, Arber S, Lüthi A (2016) Midbrain circuits for defensive behaviour. Nature 534:206–212. 10.1038/nature17996 [DOI] [PubMed] [Google Scholar]
- Varodayan FP, Soni N, Bajo M, Luu G, Madamba SG, Schweitzer P, Parsons LH, Roberto M (2016) Chronic ethanol exposure decreases CB(1) receptor function at GABAergic synapses in the rat central amygdala. Addict Biol 21:788–801. 10.1111/adb.12256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wessendorf MW (2002) Mu- and delta-opioid receptor mRNAs are expressed in periaqueductal gray neurons projecting to the rostral ventromedial medulla. Neuroscience 109:619–634. 10.1016/S0306-4522(01)00328-1 [DOI] [PubMed] [Google Scholar]
- Wolff HG, Hardy JD, Goodell H (1942) Studies on pain: measurement of the effect of ethyl alcohol on the pain threshold and on the “alarm” reaction. J Pharmacol Exp Ther 75:38–49. [Google Scholar]
- Xu W, Lundeberg T, Wang YT, Li Y, Yu LC (2003) Antinociceptive effects of calcitonin gene-related peptide in the central nucleus of the amygdala: activating opioid receptors through amygdala-periaqueductal grey pathway. Neuroscience 118:1015–1022. 10.1016/S0306-4522(03)00069-1 [DOI] [PubMed] [Google Scholar]