

Abstract
Background
IL-6 classic signaling is linked to anti-inflammatory functions while the trans-signaling is associated with pro-inflammatory responses. Classic signaling is induced via membrane-bound IL-6 receptor (IL-6R) whereas trans-signaling requires prior binding of IL-6 to the soluble IL-6R. In both cases, association with the signal transducing gp130 receptor is compulsory. However, differences in the downstream signaling mechanisms of IL-6 classic- versus trans-signaling remains largely elusive.
Methods
In this study, we used flow cytometry, quantitative PCR, ELISA and immuno-blotting techniques to investigate IL-6 classic and trans-signaling mechanisms in Human Umbilical Vein Endothelial Cells (HUVECs).
Results
We show that both IL-6R and gp130 are expressed on the surface of human vascular endothelial cells, and that the expression is affected by pro-inflammatory stimuli. In contrast to IL-6 classic signaling, IL-6 trans-signaling induces the release of the pro-inflammatory chemokine Monocyte Chemoattractant Protein-1 (MCP-1) from human vascular endothelial cells. In addition, we reveal that the classic signaling induces activation of the JAK/STAT3 pathway while trans-signaling also activates the PI3K/AKT and the MEK/ERK pathways. Furthermore, we demonstrate that MCP-1 induction by IL-6 trans-signaling requires simultaneous activation of the JAK/STAT3 and PI3K/AKT pathways.
Conclusions
Collectively, our study reports molecular differences in IL-6 classic- and trans-signaling in human vascular endothelial cells; and elucidates the pathways which mediate MCP-1 induction by IL-6 trans-signaling.
Electronic supplementary material
The online version of this article (10.1186/s12964-018-0268-4) contains supplementary material, which is available to authorized users.
Keywords: Interleukin-6 signaling, Monocyte chemoattractant Protein-1, Pro-inflammatory cytokines, Endothelium, HUVECs
Background
Vascular endothelial cells are constantly exposed to numerous types of circulating signaling molecules; and are one of the main targets for various pro- and anti-inflammatory cytokines [1]. Interleukin-6 (IL-6) is a multifunctional cytokine produced by several cell types including monocytes/macrophages, adipocytes, hematopoietic and endothelial cells [2]. IL-6 is known to regulate the synthesis of acute phase proteins and is elevated in the circulation during inflammatory conditions [2]. Furthermore, IL-6 has been shown to induce the expression of endothelial adhesion molecules and chemoattractant proteins such as the Monocyte Chemoattractant Protein-1 (MCP-1) [3, 4]. MCP-1 (also called CCL-2) is a potent monocyte chemoattractant that is secreted by numerous types of cells including endothelial cells and contributes to the initiation and progression of atherosclerotic plaques [5–7].
Numerous clinical trials using anti-IL-6 antibodies or antibodies directed against the IL-6 receptor (IL-6R) have shown therapeutic significance of blocking IL-6 signaling in chronic inflammatory diseases, including atherosclerosis [8, 9]. However, such systemic blockade of IL-6 has also been associated with adverse effects including severe infections [10, 11]. In addition, there are studies reporting a regenerative, athero-protective and anti-inflammatory role for IL-6 [12–14]. These dual roles of IL-6 in inflammation appears to originate from how IL-6 interacts with the specific target cell. IL-6 can act on target cells by binding to the membrane-bound receptor (IL-6R) and subsequent recruitment of ubiquitously expressed signal transducing gp130 receptor. This is known as IL-6 classic signaling and it is restricted to cells possessing membrane-bound IL-6R including hepatocytes, macrophages, neutrophils and some T cell subsets [15]. These cells also release the soluble receptor (sIL-6R) into the circulation predominantly through extracellular shedding or by alternative splicing [16–18]. Hence, IL-6 can alternatively bind to sIL-6R and induce intra-cellular signaling via gp130 on cells that lack the membrane-bound IL-6R, which is referred to as IL-6 trans-signaling [19, 20]. The trans-signaling is associated with pro-inflammatory functions while the classic signaling is linked to regenerative and anti-inflammatory functions [21, 22]. However, little is known about the signaling mechanisms of IL-6 classic- and trans-signaling. In this study we elucidated differences in classic- versus trans-signaling of IL-6 in human vascular endothelial cells and revealed a novel pathway by which IL-6 trans-signaling mediates its pro-inflammatory effect.
Methods
Cell culturing
Human Umbilical Vein Endothelial Cells (HUVECs, Life technologies, USA), were cultured in 75cm2 flasks (Sarstedt, Germany) containing complete endothelial medium [VascuLife basal medium supplemented with VEGF LifeFactors kit (LifeLine Cell Technologies, USA)] and antibiotics [Penicillin (0.1 U/ml) + Streptomycin (100 ng/ml)-PEST, Gibco, Life Technologies, USA]. The cultures were kept at 37 °C and 5% CO2 environment and cells were maintained until passage 10 by replacing medium every 48–72 h and/or sub-culturing upon confluence.
Treatment of HUVECs
HUVECs (3 × 105 cells/well for 6-well plates and 6 × 104 cells/well for 24-well plates) were plated overnight in complete endothelial medium containing antibiotics. The next day, the medium was replaced with fresh antibiotics free medium and cells were treated with different concentrations of IL-6, IL-6R, TNF-α and LPS (all from R&D systems, USA) and/or pharmacological inhibitors CP690550, Stattic and LY294002 (all from R&D systems, USA), and PD98059 (Santa Cruz biotechnology, USA) for different time points ranging from 5 min to 48 h. At the end of incubations, supernatants and cells were collected and kept at -80 °C until further analysis. The cells were used to extract total protein or RNA.
Gene knockdown
HUVECs were seeded in 6-well plates (2 × 105 cells/well) containing complete endothelial medium with antibiotics and incubated overnight. After washing the cells with opti-MEM (Gibco, Life Technologies, USA), incubation continued with 700 μl opti-MEM/well containing 4 μl lipofectamine (Invitrogen, USA) and a mix of 3 stealth siRNAs (10 nM of each siRNA, Invitrogen, USA) targeting same gene. The control wells instead had non-target siRNAs (30 nM, Invitrogen, USA). After 4 h of incubation, 1.3 ml of complete endothelial medium was added into each well and incubation continued. At 48 h, culture supernatants and cells were collected and kept at -80 °C until further analysis.
Total RNA isolation and cDNA synthesis
Total RNA extraction from frozen cells was achieved using E.Z.N.A® Total RNA Kit (OMEGA bio-tek inc, USA) according to manufacturer’s instructions. Briefly, TRK lysis buffer with 2% β-Mercaptoethanol was added into each well and the lysates were then mixed with 70% ethanol. The cell lysates were transferred into HiBind RNA columns and centrifuged for 1 min at 10,000 g. After washing the columns three times, RNA was eluted using RNase free water. RNA concentrations were determined using NanoDrop™ 2000 (Thermo Fisher Scientific, USA) spectrophotometer.
Extracted RNA were used to synthesize cDNA using high capacity cDNA reverse transcription kit (Thermo Fisher Scientific, USA) according to manufacturer’s instructions. Hence, 1 μg RNA extract was mixed with master mix composed of buffer, random primers, dNTPs and reverse transcriptase enzyme. The total reaction volume was adjusted to 20 μl by adding nuclease free water. A negative control containing master mix and water instead of RNA was also included. The following setup was used for thermal cycling: 10 min at 25 °C, 120 min at 37 °C, 5 min at 85 °C and kept at 4 °C before storage at − 20 °C.
Real-time PCR
mRNA expression of genes was studied using TaqMan qPCR primers/probes according to manufacturer’s instructions. In short, a standard curve was prepared by pooling equal volume of cDNA from each sample that was then serially diluted in to 6 standards. Standards, negative controls and unknown samples were run in duplicate in a 96-well PCR plate. The total reaction volume was 10 μl consisting of LuminoCt qPCR ready mix (Sigma-Aldrich, USA), TaqMan Primer/Probe (Applied Biosystems, Life technologies, USA), water and cDNA. The cycling condition used was as follows: at 95 °C for 1 s and at 60 °C for 20 s for 40 cycles in addition to one step initialization at 95 °C for 20 s in ABI 7900HT Fast Real-Time PCR system (Applied Biosystems). Then, relative quantities were recorded for each well and normalized to the expression of housekeeping gene, GAPDH.
Sandwich enzyme linked Immuno-sorbent assay (ELISA)
DuoSet® ELISA kits (R&D systems, USA) were used according to manufacturer’s instruction to determine the release of MCP-1 from HUVECs and expression of the IL-6-receptor and gp130 in HUVEC culture supernatants and cell lysates. Multiskan-Ascent (Lab Systems, Thermo Fisher Scientific, USA) Spectrophotometer was used to read absorbance at 450 nm. For IL-6R ELISA, 2 more standards to the lower end were included to cover low concentrations in samples.
Protein extraction and quantification
HUVECs were rinsed with PBS and lysed using ice-cold RIPA lysis buffer (Millipore, USA). To quantify the proteins, Micro BCA™ Protein Assay kit was used (Thermo Scientific, USA) according to manufacturer’s instructions and absorbance at 540 nm was measured using Multiskan-Ascent (Lab Systems, Thermo Fisher Scientific, USA) Spectrophotometer.
Immuno-(western) blotting
Cell lysates were mixed with 4× SDS sample buffer and denatured for 5 min at 95 °C. Next, the mixture (10 μg protein/well) was loaded on to 4–12% NuPAGE® Novex Bis-Tris gels and protein separation was achieved with MOPS/MES running buffers (both Invitrogen, USA). To determine molecular masses of the proteins, MagicMark™ XP Western Protein Standard (Invitrogen, USA) was used. Proteins were blotted onto Immobilon-FL PVDF membranes (Millipore, USA). For further steps TBS-T (10 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.1% (w/v) Tween-20) was used. For detecting signaling proteins, membranes were incubated with the following primary antibodies: anti-phospho-Stat3Tyr705 antibody (Cell Signaling Technology, USA, #9131; 1:1000 dilution), anti-phospho-AKTSer473 antibody (Cell Signaling Technology, USA, #4060; 1:2000 dilution), anti-phospho-ERK1/2 antibody (Cell Signaling Technology, USA, #9106; 1:2000 dilution), anti-STAT3 antibody (Cell Signaling Technology, USA, #4904; 1:2000 dilution), anti-AKT antibody (Cell Signaling Technology, USA, #2920; 1:2000 dilution), anti-ERK1/2 antibody (Cell Signaling Technology, USA, #4695; 1:1000 dilution), anti-phospho IκBαSer32 antibody (Cell Signaling Technology, USA, #2859; 1:1000 dilution), anti-phospho-NFκB p65Ser536 antibody (Cell Signaling Technology, USA, #3033; 1:1000 dilution), anti-NFκB p65 antibody (Cell Signaling Technology, USA, #6956; 1:1000 dilution), anti-IκBα antibody (Santa Cruz Biotechnology, USA, SC-371; 1:750 dilution), anti-p52 antibody (Millipore, USA, #05–361; 1:1000 dilution), and anti-β-Tubulin antibody (Millipore, USA, #05–661; 1:2000 dilution). This was followed by incubation with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgGs (Cell Signaling Technology, USA, #7074; 1:2000) or horse anti-mouse IgGs (Cell Signaling Technology, USA, #7076; 1:2000). Protein bands were visualized using Immobilon™ Western Chemiluniescent HRP Substrate solution from Millipore (Millipore, USA), and chemiluminiscence was recorded by a Li-Cor Odyssey Fc imager and analyzed with Image Studio Software (both Li-Cor Biotechnology UK Ltd., United Kingdom). Stripping of membranes for re-probing was done according to manufacturer’s instructions using Restore™ Plus western blot stripping buffer (Thermo Fisher Scientific, USA, #46430).
Flow cytometry
EDTA detached HUVECs were washed twice with PBS containing 1 mM EDTA and 2% FBS; and then stained with α-CD130 (gp130)-APC (clone: 2E1B02) and α-CD126 (IL-6Rα)-PE (clone: UV4) antibodies (BioLegend, UK) for 25 min at 4 °C in the dark. Fluorescence minus one (FMO) controls were used as negative control for staining. 7AAD staining was performed to remove dead cells from analyses. Stained cells were acquired using Gallios™ Flow Cytometer (Beckman Coulter Life Sciences, UK) and analyzed using Kaluza flow cytometry analysis software version 1.3 (Beckman Coulter, UK).
Statistical analysis
Data were analyzed using GraphPad Prism® statistical software version 5.0 (GraphPad Software, Inc., USA). Data are presented as mean ± standard error of the mean (SEM) of at least 3 sets of experiments. For comparison between groups, paired t-test/Wilcoxon matched paired test and one-way ANOVA for repeated measures followed by Bonferroni post-hoc test was used. p value less than 0.05 was considered as statistically significant.
Results
Expression and regulation of IL-6 receptors in vascular endothelial cells
To study whether human vascular endothelial cells express IL-6R and gp130, we used flow cytometry to detect their surface protein expression. As depicted in Fig. 1a & b, both IL-6R and gp130 proteins are expressed on surface of human vascular endothelial cells. We also assessed how different pro-inflammatory stimuli could regulate expression of IL-6R and gp130 in these cells using ELISA. Knockdown of IL-6 (80–90% knockdown efficiency, Additional file 1: Figure S1) increased the levels of IL-6R and gp130 (Fig. 1c & d). In addition, treatment of endothelial cells with pro-inflammatory stimuli (TNF-α or LPS) resulted in downregulation of both IL-6R and sIL-6R while upregulating gp130 and sgp130 levels (Fig. 1e & f). Overall, these findings demonstrate that IL-6R and gp130 are expressed on human vascular endothelial cells, allowing for the activation of both IL-6 classic- and trans-signaling in these cells.
Fig. 1.

Expression and regulation of IL-6R and gp130 in human vascular endothelial cells. Flow cytometry analyses of EDTA detached endothelial cells stained for a IL-6R and b gp130. The light grey histograms show respective FMO (fluorescence minus one) controls and the dark grey histograms show IL-6R or gp130 stained cells. ELISA analyses of the levels of c IL-6R and d gp130 in cell lysates and medium after 48 h of IL-6 knockdown. ELISA analyses of the levels of e IL-6R and f gp130 in cell lysates and medium after 48 h of treatment with TNF-α (50 ng/ml) and LPS (100 ng/ml). Data is presented as mean ± SEM of at least 3–4 experiments each run-in duplicate. *p < 0.05, **p < 0.01, ***p < 0.001 compared to control
MCP-1 induction in human vascular endothelial cells in response to IL-6
Next, we investigated the response of human vascular endothelial cells to the treatment with IL-6 alone or in combination with sIL-6R on the expression and release of MCP-1. We found that IL-6 in combination with sIL-6R induced a prominent increase in MCP-1 mRNA that peaked after 60 min, while IL-6 alone had a very limited effect (Fig. 2a). In addition, we show that stimulation with IL-6 in combination with sIL-6R, but not IL-6 alone, induced a dose-dependent release of MCP-1 into the medium (Fig. 2b). Further, stimulation of human vascular endothelial cells with combination of IL-6 and sIL-6R, but not IL-6 alone, upregulated cell adhesion molecules VCAM-1 and ICAM-1 (Additional file 2: Figure S2A and 2B). These findings indicate that IL-6 requires the sIL-6R to induce pro-inflammatory responses in human vascular endothelial cells.
Fig. 2.
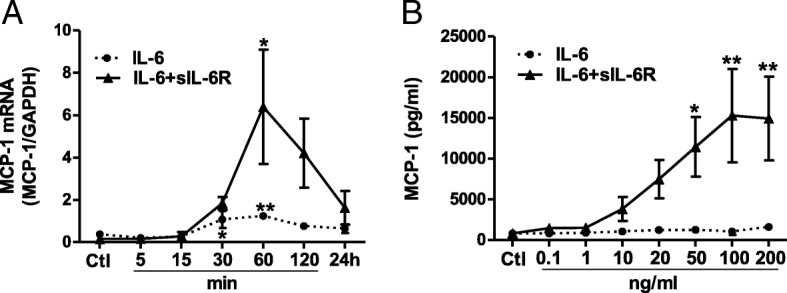
MCP-1 mRNA expression and release from human vascular endothelial cells. a qPCR analyses depicting the relative expression of MCP-1 mRNA after exposure to IL-6 (100 ng/ml) alone or in combination with sIL-6R (200 ng/ml). b ELISA data showing the MCP-1 release into medium (48 h) induced by increasing concentrations of IL-6 alone or in combination with sIL-6R. Data is presented as mean ± SEM of 3 experiments each run-in duplicate. *p < 0.05, **p < 0.01 compared to its respective control
Activation of signaling pathways by IL-6 with or without sIL-6R
In order to investigate the different signaling pathways engaged, we treated human vascular endothelial cells with IL-6 alone (classic signaling) or together with sIL-6R (trans-signaling) for increasing durations. We then analyzed activation of different signaling pathways using immuno-(western) blotting. We found that phosphorylation of STAT3Tyr705 is induced by both IL-6 alone and in combination with sIL-6R (Fig. 3a, left) in a dose-dependent manner (Additional file 3: Figure S3). When applied together with sIL-6R, very low concentrations of IL-6 (i.e. 1 ng/ml) were sufficient to induce STAT3Tyr705 phosphorylation (Additional file 3: Figure S3), while higher concentrations (i.e. 50 ng/ml) were required to induce similar degree of phosphorylation when IL-6 is added without sIL-6R. Although IL-6 in combination with sIL-6R provoked a stronger phosphorylation of STAT3Tyr705 compared to IL-6 alone, both treatments follow similar kinetics in which the phosphorylation peaked after 10-15 min of stimulation (Fig. 3a, right).
Fig. 3.

Western blot analyses showing phosphorylation of a STAT3Tyr705, b AKTSer473 and c ERK1/2Thr202/Tyr204 in human vascular endothelial cells treated with IL-6 alone (50 ng/ml) or in combination with sIL-6R (100 ng/ml). One representative blot containing the phosphorylated protein, total protein and β-tubulin (loading control) is shown for each pathway (left column). The signals from the phosphorylated proteins and total proteins are first normalized to β-tubulin, and the ratio of the phosphorylated proteins and the total proteins are calculated. The graphs show arbitrary units (a.u., control is set to 1) compiled from 3 independent experiments presented as mean ± SEM for each pathway (right column). *p < 0.05, **p < 0.01, ***p < 0.001 compared to control
In addition, we found that IL-6 alone does not induce phosphorylation of AKTSer473 (Fig. 3b, left) or ERK1/2Thr202/Tyr204 (Fig. 3c, left). However, when applied in combination with sIL-6R, IL-6 causes a strong AKTSer473 (Fig. 3b, right) as well as ERK1/2Thr202/Tyr204 (Fig. 3c, left) phosphorylation which peaked at 30 min after treatment. We also investigated whether IL-6 induces activation of NFκB signaling pathways by determining the level of p-p65Ser32, pIκBSer536 and IκB for canonical NFκB activation; and the level of p100 and p52 for non-canonical NFκB activation. The treatment of human vascular endothelial cells with IL-6 alone or in combination with sIL-6R did not cause phosphorylation of p65Ser32 or IκBSer536 nor did it reduce the level of IκB (Additional file 4: Figure S4), unlike the case for TNF-α stimulation (data not shown). Moreover, the levels of p100 and p52 were not affected by neither IL-6 alone nor in combination with sIL-6R (Additional file 4: Figure S4). These findings indicate that IL-6 signaling pathways are distinct; in which classic signaling engages the JAK/STAT3 pathway while trans-signaling employs the JAK/STAT3, PI3K/AKT, and the MEK/ERK signaling pathways. In addition, neither canonical- nor non-canonical NFκB signaling pathways seem to be activated by either IL-6 classic or trans-signaling.
Pathways mediating IL-6 trans-signaling induced MCP-1 in human vascular endothelial cells
To further elucidate the pathways accounting for the regulation of MCP-1 by IL-6 trans-signaling, we used pharmacological inhibitors or siRNA to interfere with the different pathways and assessed MCP-1 expression after exposure to IL-6 in combination with sIL-6R. We found that pre-treatment of human vascular endothelial cells with the JAK inhibitor CP690550 prevents the trans-signaling induced phosphorylation of STAT3Tyr705, AKTSer473 and ERK1/2Thr202/Tyr204 (Fig. 4a). In addition, CP690550 pre-treatment downregulated the MCP-1 expression evoked via trans-signaling in these cells (Fig. 4a). These findings indicate that JAK is upstream of STAT3, AKT and ERK1/2 in the signaling pathway induced by IL-6 trans-signaling, and that JAK is crucial for the induction of MCP-1.
Fig. 4.

The effect of different inhibitions on IL-6 trans-signaling induced MCP-1 mRNA expression. The effect of a CP960550 (JAK inhibitor, 10 μM), b STAT3 knockdown, c LY294002 (PI3K inhibitor, 50 μM) and d PD98059 (MEK inhibitor, 10 μM) on trans-signaling (IL-6 = 100 ng/ml, sIL-6R = 200 ng/ml) induced MCP-1 expression is presented as mean ± SEM (n = 3 for each). Western blots show effect of CP960550, STAT3 knockdown, LY294002 and PD98059 on IL-6 trans-signaling (IL-6 = 50 ng/ml, sIL-6R = 100 ng/ml) induced phosphorylation of downstream pathway proteins. *p < 0.05, **p < 0.01, ***p < 0.001 compared to control
Knockdown of STAT3 in human vascular endothelial cells using siRNA (knockdown efficiency 80–90%) abolished the phosphorylation of STAT3Tyr705 caused by IL-6 trans-signaling (Fig. 4b). In line with that, qPCR analyses revealed that MCP-1 induction by IL-6 trans-signaling was eliminated in STAT3 knockdown cells (Fig. 4b). Similarly, treating human vascular endothelial cells with the PI3K inhibitor LY294002 prior to stimulation with IL-6 combined with sIL-6R resulted in the suppression of AKTSer473 phosphorylation (Fig. 4c). Furthermore, MCP-1 induction by IL-6 trans-signaling was inhibited by LY294002 pre-treatment (Fig. 4c). Pre-treatment with the MEK inhibitor PD98059 blocked IL-6 trans-signaling induced phosphorylation of ERK1/2Thr202/Tyr204(Fig. 4d), but not MCP-1 expression in human vascular endothelial cells (Fig. 4d). Overall, our findings demonstrate that IL-6 trans-signaling employs the JAK/STAT3 and PI3K/AKT pathways to provoke MCP-1 expression in human vascular endothelial cells.
Discussion
In this study, we elucidated differences in classic- versus trans-signaling of IL-6 in human vascular endothelial cells. Our results show that human vascular endothelial cells express IL-6R and gp130 on their surface allowing for the activation of both IL-6 classic- and trans-signaling in these cells. We demonstrate that IL-6 classic signaling causes transient phosphorylation of STAT3Tyr705, while trans-signaling also leads to phosphorylation of AKTSer473 and ERK1/2Thr202/Tyr204. Further, we report that IL-6 trans-signaling induces the expression and release of the pro-inflammatory chemokine MCP-1 in human vascular endothelial cells, and that this is mediated via JAK/STAT3 and PI3K/AKT pathways.
The general assumption is that the expression of IL-6R is restricted to only a few cell types, such as hepatocytes and immune cells. However, we provide evidence that IL-6R is expressed on the surface of human vascular endothelial cells. This finding is in contrast to previous studies which could not to determine IL-6R expression on these cells [23], and indirect implications from the lack in responsiveness of the cells to IL-6 [24]. We also show that vascular endothelial cells release IL-6R in its soluble form, and that its expression is regulated by pro-inflammatory agents. In agreement with findings in monocytes, TNF-α [25] and LPS [26] treatment repress the level of both membrane-bound and soluble IL-6R in vascular endothelial cells. On the other hand, both TNF-α and LPS upregulate gp130 surface expression and its release from vascular endothelial cells. This indicates that pro-inflammatory stimuli render endothelial cells to become more responsive to trans-signaling and may favor a pro-inflammatory response. However, it is important to note that gp130 is a common signal transducing receptor for several cytokines and might also initiate other responses. Our results show that activation of classic signaling induces a minor induction of MCP-1 mRNA although it does not affect the protein release. The trans-signaling, however, induces a prominent increase in MCP-1 mRNA which corresponds to significantly higher level of MCP-1 release. These latter findings are consistent with previous reports [23, 24].
The IL-6R has only a short intracellular domain and its signal transduction is based on ligand-binding dependent recruitment of gp130 to which JAKs (JAK1, JAK2 and TYK2) are closely associated [27, 28]. Hence, IL-6 is generally known to activate JAK/STAT3 pathway regardless of binding to soluble or membrane bound IL-6R. In line with this, we show that both classic- and trans-signaling of IL-6 trigger STAT3Tyr705 phosphorylation in a dose- and time-dependent manner. However, the trans-signaling causes a markedly increased STAT3Tyr705 phosphorylation compared to classic signaling. Furthermore, we report that IL-6 trans-signaling, but not classic signaling, leads to an activation of the PI3K/AKT and MEK/ERK pathways. It has previously been suggested that gp130-mediated activation of these pathways is achieved by the recruitment of SHP-2 (SH2 domain-containing protein-tyrosine phosphatase) to intracellular domain of gp130 (Tyr759) [29]. The same phospho-site also serves as docking site for SOCS3 (suppressor of cytokine signaling 3), a key negative regulator of IL-6 signaling pathways [30]. MCP-1 production by human vascular endothelial cells in response to pro-inflammatory mediators has been shown to involve NFκB pathways [31]. Moreover, IL-6 has been shown to trigger NFκB activation in intestinal epithelial cells thereby promoting inflammation [32]. We found that neither IL-6 classic- nor trans-signaling induce activation of the canonical or non-canonical NFκB pathways, indicating that the induction of MCP-1 by IL-6 trans-signaling is independent of NFκB pathways in vascular endothelial cells.
The involvement of STAT3 in IL-6 trans-signaling induced MCP-1 production from vascular endothelial cells has been previously reported by the use of STAT3 inhibitors such as Stattic [24]. In our hands, however, this inhibitor strongly affected cell viability (data not shown) and therefore we employed siRNA guided STAT3 knockdown instead. In line with previous findings [24], we determined that STAT3 is essential for the trans-signaling mediated MCP-1 expression in vascular endothelial cells. Nevertheless, blockade of PI3K/AKT pathway also resulted in a complete inhibition of trans-signaling caused AKTSer473 phosphorylation as well as MCP-1 induction. This indicates that MCP-1 induction by IL-6 trans-signaling requires simultaneous activation of the JAK/STAT3 and PI3K/AKT pathways. It is well known that activation of the JAK/STAT3 pathway leads to STAT3 dimerization and translocation into the nucleus where it initiates gene transcription [33]. However, the downstream effects of PI3K signaling are mostly associated with activation of S6 kinases (e.g. S6 K1) and inhibition of 4E-binding protein 1 (4E-BP1), both participating in post-transcriptional regulation [34]. Given the significant reduction in MCP-1 mRNA expression by PI3K signaling inhibition, it is likely that activation of the PI3K pathway also activates downstream transcription factors. Several mechanisms by which PI3K regulates gene transcription have been shown previously [35–37]. Furthermore, we show that the blockade of the MEK/ERK pathway abolishes trans-signaling induced ERK1/2Thr202/Tyr204 phosphorylation while the MCP-1 induction remains unaffected. These findings suggest that ERK1/2 activation in response to IL-6 trans-signaling has a minor role in MCP-1 induction in human vascular endothelial cells. In contrast to our findings, the MEK/ERK pathway has been shown to be involved in IL-6 induced regulation of MCP-1 in fibroblasts [38]. This may be explained by a cell type specific and distinct activation of pathways and responses caused by IL-6 trans-signaling. As expected, inhibition of JAK abrogated trans-signaling induced phosphorylation of STAT3Tyr705, AKTSer473, and ERK1/2Thr202/Tyr204 and abolished MCP-1 induction.
Conclusions
Put together, this study sheds light on differences in IL-6 classic- and trans-signaling on molecular level in human vascular endothelial cells, demonstrating that concurrent activation of the JAK/STAT3 and PI3K/AKT pathways is essential for trans-signaling induced MCP-1 production.
Additional files
Figure S1. Bar graph showing ELISA analyses on IL6 release from human vascular endothelial cells during IL6 knockdown compared to control. Data is presented as mean ± SEM of 8 experiments each run-in duplicate. ***p < 0.01 compared to control. (PDF 14 kb)
Figure S2. Bar graphs showing mRNA expression of cell adhesion molecule (A) VCAM-1 and (B) ICAM-1 by human vascular endothelial cells in response to stimulation with IL-6 and sIL-6R (24 h). Data is presented as mean ± SEM of 3 experiments each run-in duplicate. **p < 0.01, ***p < 0.001 compared to control. (PDF 19 kb)
Figure S3. Phosphorylation of STAT3Tyr705 in response to increasing concentration of (A) IL-6 alone or (B) in combination with sIL-6R. One representative blot and total STAT3 (loading control) is shown (left column). The graphs show arbitrary units (a.u., control is set to 1) compiled from 2 to 3 independent experiments presented as mean ± SEM for each pathway (right column). *p < 0.05, **p < 0.01 compared to control. (PDF 235 kb)
Figure S4. Western blot analyses showing the levels of pIκBSer32/IκB and -p65Ser32/p65 (canonical NFκB), and p100/p52 (non-canonical NFκB) in vascular endothelial cells stimulated with IL-6 alone (50 ng/ml, top section) or in combination with sIL-6R (100 ng/ml, bottom section). (PDF 177 kb)
Acknowledgements
The authors would like to thank the Knowledge Foundation, Längmanska Foundation, and the Foundation for Old Servants (Stiftelsen Gamla Tjänarinnor) for supporting this study.
Funding
This work was supported by the Knowledge Foundation, Längmanska Foundation, and the Foundation for Old Servants (Stiftelsen Gamla Tjänarinnor).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- a.u
Arbitrary units
- FMO
Fluorescence minus one
- gp130
Glyco-protein-130
- HUVECs
Human Umbilical Vein Endothelial Cells
- ICAM-1
Intercellular adhesion molecule-1
- IL-6
Interleukin-6
- IL-6R
IL-6 Receptor
- MCP-1
Monocyte Chemo-attractant Protein-1
- sgp130
Soluble-gp130
- SHP-2
SH2 domain-containing protein-tyrosine phosphatase
- sIL-6R
Soluble-IL-6R
- SOCS3
Suppressor of Cytokine Signaling-3
- VCAM-1
Vascular cell adhesion molecule-1
Authors’ contributions
MMZ, AKK, AS and LUL designed the experiments. MMZ, ML, KF, AKK, GP and LUL performed the experimental work. MMZ, AKK, AS and LUL analyzed the data and interpreted the results. MMZ wrote the manuscript with help from AS and LUL. MMZ, KF, AKK, MG, AS and LUL provided scientific input, and all authors read and approved the final manuscript.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Mulugeta M. Zegeye, Email: mulugeta.m.zegeye@oru.se
Madelene Lindkvist, Email: madelene.lindkvist@oru.se.
Knut Fälker, Email: knut.falker@oru.se.
Ashok K. Kumawat, Email: ashok.kumawat@oru.se
Geena Paramel, Email: geenapvarghese@gmail.com.
Magnus Grenegård, Email: magnus.grenegard@oru.se.
Allan Sirsjö, Email: allan.sirsjo@oru.se.
Liza U. Ljungberg, Email: liza.ljungberg@oru.se
References
- 1.Gimbrone MA, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118:620–636. doi: 10.1161/CIRCRESAHA.115.306301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ataie-Kachoie P, Pourgholami MH, Richardson DR, Morris DL. Gene of the month: interleukin 6 (IL-6) J Clin Pathol. 2014;67:932–937. doi: 10.1136/jclinpath-2014-202493. [DOI] [PubMed] [Google Scholar]
- 3.Suzuki M, Hashizume M, Yoshida H, Mihara M. Anti-inflammatory mechanism of tocilizumab, a humanized anti-IL-6R antibody: effect on the expression of chemokine and adhesion molecule. Rheumatol Int. 2010;30:309–15. 10.1007/s00296-009-0953-0. [DOI] [PubMed]
- 4.Wung BS, Ni CW, Wang DL. ICAM-1 induction by TNFalpha and IL-6 is mediated by distinct pathways via Rac in endothelial cells. J Biomed Sci. 2005;12:91–101. doi: 10.1007/s11373-004-8170-z. [DOI] [PubMed] [Google Scholar]
- 5.Dawson TC, Kuziel WA, Osahar TA, Maeda N. Absence of CC chemokine receptor-2 reduces atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis. 1999;143:205–211. doi: 10.1016/S0021-9150(98)00318-9. [DOI] [PubMed] [Google Scholar]
- 6.Öhman MK, Wright AP, Wickenheiser KJ, Luo W, Russo HM, Eitzman DT. Monocyte chemoattractant protein-1 deficiency protects against visceral fat-induced atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30:1151–1158. doi: 10.1161/ATVBAHA.110.205914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang X, Liu X, Shang H, Xu Y, Qian M. Monocyte chemoattractant protein-1 induces endothelial cell apoptosis in vitro through a p53-dependent mitochondrial pathway. Acta Biochim Biophys Sin. 2011;43:787–95. 10.1093/abbs/gmr072.Advance. [DOI] [PubMed]
- 8.Yao X, Huang J, Zhong H, Shen N, Faggioni R, Fung M, et al. Targeting interleukin-6 in inflammatory autoimmune diseases and cancers. Pharmacol Ther. 2014;141:125–129. doi: 10.1016/j.pharmthera.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 9.Tanaka T, Kishimoto T. Targeting interleukin-6: all the way to treat autoimmune and inflammatory diseases. Int J Biol Sci. 2012;8:1227–1236. doi: 10.7150/ijbs.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Md Yusof MY, Emery P. Targeting interleukin-6 in rheumatoid arthritis. Drugs. 2013;73:341–356. doi: 10.1007/s40265-013-0018-2. [DOI] [PubMed] [Google Scholar]
- 11.Schiff MH, Kremer JM, Jahreis A, Vernon E, Isaacs JD, van Vollenhoven RF. Integrated safety in tocilizumab clinical trials. Arthritis Res Ther. 2011;13:R141. doi: 10.1186/ar3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xing Z, Gauldie J, Cox G, Baumann H, Jordana M, Lei X-F, et al. IL-6 is an Antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J Clin Invest. 1998;101:311–320. doi: 10.1172/JCI1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Madan M, Bishayi B, Hoge M, Amar S. Atheroprotective role of interleukin-6 in diet- and/or pathogen-associated atherosclerosis using an ApoE heterozygote murine model. Atherosclerosis. 2008;197:504–514. doi: 10.1016/j.atherosclerosis.2007.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leibinger M, Müller A, Gobrecht P, Diekmann H, Andreadaki A, Fischer D. Interleukin-6 contributes to CNS axon regeneration upon inflammatory stimulation. Cell Death Dis. 2013;4 10.1038/cddis.2013.126. [DOI] [PMC free article] [PubMed]
- 15.Schaper F, Rose-John S. Interleukin-6: biology, signaling and strategies of blockade. Cytokine Growth Factor Rev. 2015;26:475–487. doi: 10.1016/j.cytogfr.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 16.Wolf J, Rose-John S, Garbers C. Interleukin-6 and its receptors: a highly regulated and dynamic system. Cytokine. 2014;70:11–20. doi: 10.1016/j.cyto.2014.05.024. [DOI] [PubMed] [Google Scholar]
- 17.Schumacher N, Meyer D, Mauermann A, Von Der Heyde J, Wolf J, Schwarz J, et al. Shedding of endogenous interleukin-6 receptor (IL-6R) is governed by a disintegrin and metalloproteinase (ADAM) proteases while a full-length IL-6R isoform localizes to circulating microvesicles. J Biol Chem. 2015;290:26059–26071. doi: 10.1074/jbc.M115.649509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Riethmueller S, Somasundaram P, Ehlers JC, Hung CW, Flynn CM, Lokau J, et al. Proteolytic Origin of the Soluble Human IL-6R In Vivo and a Decisive Role of N-Glycosylation. PLOS Biology. 2017;15. 10.1371/journal.pbio.2000080. [DOI] [PMC free article] [PubMed]
- 19.Rose-John S. Il-6 trans-signaling via the soluble IL-6 receptor: importance for the proinflammatory activities of IL-6. Int J Biol Sci. 2012;8:1237–1247. doi: 10.7150/ijbs.4989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones SA, Rose-John S. The role of soluble receptors in cytokine biology: the agonistic properties of the sIL-6R/IL-6 complex. Biochim Biophys Acta, Mol Cell Res. 2002;1592:251–263. doi: 10.1016/S0167-4889(02)00319-1. [DOI] [PubMed] [Google Scholar]
- 21.Reiss AB, Siegart NM, De Leon J. Interleukin-6 in atherosclerosis: atherogenic or atheroprotective? Clin Lipidol. 2017;12:14–23. [Google Scholar]
- 22.Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta, Mol Cell Res. 2011;1813:878–888. doi: 10.1016/j.bbamcr.2011.01.034. [DOI] [PubMed] [Google Scholar]
- 23.Romano M, Sironi M, Toniatti C, Polentarutti N, Fruscella P, Ghezzi P, et al. Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity. 1997;6:315–325. doi: 10.1016/S1074-7613(00)80334-9. [DOI] [PubMed] [Google Scholar]
- 24.Chatterjee PK, Al-Abed Y, Sherry B, Metz CN. Cholinergic agonists regulate JAK2/STAT3 signaling to suppress endothelial cell activation. Am J Physiol Physiol. 2009;297:C1294–C1306. doi: 10.1152/ajpcell.00160.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanceau J, Wijdenes J, Revel M, Wietzerbin J. IL-6 and IL-6 receptor modulation by IFN-gamma and tumor necrosis factor-alpha in human monocytic cell line (THP-1). Priming effect of IFN-gamma. J Immunol. 1991;147:2630–2637. [PubMed] [Google Scholar]
- 26.Bauer J, Lengyel G, Bauer TM, Acs G, Gerok W. Regulation of interleukin-6 receptor expression in human monocytes and hepatocytes. FEBS Lett. 1989;249:27–30. doi: 10.1016/0014-5793(89)80008-0. [DOI] [PubMed] [Google Scholar]
- 27.Yamasaki K, Taga T, Hirata Y, Yawata H, Kawanishi Y, Seed B, et al. Cloning and expression of the human interleukin-6 (BSF-2/IFN beta 2) receptor. Science. 1988;241:825–828. doi: 10.1126/science.3136546. [DOI] [PubMed] [Google Scholar]
- 28.HEINRICH PC, BEHRMANN I, MÜLLER-NEWEN G, SCHAPER F, GRAEVE L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J. 1998;334:297–314. doi: 10.1042/bj3340297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eulenfeld R, Dittrich A, Khouri C, Müller PJ, Mütze B, Wolf A, et al. Interleukin-6 signalling: more than Jaks and STATs. Eur J Cell Biol. 2012;91:486–495. doi: 10.1016/j.ejcb.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 30.Lang R, Pauleau AL, Parganas E, Takahashi Y, Mages J, Ihle JN, et al. SOCS3 regulates the plasticity of gp130 signaling. Nat Immunol. 2003;4:546–550. doi: 10.1038/ni932. [DOI] [PubMed] [Google Scholar]
- 31.Martin T, Cardarelli PM, Parry GC, Felts KA, Cobb RR. Cytokine induction of monocyte chemoattractant protein-1 gene expression in human endothelial cells depends on the cooperative action of NF-kappa B and AP-1. Eur J Immunol. 1997;27:1091–1097. doi: 10.1002/eji.1830270508. [DOI] [PubMed] [Google Scholar]
- 32.Wang L, Walia B, Evans J, Gewirtz AT, Merlin D, Sitaraman SV. IL-6 induces NF- B activation in the intestinal epithelia. J Immunol. 2003;171:3194–3201. doi: 10.4049/jimmunol.171.6.3194. [DOI] [PubMed] [Google Scholar]
- 33.Kiu H, Nicholson SE. Biology and significance of the JAK/STAT signalling pathways. Growth Factors. 2012;30:88–106. doi: 10.3109/08977194.2012.660936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saleiro D, Platanias LC. Intersection of mTOR and STAT signaling in immunity. Trends Immunol. 2015;36:21–29. doi: 10.1016/j.it.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yokogami K, Wakisaka S, Avruch J, Reeves SA. Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr Biol. 2000;10:47–50. doi: 10.1016/S0960-9822(99)00268-7. [DOI] [PubMed] [Google Scholar]
- 36.Kim JH, Jae EK, Liu HY, Cao W, Chen J. Regulation of interleukin-6-induced hepatic insulin resistance by mammalian target of rapamycin through the STAT3-SOCS3 pathway. J Biol Chem. 2008;283:708–715. doi: 10.1074/jbc.M708568200. [DOI] [PubMed] [Google Scholar]
- 37.Kim J-H, Yoon M-S, Chen J. Signal transducer and activator of transcription 3 (STAT3) mediates amino acid inhibition of insulin signaling through serine 727 phosphorylation. J Biol Chem. 2009;284:35425–35432. doi: 10.1074/jbc.M109.051516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sobota RM, Müller PJ, Heinrich PC, Schaper F. Prostaglandin E1 inhibits IL-6-induced MCP-1 expression by interfering specifically in IL-6-dependent ERK1/2, but not STAT3, activation. Biochem J. 2008;412:65–72. doi: 10.1042/BJ20071572. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Bar graph showing ELISA analyses on IL6 release from human vascular endothelial cells during IL6 knockdown compared to control. Data is presented as mean ± SEM of 8 experiments each run-in duplicate. ***p < 0.01 compared to control. (PDF 14 kb)
Figure S2. Bar graphs showing mRNA expression of cell adhesion molecule (A) VCAM-1 and (B) ICAM-1 by human vascular endothelial cells in response to stimulation with IL-6 and sIL-6R (24 h). Data is presented as mean ± SEM of 3 experiments each run-in duplicate. **p < 0.01, ***p < 0.001 compared to control. (PDF 19 kb)
Figure S3. Phosphorylation of STAT3Tyr705 in response to increasing concentration of (A) IL-6 alone or (B) in combination with sIL-6R. One representative blot and total STAT3 (loading control) is shown (left column). The graphs show arbitrary units (a.u., control is set to 1) compiled from 2 to 3 independent experiments presented as mean ± SEM for each pathway (right column). *p < 0.05, **p < 0.01 compared to control. (PDF 235 kb)
Figure S4. Western blot analyses showing the levels of pIκBSer32/IκB and -p65Ser32/p65 (canonical NFκB), and p100/p52 (non-canonical NFκB) in vascular endothelial cells stimulated with IL-6 alone (50 ng/ml, top section) or in combination with sIL-6R (100 ng/ml, bottom section). (PDF 177 kb)
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.