

Stem cells possess the ability to divide symmetrically or asymmetrically to allow for maintenance of the stem cell pool or become committed progenitors and differentiate into various cell lineages. The unique self-renewal capabilities and pluripotency of stem cells are integral to tissue regeneration and repair (Oh et al., 2014). Multiple mechanisms including intracellular programs and extrinsic cues are reported to regulate neural stem cell (NSC) fate (Bond et al., 2015). A recent study, published in Cell Stem Cell, identified a novel mechanism whereby mitochondrial dynamics drive NSC fate (Khacho et al., 2016).
Mitochondrial dynamics describes the interplay between mitochondrial fission and fusion. The balance between these two events determines mitochondrial shape and function. Three key proteins involved in the regulation of mitochondrial fission and fusion in mammals are dynamin-related protein 1 (Drp1), mitofusins 1 and 2 (Mfn1/2) and the dynamin-like GTPase, optic atrophy 1 (Opa1) (Liesa and Shirihai, 2013). Drp1 regulates mitochondrial fission while Mfn1/2 and Opa1 regulate mitochondrial fusion. Mitochondrial fission and fusion proteins play important roles in the nervous system. For example, Mfn2 and Opa1 promote neuronal survival following injury (Jahani-Asl et al., 2007; Jahani-Asl et al., 2011), and Drp1 promotes brain tumor stem cell survival and tumorigenesis (Xie et al., 2015). However, the role of mitochondrial fission and fusion proteins in the regulation of NSC fate and neuronal regeneration has remained unexplored. This study for the first time provides important mechanistic insights on how mitochondrial fission and fusion proteins modify the transcriptomic landscape of NSCs towards self-renewal or differentiation (Khacho et al., 2016).
Khacho et al. (2016) documented a distinct change in mitochondrial morphology between uncommitted Sox2-positive NSCs and committed Tbr2-positive neural progenitor cells (NPCs). Sox2 positive NSCs showed elongated mitochondria while the Tbr2 positive NPCs displayed fragmented mitochondria. With these observations, authors asked how mitochondrial fission and fusion machineries impact stem cell fate. They showed that inducing mitochondrial fragmentation in the developing cortex via conditional genetic deletion of Mfn1/2 in uncommitted embryonic day 9.5–10.5 (E9.5–10.5) cells of the telencephalon impaired NSC self-renewal causing a long-term depletion in the pool, as typically seen in aging or neurodegeneration. They further assessed the role of fission and fusion proteins in NSC self-renewal in vitro using E11.5 embryonic cortical cells. While loss of fusion proteins, Mfn1/2 and Opa1, impaired neurosphere formation, loss of fission protein, Drp1, increased neurosphere numbers. Interestingly, neither the acute knock-down of Mfn1/2 nor Drp1 resulted in a change in cell viability or number. Together these data suggest that defective mitochondrial fusion impairs NSC self-renewal. In other studies, Khacho et al. (2016) performed in utero electroporation of shOpa1 on E13.5 pups and were able to show increased differentiation of NSCs upon loss of mitochondrial fusion.
Next, authors determined if mitochondrial dysfunction due to dysregulation of mitochondrial dynamics contributes to specification of NSC fate. Khacho et al. (2016) assessed the metabolic capabilities of E12.5 Sox2-expressing uncommitted cells following the knockdown of Mfn1/2, Drp1 or, Opa1. Total ATP levels and mitochondrial membrane potential were measured in multiple conditions to determine how expression levels of Drp1, Opa1, and Mfn1/2 affect ATP production. No differences were seen in total cellular ATP levels or in mitochondrial membrane potential upon loss of Mfn1/2, Drp1, or Opa1. This indicates that differences in energy production are not the underlying cause of impairments in NSC self-renewal in cells with defective fusion machinery. Using cells from E11.5 cortical tissue, Khacho et al. (2016) assessed levels of key metabolic proteins to determine the metabolic phenotype of NSCs vs. NPCs. Sox2 positive NSCs showed elevated levels of hexokinase-II (HXKII) and pyruvate dehydrogenase kinase 1 (PDK1), key regulators of glycolysis. Additionally, ATP measurements following oligomycin treatment (an ATP synthase inhibitor) showed little variation compared to control, suggesting Sox2 positive cells are largely glycolytic. Using the same tests, Khacho et al. (2016) were able to demonstrate that committed NPCs displayed the opposite profile, transitioning to a reliance on oxidative phosphorylation (OXPHOS). This was further supported by measuring oxygen consumption rates (OCR) in both cell types, as well as fluorescence activated cell sorting (FACS)-sorted CD15 positive uncommitted and CD15 negative committed cells. Uncommitted cells were shown to have lower basal OCR as well as ATP-linked OCR. As cells transitioned to a committed fate, basal and ATP-linked OCR increased, supporting a shift towards OXPHOS and away from glycolysis. Together, these results describe two distinct metabolic phenotypes which are independent of total cellular ATP-levels. As cells become committed, mitochondrial fragmentation is seen in conjunction with a transition away from glycolysis and toward OXPHOS, in an ATP-independent manner. Khacho et al. (2016) determined that as asymmetric division generates neural progenitors which commit to a differentiated fate, fragmented mitochondria are generated by increased mitochondrial fission resulting in elevated levels of reactive oxygen species (ROS).
ROS are known signaling molecules that have been implicated in several cellular processes including the regulation of stem cell self-renewal and differentiation (Bigarella et al., 2014). As such, Khacho et al. (2016) asked if ROS mediate the impact of mitochondrial dynamics on NSC fate. To assess the effects of ROS signaling, Khacho et al performed FACS on Sox2-GFP expressing cortical cells from E10.5 embryos. Sox2 negative cells showed increased mitochondrial superoxide (mtROS) and cytoplasmic ROS compared to Sox2 positive uncommitted cells. This difference was also seen in CD15 positive vs CD15 negative cells. Inducing mitochondrial fragmentation by knockdown of Mfn1/2 or Opa1 was able to mimic the increase in ROS as seen in committed cells without inducing any signs of oxidative damage. These effects could be rescued with the addition of antioxidants. Similarly, Knockdown of Drp1 resulted in the opposite effect, with decreased mtROS. These data show that mitochondrial dynamics regulates NSC self-renewal or differentiation via modification of ROS levels. In additional studies, authors used genetic and pharmacological methods to further investigate that complex-1-mediated generation of ROS following mitochondrial fragmentation regulates NSC fate. Addition of rotenone, a complex I inhibitor, to uncommitted cells or deletion of apoptosis inducing factor (AIF), a classical model of complex I deficiency, caused a significant decrease in neurosphere formation, confirming that the increased ROS as a result of altered mitochondrial dynamics impacts stem cell fate decisions (Khacho et al., 2017).
To identify signaling targets of ROS and mitochondrial dynamics Khacho et al. (2017) then performed RNA-sequencing to establish differentially expressed genes on E12.5 Sox2 positive cells from AIF wild type and knockout from cortical tissue as a model for complex I deficiency and increased ROS mimicking loss of fusion. This screening was validated by quantitative reverse transcription polymerase chain reaction (RT-qPCR), resulting in the identification of gene networks involved in neuronal differentiation, redox response, and the Notch pathway, with a marked upregulation of Botch, a suppressor of Notch signaling. Botch upregulation can be induced by inhibition of complex I with rotenone and can be countered by the addition of antioxidants, confirming this upregulation is mediated by ROS levels that can be dictated by mitochondrial dynamics. Finally, activity of the transcription factor NF-E2-related factor 2 (NRF2) was identified as highly upregulated in AIF knockout, Mfn1/2 knockout, shAIF, and shOpa1 cells. The authors found NRF2 knockout in E15.5 brains resulted in abnormal cortical development and a reduction in the number of cells committed to a neuronal fate. Similarly, in utero electroporation of dnNRF2 also reduces commitment to differentiation. Furthermore, NRF2 knockdown causes a loss in Botch signaling, restoring notch signaling and decreasing the expression of pro-neuronal transcription factors. In turn, the elevated levels of ROS stimulate activation of the nuclear factor erythroid 2-related factor 2 (NRF2). NRF2 then induces differentiation through upregulation of the differentiation factors Isl1, Olig2, Lhx5, Nkx2.1, Sim1, and Six3 and inhibits self-renewal through disruption of Notch signaling. Khacho et al. (2017) describe a system whereby mitochondrial dynamics regulate stem cell fate through ROS-NRF2 signaling. As uncommitted cells transition into committed neural progenitors, a distinct phenotypic change is observed, mitochondria fragmentation begins, and the cells upregulate OXPHOS activity and reduce glycolytic activity. This metabolic change results in a subsequent increase in ROS which in turn induces the activation of NRF2 to initiate a transcriptional program leading to differentiation (Figure 1).
Figure 1.
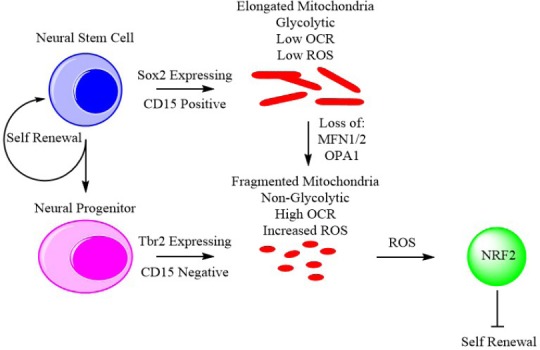
Mitochondrial dynamics regulate stem cell fate through reactive oxygen species (ROS)-mediated activation of NF-E2-related factor 2 (NRF2).
Khacho et al. (2016) observed that neural stem cells and neural progenitors display distinct alterations in mitochondrial dynamics. Neural stem cells identified by Sox2 or CD15 expression showed elongated mitochondria as well as reduced oxygen consumption rate and ATP production. Conversely, neural progenitors identified by Tbr2 expression or the absence of CD15 showed fragmented mitochondria, elevated oxygen consumption rate and heightened levels of ROS. Knockdown of mitofusins 1 and 2 (Mfn1/2) or optic atrophy 1 (Opa1) in neural stem cells resulted in mitochondrial fragmentation and induction of a metabolic phenotype similar to that of neural progenitors. Elevated ROS due to mitochondrial fragmentation then activates NRF2 signaling which represses neural stem cell self renewal, driving differentiation and reducing the stem cell pool.
Finally, having demonstrated that mitochondrial dynamics function upstream to regulate stem cell differentiation and self renewal, authors investigated the in vivo effects of disrupting mitochondrial dynamics on cognition. An inducible Mfn1/2 knockout using a NestinCreERT2 model was generated to assess neurogenesis and cognition, particularly fear conditioning and spatial learning with reversal in a Morris water maze. The authors were able to identify reduced populations of doublecortin positive neurons in conjunction with reduced recall of fear conditioning and impaired performance in recall in the Morris water maze test. This indicates reduced neurogenesis as well as distinct impairment of hippocampal memory (Yau et al., 2015). These analyses suggest that changes in mitochondrial dynamics can impact learning and memory through the regulation of stem cell fate which may have major implications in conditions where mitochondria become dysfunctional such as during aging and neurodegenerative diseases. These studies suggest, for the first time a potential mechanism for the link between mitochondrial dynamics and the cognitive symptoms of aging and neurodegenerative disease.
A related previous study investigated mitochondrial dynamics in brain tumor stem cells (Xie et al., 2015). These are a small population of brain stem cells with the ability to undergo self-renewal and long-term proliferation. Brain tumor stem cells (BTSCs) and NSCs share common characteristics in that they express neural stem cell markers and differentiate into neural lineages. In contrast to the model presented by Khacho et al. (2017), Xie et al. (2015) describe a role for Drp1 in the regulation of brain tumor stem cells whereby Drp1 activity is upregulated in comparison to differentiated cells. Upon shRNA mediated silencing of Drp1, BTSC display decreased cell growth, tumorigenicity, neurosphere formation, and OCR while showing increased survival and tumor latency. So, while Drp1 loss promotes mitochondrial elongation and induces a stem cell like phenotype and self renewal in NSC, the opposite is observed in BTSC. Together these findings provide a platform to appreciate differences in NSC and tumorigenic BTSC. Further research must be done to elucidate the precise mechanisms by which mitochondrial dynamics can differentially regulate cancer stem cells versus neural stem cells, particularly in the extremely heterogeneous tumors of glioblastoma.
While the link between mitochondrial dynamics, aging, and neurodegenerative disease has been known, the underlying molecular mechanism remained unclear. The discovery of mitochondrial dynamics as an upstream regulator of neural stem cell fate provides new promising approaches to maintain and enhance the stem cell pool in degenerative diseases.
AJ-A is a Fonds de recherche du Québec - Santé (FRQS) scholar and supported by a grant from Natural Sciences and Engineering Research Council of Canada (NSERC RGPIN-2016-06605).
Additional file: Open peer review reports 1 (101.9KB, pdf) , 2 (99.1KB, pdf) .
Footnotes
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewers: Saravana Kumar, Saveetha Dental College, India; L. Buzanska, Mossakowski Medical Research Centre, Poland.
References
- Bigarella CL, Liang R, Ghaffari S. Stem cells and the impact of ROS signaling. Development. 2014;141:4206–4218. doi: 10.1242/dev.107086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond AM, Ming GL, Song H. Adult mammalian neural stem cells and neurogenesis: five decades Later. Cell Stem Cell. 2015;17:385–395. doi: 10.1016/j.stem.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahani-Asl A, Pilon-Larose K, Xu W, MacLaurin JG, Park DS, McBride HM, Slack RS. The mitochondrial inner membrane GTPase, optic atrophy 1 (Opa1), restores mitochondrial morphology and promotes neuronal survival following excitotoxicity. J Biol Chem. 2011;286:4772–4782. doi: 10.1074/jbc.M110.167155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahani-Asl A, Cheung EC, Neuspiel M, MacLaurin JG, Fortin A, Park DS, McBride HM, Slack RS. Mitofusin 2 protects cerebellar granule neurons against injury-induced cell death. J Biol Chem. 2007;282:23788–23798. doi: 10.1074/jbc.M703812200. [DOI] [PubMed] [Google Scholar]
- Khacho M, Clark A, Svoboda DS, MacLaurin JG, Lagace DC, Park DS, Slack RS. Mitochondrial dysfunction underlies cognitive defects as a result of neural stem cell depletion and impaired neurogenesis. Hum Mol Genet. 2017;26:3327–3341. doi: 10.1093/hmg/ddx217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khacho M, Clark A, Svoboda DS, Azzi J, MacLaurin JG, Meghaizel C, Sesaki H, Lagace DC, Germain M, Harper ME, Park DS, Slack RS. Mitochondrial dynamics impacts stem cell identity and fate decisions by regulating a nuclear transcriptional program. Cell Stem Cell. 2016;19:232–247. doi: 10.1016/j.stem.2016.04.015. [DOI] [PubMed] [Google Scholar]
- Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013;17:491–506. doi: 10.1016/j.cmet.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Lee YD, Wagers AJ. Stem cell aging: mechanisms, regulators and therapeutic opportunities. Nat Med. 2014;20:870–880. doi: 10.1038/nm.3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Q, Wu Q, Horbinski CM, Flavahan WA, Yang K, Zhou W, Dombrowski SM, Huang Z, Fang X, Shi Y, Ferguson AN, Kashatus DF, Bao S, Rich JN. Mitochondrial control by DRP1 in brain tumor initiating cells. Nat Neurosci. 2015;18:501–510. doi: 10.1038/nn.3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau SY, Li A, So KF. Involvement of adult hippocampal neurogenesis in learning and forgetting. Neural Plast. 2015;2015:717958. doi: 10.1155/2015/717958. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.