

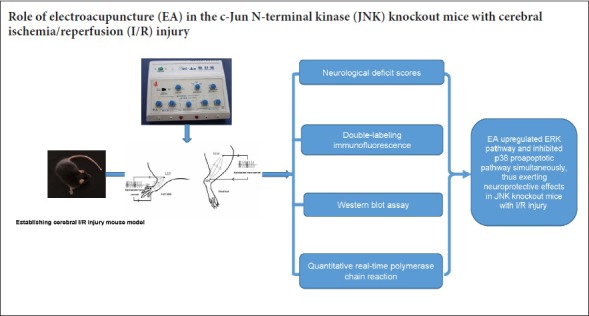
Keywords: nerve regeneration, MAPKs signaling pathway, cerebral ischemia/reperfusion, stress-activated pathway, growth factor-activated pathway, electroacupuncture, apoptosis
Abstract
Simple regulation of c-Jun N-terminal kinase (JNK) or p38 mitogen-activated protein kinase (MAPK) pathways is not enough to trigger cell apoptosis. However, activation of the stress activated pathway (JNK/p38 MAPK) together with inhibition of the growth factor activated extracellular signal-regulated kinase (ERK) pathway can promote cell apoptosis. We hypothesized that inhibition of the JNK or p38 pro-apoptotic pathway and activating the ERK pathway could be the mechanism of anti-apoptosis following cerebral ischemia/reperfusion injury. To investigate the mechanism of the protective effect of electroacupuncture on cerebral ischemia/reperfusion injury in JNK knockout mice, mouse models of cerebral ischemia/reperfusion injury were established by Longa’s method. Electroacupuncture was conducted at acupoints Chize (LU5), Hegu (LI4), Sanyinjiao (SP6) and Zusanli (ST36) 1.5 hours after ischemia/reperfusion injury for 20 minutes, once a day. The neurological function was evaluated using neurological deficit scores. The expression of phospho-extracellular signal-regulated kinase (p-ERK) and phospho-p38 (p-p38) in JNK knockout mice was detected using double-labeling immunofluorescence and western blot assay. The mRNA expression of ERK and p38 was measured by quantitative real-time polymerase chain reaction. Electroacupuncture improved neurological function, increased the immunoreactivity and relative expression of p-ERK and reduced that of p-p38 in the cerebral cortex and hippocampus on the injured side. Electroacupuncture increased mRNA expression of ERK, but decreased that of p38 in the cerebral cortex and hippocampus on the injured side. In conclusion, electroacupuncture upregulated the protective ERK pathway and inhibited the pro-apoptotic p38 pathway, thereby exerting a neuroprotective effect and improving the neurological function in JNK knockout mice.
Introduction
Cerebral ischemia leads to inflammation, oxidative damage and programmed cell death that aggravate brain tissue damage and seriously influence the daily life of stroke patients (Dirnagl, 2012). The mechanism of cerebral ischemia/reperfusion (I/R) injury is mainly associated with inflammation and apoptosis (Broughton et al., 2009; Eltzschig and Eckle, 2011). Reducing apoptosis helps the recovery of brain functions (Bhuiyan and Fukunaga, 2009).
Mitogen-activated protein kinases (MAPKs) play a role in neural cell growth and proliferation (Irving and Bamford, 2002). The MAPKs signaling pathway is widely expressed in the nervous system and regulates the repair or apoptosis of nerve cells after cerebral I/R injury (Abe et al., 2000; Shaw and Kirshenbaum, 2006; Kovalska et al., 2012). The main three subfamilies of MAPKs; extracellular signal-regulated kinases (ERKs), c-Jun N-terminal kinases (JNKs), and p38 kinases, are involved in regulating dysfunction and apoptosis of neuronal cells after cerebral I/R injury (Yang et al., 2003; Hu et al., 2012). The ERK signaling pathway starts the self-repair process, which plays an important role in recovering from cerebral I/R injury (Shioda et al., 2009). p38 and JNK serve as mediators of cellular stresses, which are involved in inflammation and neuronal cell apoptosis (Yoshida et al., 2010; Kyriakis and Avruch, 2012). A previous study also proved that cell survival or apoptosis was dependent on the dynamic balance of the growth factor-activated (ERK pathway) and stress-activated (JNK/P38 signaling pathway) pathways (Junttila et al., 2008). Broad “crosstalk” between several signaling pathways has been reported and could lead to mutual synergy or inhibition between pathways (Junttila et al., 2008). Another study suggested that simple JNK or p38 MAPK was not enough to trigger the process of apoptosis. Only the concurrent activation of stress-activated pathway (JNK/p38 MAPK) coupled with the restraint of the growth factor-activated ERK pathway could promote cell apoptosis (Xia et al., 1995). We propose that inhibiting the JNK or p38 pro-apoptotic pathway and simultaneously activating the ERK protective pathway could result in an anti-apoptotic mechanism after cerebral I/R injury. Therefore, we knocked out one of the stress-activated pathways (JNK) and investigated the effect of electroacupuncture (EA) on ERK and p38 kinase pathways in the JNK knockout mice after I/R injury.
EA therapy is advantageous in improving neurological deficit symptoms and rehabilitating limb motor function after cerebral I/R injury (Chavez et al., 2017). “Chize (LU5), Hegu (LI4)”, “Sanyinjiao (SP6) and Zusanli (ST36)” served as common acupoints for the clinical treatment of stroke (Xiong et al., 2008; Yang et al., 2015). A previous study found that EA could reduce the area of cerebral infarction and neuronal apoptosis in JNK knockout mice with cerebral I/R injury (Feng, 2015). However, the role of EA in the anti-apoptotic mechanism of JNK knockout mice with cerebral I/R injury needs further investigation. This study aims to elucidate the possible mechanisms of EA action in JNK knockout mice with cerebral I/R injury.
Materials and Methods
Animals
A total of 54 JNK knockout mice (half males and half females), weighing 20–25 g, were purchased from Cyagen Biosciences Co. Ltd. (Guangzhou, China; license number: SCXK (Yue) 2013-0002). The experimental procedure followed the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 8023, revised 1986), and was approved by the Animal Ethics and Welfare Committee of Southern Medical University of China (approval No. 2013-032). The mice were fed standard rodent chow, and allowed free access to water. The mice were acclimatized to the animal house that was maintained at a room temperature between 20°C and 22°C and a relative humidity of 65–70%. The JNK knockout mice were randomized into sham group (sham control), I/R group (model), and EA group (I/R + EA) (n = 18 per group).
Establishing cerebral I/R injury mouse model
The I/R injury model was established by middle cerebral artery occlusion, as described previously (Longa et al., 1989). Briefly, after anesthetizing the mice with 10% chloral hydrate (300 mg/kg) by intraperitoneal injection, the left common carotid artery, left internal carotid artery, external carotid artery, and vagus nerve were carefully exposed via a midline incision under a surgical microscope. Approximately 11 ± 0.5 mm of a nylon suture was introduced into the internal carotid artery to occlude the middle cerebral artery until a slight resistance was observed during insertion. Reperfusion was achieved by withdrawing the thread slowly to restore the blood supply to the ischemic area after 30 minutes of occlusion. The mice in the sham group underwent the same surgical procedures as described earlier but arterial occlusion was not performed.
Electroacupuncture intervention
The mice in the EA group underwent EA stimulation 1.5 hours after I/R injury (then once a day until sacrifice). Stainless steel acupuncture needles (diameter: 0.16 × 13 mm2; Suzhou Universal Acupuncture Medical Devices Co., Ltd., Suzhou, China) were inserted 2–3 mm into LU5, LI4, ST36 and SP6 acupoints of the paralyzed limb. They were located according to the Experimental Acupuncture Science edited by Li (2007). The location of the selected acupoints were as follows: LU5, in the depression of outer end of the transverse cubital crease; LI4, between 1st metacarpal bone and 2nd metacarpal bone; ST36, 3.5 mm below the fibular head at the outer lateral posterior knee; SP6, 5 mm above the tip of the inner ankle of the posterior limb. The acupoints were stimulated for 20 minutes with a dilatational wave of frequency 5/10 Hz and intensity 2 mA using an EA instrument (Model KWD-808I, Suzhou Universal Acupuncture Medical Devices Co., Ltd.).
Neurological deficit scores
The neurological deficit scores were evaluated in six mice at 2 hours, 1 and 3 days after the operation using a 5-point neurological scale according to a previous method (Longa et al., 1989). The neurological deficit scores were defined as follows: score 0, no observable deficit; score 1, failure to fully extend the right forepaw; score 2, circling to the contralateral side; score 3, falling to the contralateral side; and score 4, loss of walking or consciousness. The mice with scores 0 or 4 were excluded.
Tissue sampling
After terminal anesthesia, rapid perfusion with 0.9% normal saline (NaCl) and 4% paraformaldehyde in phosphate buffered saline for 3–5 minutes eliminated the influence of blood factors. The whole brain were dissected out of the cranial cavity quickly and washed with ice-cold saline. The separated brain tissue was frozen in liquid nitrogen and stored at −80°C pending subsequent double-labeling immunofluorescence, quantitative real-time polymerase chain reaction (PCR) and western blot assay.
Double-labeling immunofluorescence
The expression of phospho-ERK (p-ERK) and phospho-p38 (p-p38) on the injured side of the cortex and hippocampus at 2 hours, 1 day and 3 days (Shipp, 2007) were measured using the double-labeling immunofluorescence in six mice at each time point. Frozen sections were boiled in EDTA antigen repair solution (pH 8.0) (Cell Signaling Technology, Danvers, MA, USA) for antigen retrieval and blocked with 3% bovine serum albumin for 30 minutes at room temperature. For double-labeling immunofluorescence, the sections were incubated with a primary antibody against p-p38 monoclonal antibody (anti-rabbit, 1:100; Cell Signaling Technology, Boston, USA) diluted in phosphate-buffered saline at 4°C overnight. After washing, the sections were incubated with the corresponding secondary antibody (cy3-goat anti-rabbit, 1:300; Google Biological Technology Co., Ltd., Wuhan, China). Next, the sections were incubated with an anti-p-ERK primary monoclonal antibody (anti-mouse, 1:100; Abcam, Cambridge, UK) and a secondary antibody (488 goat anti-mouse, 1:400; Google Biological Technology Co., Ltd.). The sections were observed and imaged under a laser scanning confocal microscope (LSM510META; Zeiss, Jena, Germany). Positive integrated optical densities (IODs) of p-ERK or p-p38 were evaluated using the Image-Pro Plus 6.0 software (Media Cybernetics, Bethesda, MD, USA). IOD values = Positive area in immunofluorescence image × optical density. A higher IOD value corresponded to a higher expression of p-ERK, or p-p38.
Quantitative real-time PCR
Quantitative real-time PCR was performed to measure the mRNA expression of ERK and p38 in the injured brain in three samples in each group. The total RNA was extracted from the left (injured) side of brain at 2 hours, 1 day and 3 days using the TRIzol reagent (Life Technologies, Carlsbad, CA, USA). Afterwards, 2.0 µg of total RNA was reverse-transcribed to cDNA using a Toyobo First Strand cDNA Synthesis Kit (Qiagen, Hilden, Germany). Quantitative real-time PCR was performed using a THUNDERBIRD SYBR q-PCRMix (Toyobo, Osaka, Japan). The PCR primer sequence was synthesized by the Invitrogen Company (Shanghai, China) as follows: β-actin: forward 5′-GTG ACG TTG ACA TCC GTA AAG A-3′, reverse 5′-GTA ACA GTC CGC CTA GAA GCA C-3′; ERK: forward 5′-TGG AGA CGG ACC TTT ACA AGC-3′, reverse 5′-CAA GTG GTG TTC AGC AGG AGG-3′; and p38: forward 5′-GAC CTA CTG GAG AAG ATG CTC GTT-3′, reverse 5′-TTT CAA AGG ACT GGT CAT AAG GGT-3′. cDNA was amplified under the following cyclic conditions: 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for 60 seconds. The mRNA expression of ERK and p38 was normalized to the expression level of β-actin and calculated using the following equation: fold change = 2–ΔΔCT.
Western blot assay
Proteins from the brain tissue of three samples in each group, 3 days after I/R, were extracted and homogenized in a radioimmunoprecipitation assay lysis buffer. The proteins were electrophoresed using 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto polyvinylidene fluoride membranes (Millipore, Boston, USA). The membranes were blocked with 5% nonfat dry milk for 1 hour and incubated with rabbit anti-ERK polyclonal antibody (1:1000; Affinity Biosciences, OH, USA), p-ERK polyclonal antibody (1:1000; Affinity Biosciences), p38 monoclonal antibody (Abcam), p-p38 monoclonal antibody (1:1000; Cell Signaling Technology, Boston, MA, USA), and mouse anti-glyceraldehyde-3-phosphate dehydrogenase monoclonal antibody (1:1000; Google Biological Technology Co., Ltd.) at 4°C overnight. The membranes were incubated with a corresponding secondary antibody (1:3000; goat anti-rabbit/mouse IgG; Google Biological Technology Co., Ltd.) in Tris-buffered saline and Tween 20 for 30 minutes. The blots were developed using an enhanced chemiluminescence, and the intensity of the bands was measured using the AlphaEaseFC analyzer software (AlphaInnotech, San Leandro, CA, USA). The optical density value ratio of the target band to the internal reference served as the relative expression of the target protein.
Statistical analysis
All data, presented as the mean ± SD, were analyzed using SPSS 21.0 software (SPSS, Chicago, IL, USA). The statistical analysis of the data was conducted using the one-way analysis of variance via least significant difference post hoc comparison when comparing more than two groups. If the tested data were not normally distributed, the nonparametric test was adopted. A P value less than 0.05 was considered statistically significant.
Results
Effects of EA on the neurological function of JNK knockout mice with I/R injury
The neurological deficit scores were evaluated in the sham, I/R, and EA groups. The mice in the I/R and EA groups presented with obvious neurological deficits by two hours after I/R, while the JNK knockout mice in the sham group did not manifest any signs of cerebral damage. Figure 1 shows that there were no significant differences between the neurological deficit scores of the I/R and EA groups at 2 hours and 1 day after I/R injury (P > 0.05). However, the neurological deficit scores of the EA group were significantly lower than those of the I/R group 3 days after I/R injury (P < 0.05).
Figure 1.
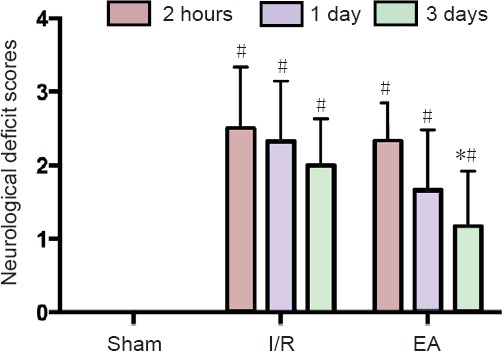
Effects of Electroacupuncture on the neurological deficit of the JNK knockout mice with I/R injury.
Data are presented as the mean ± SD (n = 6); one-way analysis of variance followed by least significant difference post hoc test was used. *P < 0.05, vs. I/R group; #P < 0.05, vs. sham group. EA: Electroacupuncture; I/R: ischemia/reperfusion; JNK: c-Jun N-terminal kinase.
Effects of EA on the p-ERK and p-p38 immunoreactivities in the hippocampus and cortex of the JNK knockout mice with I/R injury
The expression of p-ERK and p-p38 signaling pathways in the JNK knockout mice with I/R injury was detected using double-labeling immunofluorescence (Figure 2C, F). Figure 2A, B show that both p-ERK and p-p38 immunoreactivities in the hippocampus increased to a peak at 1 day after I/R injury, and stabilized at 3 days. EA increased the p-ERK immunoreactivity in the JNK knockout mice with I/R injury at 1 day and 3 days (P < 0.05). However, the p-p38 immunoreactivity was lower in the EA group than in the I/R group at 1 day and 3 days (P < 0.05). Similar results were found in the cortex (Figure 2D, E).
Figure 2.

The p-ERK and p-p38 immunoreactivity in the hippocampus and cortex of JNK knockout mice with cerebral I/R injury.
(A, D) The p-ERK immunoreactivity in the hippocampus and cortex of JNK knockout mice. (B, E) The p-p38 immunoreactivity in the hippocampus and cortex of JNK knockout mice. Data are representative of six individual mice in each group. Data are expressed as the mean ± SD (n = 6); one-way analysis of variance followed by least significant difference post hoc test. *P < 0.05, vs. I/R group; #P < 0.05, vs. sham group. (C) Expression of p-ERK and p-p38 in the hippocampus of JNK knockout mice at 3 days under a laser scanning confocal microscope (scale bars: 9 μm). (F) Expression of p-ERK and p-p38 in the cortex of JNK knockout mice at 3 days under a laser scanning confocal microscope (original magnification, 400×). Arrows indicate p-ERK and p-p38 immunoreactivity. EA: Electroacupuncture; I/R: ischemia/reperfusion; JNK: c-Jun N-terminal kinase; p-ERK: phospho-extracellular signal-regulated kinase; DAPI: 4′,6-diamidino-2-phenylindole; IOD: integrated optical density.
Effects of EA on the mRNA expression levels of ERK and p38 in the JNK knockout mice with I/R injury
To investigate further the mechanism underlying the protective effect of EA treatment on the expression of ERK and p38, the mRNA expressions of ERK and p38 were evaluated in the JNK knockout mice using quantitative real-time PCR. Figure 3 shows that the mRNA expressions of ERK and p38 were upregulated, peaking at 1 day, then decreasing by 3 days. However, the mRNA expressions of ERK and p38 were not significantly different between the I/R and EA groups at the 2-hour time point (both P > 0.05). After 1 and 3 days of I/R injury, the mRNA expression of ERK was enhanced and that of p38 diminished in the EA group compared with the I/R group (all P < 0.05; Figure 3).
Figure 3.

The mRNA expression of ERK and p38 in the injured brain tissues of JNK knockout mice with cerebral I/R injury.
(A, B) The mRNA expression of ERK (A) and p38 (B) in each group at different time points was assessed by quantitative real-time polymerase chain reaction. Data are presented as the mean ± SD (n = 3); one-way analysis of variance followed by least significant difference post hoc test was used. *P < 0.05, vs. I/R group; #P < 0.05, vs. sham group. EA: Electroacupuncture; I/R: ischemia/reperfusion; ERK: extracellular signal-regulated kinase.
Effects of EA on the protein expression of p-ERK and p-P38 in the JNK knockout mice with I/R injury
Western blot assay results showed the target protein expression of p-ERK and p-p38 at 3 days post I/R. Compared with the I/R group, the protein expression of p-ERK was upregulated and that of p-p38 was downregulated in the EA group (both P < 0.05), which was consistent with the finding of double-labeling immunofluorescence. However, the protein expression of p-ERK in the sham group was similar to that in the I/R group, and the protein expression of p-p38 in the EA group was not significantly different from that in the sham group (P > 0.05; Figure 4).
Figure 4.

Protein expression of p-ERK and p-P38 in the JNK knockout mice with I/R injury.
(A) Relative expression of p-ERK in each group at 3 days. (B) Relative expression p-p38 in each group at 3 days. Data are presented as the mean ± SD (n = 3); one-way analysis of variance followed by least significant difference post hoc test was used. *P < 0.05, vs. I/R group; #P < 0.05, vs. sham group. (C) Bands of p-ERK and p-p38 assessed by western blot assay. EA: Electroacupuncture; I/R: ischemia/reperfusion; JNK: c-Jun N-terminal kinase; ERK: extracellular signal-regulated kinase; p-ERK: phospho-extracellular signal-regulated kinase; GAPDH: glyceraldehyde-3-phosphate dehydrogenase.
Discussion
Cerebral ischemia involves a series of pathological changes, such as inflammation, oxygen deficit, brain edema and nerve cell apoptosis, leading to brain injury and reduced limb motor function. Reperfusion causes further brain injury and apoptosis. The neurological deficit symptoms we observed indicated neurological dysfunction and brain damage following I/R injury (Vaibhav et al., 2013; Yaidikar and Thakur, 2015). We found that EA could alleviate the neurological deficits in JNK knockout mice with cerebral I/R injury, especially after 3 days. Other studies have also indicated that EA could improve the limb locomotor function and provide some neuroprotective effects against I/R injury (Zhang et al., 2014b; Lu et al., 2015; Liu et al., 2016). EA has been shown to reduce cerebral infarct areas and inhibit apoptosis of nerve cells in the JNK knockout mice with I/R injury (Feng, 2015).
The possible mechanisms underlying the neuroprotective effects of EA against I/R injury may be associated with the regulation of MAPK family. The MAPK family plays an important role in I/R injury; it regulates neural cell death and survival via signaling pathways related to cell apoptosis (Nozaki et al., 2001). The MAPK family mainly consists of ERK, p38, and JNK signaling pathways. Although the JNK signaling pathway had been knocked out in our study, double-labeling immunofluorescence showed that the expression of p-ERK was still upregulated and the expression of p-p38 was downregulated in the EA group compared with the I/R group at the same time points. These results indicate that EA might inhibit neuronal apoptosis and improve neurological deficit symptoms by activating the expression of p-ERK and inhibiting the expression of p-p38. Our western blot assay and quantitative PCR data further verified the double-labeling immunofluorescence findings. The MAPK/ERK pathway is mainly involved in regulating cell proliferation, migration, differentiation, and reducing apoptosis (Sun et al., 2015), whereas the activation of p38 promotes neuronal apoptosis (Kim and Choi, 2015). The ERK activated by phosphorylation can inhibit caspase-9 processing and activation, blocking the caspase cascade and inhibit apoptosis (Allan et al., 2003). A related study has indicated that the inhibition of ERK activation by ERK inhibitors in vitro and in vivo can weaken the expression of cleaved caspase-3 to protect neural cells from brain injury-induced apoptosis (Zhao et al., 2012). Previous studies have also found that EA can inhibit neural cell apoptosis during I/R injury by activating the ERK signaling pathway (Cheng et al., 2014b; Wu et al., 2015), which is consistent with the present findings. However, p38 mediates inflammation and promotes apoptosis.
Studies have shown that the activation of p38 induces further downstream transcription factors and activates caspase-3, caspase-6, and caspase-7, thereby activating the genes associated with apoptosis, resulting in nerve cell apoptosis (Morel et al., 2005; Chan et al., 2009). Inhibition of p38 phosphorylation can promote the phosphorylation of ERK1/2, alleviate brain injury through restraining autophagic cell death and rescue degenerative neural cells in ischemic penumbra (Zhang et al., 2014a). Cheng et al. (2014a) reported that EA at acupoints can inhibit the expression of p38, thereby reducing the oxidative/nitrative stress and downregulating the tumor necrosis factor-α/TRADD/FADD/cleaved caspase-8/cleaved caspase-3 apoptotic pathway to attenuate neural cell apoptosis.
The findings of previous studies and the present study suggest that EA stimulation exerts an anti-apoptotic effect against I/R injury, which involves upregulating the expression of p-ERK and inhibiting the expression of p-p38 at the same time. The p-ERK signaling pathway was significantly upregulated and the p-p38 signaling pathway was downregulated in the same brain section from the EA group compared with a similar section from the I/R group, and at both 1 and 3 days. This indicates that EA could synchronously regulate ERK and p38 signaling pathways in the JNK knockout mice with I/R injury. It confirms the previous assumption that EA treatment exerts a neuroprotective effect against cerebral I/R injury, which may not simply rely on one of the MAPK signaling pathways, but requires simultaneous regulation of several MAPK signaling pathways. EA may regulate cerebral I/R injury in JNK knockout mice by activating the neuroprotective pathway (ERK) and inhibiting the p38 pro-apoptotic pathway (MAPK signaling pathway). This suggests some “crosstalk” exists between ERK and p38. Other research has indicated that inhibiting the expression of p38 might stimulate the phosphorylation of ERK signaling pathway (Lee et al., 2002; Li et al., 2003), and that the activation of p38/JNK might shut off the ERK pathway-mediated survival signaling and induce apoptosis (Junttila et al., 2008). Therefore, EA may upregulate the expression of p-ERK and downregulate the expression of p-p38 at the same time to promote the crosstalk between the two signaling pathways that can exert a better neuroprotective effect against cerebral I/R injury.
Early EA intervention after cerebral ischemia can increase cerebral blood flow, reduce the extent of cerebral I/R injury, and expand the therapeutic time window (Ren et al., 2010; Kim et al., 2013). This indicated that the best time for acupuncture treatment is within 3 hours of ischemia (Wang et al., 2012). We chose 1.5 hours after cerebral I/R injury for EA treatment. The double-labeling immunofluorescence and quantitative real-time PCR results indicated that the expression of ERK was upregulated and the p38 expression was suppressed at 1 and 3 days in the EA group compared with the I/R group. However, no significant difference was found between the EA and I/R groups at 2 hours. Nerve cell apoptosis usually reaches a peak at 24–48 hours after cerebral I/R injury (Zhang and Li, 2012). The EA-regulated ERK and p38 signaling pathways in JNK knockout mice with cerebral I/R injury were obvious 1 day after cerebral I/R injury, which was the peak point of apoptosis. The aftereffects of EA and self-repair mechanisms during the increase in apoptosis would promote the ERK protective pathway and inhibit the p38 pro-apoptotic pathway simultaneously to exert a neuroprotective effect. Therefore, EA would not have an immediate effect after I/R injury in the JNK knockout mice. Rather, EA can exert a neuroprotective effect 1 day after I/R injury compared with the I/R group. A significant difference was found between the EA and I/R groups after 3 days, indicating that EA intervention once a day continued to exert an effect and regulate the expression of ERK and p38 in JNK knockout mice to alleviate cerebral I/R injury.
However, this study still has limitations. First, the I/R injury model was established by middle cerebral artery occlusion, which may have a high death rate and model establishment takes a long time. A better model for I/R injury established by an optimized method is needed in future research. Second, we used double-labeling immunofluorescence to detect the expression of p-ERK and p-p38. The crosstalk between antibodies may affect the expression of the target pathway to some extent. Therefore, a comprehensive method with less interference is needed to measure the expression of p-ERK and p-p38 simultaneously in a future study.
EA at acupoints “LU5,” “LI4,” “ST36,” and “SP6” could alleviate neurological deficit symptoms and reduce apoptosis in the JNK knockout mice with I/R injury. Enhancing the ERK protective pathway and inhibiting the p38 pro-apoptotic pathway simultaneously may be one of the key mechanisms of EA treatment for cerebral I/R injury. EA treatment may be most effective against apoptosis one day after I/R. Three days afterwards maybe a good time point to measure the neuroprotective effect of accumulative acupuncture.
Acknowledgments
The authors are very grateful to all staff from Experimental Animal Center of Southern Medical University of China and thank all the researchers involved in animal experiments.
Footnotes
Conflicts of interest: We declare that we have no conflict of interest.
Financial support: This study was supported by the National Natural Science Foundation of China, No. 81173355. The funding body played no role in the study design, in the collection, analysis and interpretation of data, in the writing of the report, or in the decision to submit the article for publication.
Institutional review board statement: All experimental procedures were approved by the Animal Ethics and Welfare Committee of Southern Medical University of China (approval No. 2013-032). The experimental procedure followed the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 8023, revised 1985).
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewers: Clarissa Cavarsan, Federal University of Paraná, Brazil; Lijie Zhai, University of Illinois at Chicago, USA.
Funding: This study was supported by the National Natural Science Foundation of China, No. 81173355.
(Copyedited by Yu J, Li CH, Qiu Y, Song LP, Zhao M)
References
- Abe J, Baines CP, Berk BC. Role of mitogen-activated protein kinases in ischemia and reperfusion injury: the good and the bad. Circ Res. 2000;86:607–609. doi: 10.1161/01.res.86.6.607. [DOI] [PubMed] [Google Scholar]
- Allan LA, Morrice N, Brady S, Magee G, Pathak S, Clarke PR. Inhibition of caspase-9 through phosphorylation at Thr 125 by ERK MAPK. Nat Cell Biol. 2003;5:647–654. doi: 10.1038/ncb1005. [DOI] [PubMed] [Google Scholar]
- Bhuiyan MS, Fukunaga K. Mitochondrial serine protease HtrA2/Omi as a potential therapeutic target. Curr Drug Targets. 2009;10:372–383. doi: 10.2174/138945009787846399. [DOI] [PubMed] [Google Scholar]
- Broughton BR, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke. 2009;40:e331–339. doi: 10.1161/STROKEAHA.108.531632. [DOI] [PubMed] [Google Scholar]
- Chan PS, Koon HK, Wu ZG, Wong RN, Lung ML, Chang CK, Mak NK. Role of p38 MAPKs in hypericin photodynamic therapy-induced apoptosis of nasopharyngeal carcinoma cells. Photochem Photobiol. 2009;85:1207–1217. doi: 10.1111/j.1751-1097.2009.00572.x. [DOI] [PubMed] [Google Scholar]
- Chavez LM, Huang SS, MacDonald I, Lin JG, Lee YC, Chen YH. Mechanisms of acupuncture therapy in ischemic stroke rehabilitation: a literature review of basic studies. Int J Mol Sci. 2017;18:E2270. doi: 10.3390/ijms18112270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CY, Lin JG, Tang NY, Kao ST, Hsieh CL. Electroacupuncture-like stimulation at the Baihui (GV20) and Dazhui (GV14) acupoints protects rats against subacute-phase cerebral ischemia-reperfusion injuries by reducing S100B-mediated neurotoxicity. PLoS One. 2014a;9:e91426. doi: 10.1371/journal.pone.0091426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CY, Lin JG, Su SY, Tang NY, Kao ST, Hsieh CL. Electroacupuncture-like stimulation at Baihui and Dazhui acupoints exerts neuroprotective effects through activation of the brain-derived neurotrophic factor-mediated MEK1/2/ERK1/2/p90RSK/bad signaling pathway in mild transient focal cerebral ischemia in rats. BMC Complement Altern Med. 2014b;14:92. doi: 10.1186/1472-6882-14-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U. Pathobiology of injury after stroke: the neurovascular unit and beyond. Ann N Y Acad Sci. 2012;1268:21–25. doi: 10.1111/j.1749-6632.2012.06691.x. [DOI] [PubMed] [Google Scholar]
- Eltzschig HK, Eckle T. Ischemia and reperfusion--from mechanism to translation. Nat Med. 2011;17:1391–1401. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng YH. Changsha: Hunan University of Chinese Medicine; 2015. The experiment research of electro-acupuncture for the apoptosis and the influence of MAPK signaling pathway by JNK gene knock-out mice on cerebral ischemia re-perfusion injury. [Google Scholar]
- Hu SQ, Ye JS, Zong YY, Sun CC, Liu DH, Wu YP, Song T, Zhang GY. S-nitrosylation of mixed lineage kinase 3 contributes to its activation after cerebral ischemia. J Biol Chem. 2012;287:2364–2377. doi: 10.1074/jbc.M111.227124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irving EA, Bamford M. Role of mitogen- and stress-activated kinases in ischemic injury. J Cereb Blood Flow Metab. 2002;22:631–647. doi: 10.1097/00004647-200206000-00001. [DOI] [PubMed] [Google Scholar]
- Junttila MR, Li SP, Westermarck J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J. 2008;22:954–965. doi: 10.1096/fj.06-7859rev. [DOI] [PubMed] [Google Scholar]
- Kim EK, Choi EJ. Compromised MAPK signaling in human diseases: an update. Arch Toxicol. 2015;89:867–882. doi: 10.1007/s00204-015-1472-2. [DOI] [PubMed] [Google Scholar]
- Kim JH, Choi KH, Jang YJ, Bae SS, Shin BC, Choi BT, Shin HK. Electroacupuncture acutely improves cerebral blood flow and attenuates moderate ischemic injury via an endothelial mechanism in mice. PLoS One. 2013;8:e56736. doi: 10.1371/journal.pone.0056736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalska M, Kovalska L, Pavlikova M, Janickova M, Mikuskova K, Adamkov M, Kaplan P, Tatarkova Z, Lehotsky J. Intracellular signaling MAPK pathway after cerebral ischemia-reperfusion injury. Neurochem Res. 2012;37:1568–1577. doi: 10.1007/s11064-012-0752-y. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev. 2012;92:689–737. doi: 10.1152/physrev.00028.2011. [DOI] [PubMed] [Google Scholar]
- Lee J, Hong F, Kwon S, Kim SS, Kim DO, Kang HS, Lee SJ, Ha J, Kim SS. Activation of p38 MAPK induces cell cycle arrest via inhibition of Raf/ERK pathway during muscle differentiation. Biochem Biophys Res Commun. 2002;298:765–771. doi: 10.1016/s0006-291x(02)02562-7. [DOI] [PubMed] [Google Scholar]
- Li SP, Junttila MR, Han J, Kahari VM, Westermarck J. p38 Mitogen-activated protein kinase pathway suppresses cell survival by inducing dephosphorylation of mitogen-activated protein/extracellular signal-regulated kinase kinase1, 2. Cancer Res. 2003;63:3473–3477. [PubMed] [Google Scholar]
- Li ZR. Experimental Acupuncture Science. Beijing: China Press of Traditional Chinese Medicine; 2007. [Google Scholar]
- Liu W, Wang X, Zheng Y, Shang G, Huang J, Tao J, Chen L. Electroacupuncture inhibits inflammatory injury by targeting the miR-9-mediated NF-kappaB signaling pathway following ischemic stroke. Mol Med Rep. 2016;13:1618–1626. doi: 10.3892/mmr.2015.4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- Lu Y, Zhao H, Wang Y, Han B, Wang T, Zhao H, Cui K, Wang S. Electro-acupuncture up-regulates astrocytic MCT1 expression to improve neurological deficit in middle cerebral artery occlusion rats. Life Sci. 2015;134:68–72. doi: 10.1016/j.lfs.2015.05.014. [DOI] [PubMed] [Google Scholar]
- Morel C, Ibarz G, Oiry C, Carnazzi E, Berge G, Gagne D, Galleyrand JC, Martinez J. Cross-interactions of two p38 mitogen-activated protein (MAP) kinase inhibitors and two cholecystokinin (CCK) receptor antagonists with the CCK1 receptor and p38 MAP kinase. J Biol Chem. 2005;280:21384–21393. doi: 10.1074/jbc.M408851200. [DOI] [PubMed] [Google Scholar]
- Nozaki K, Nishimura M, Hashimoto N. Mitogen-activated protein kinases and cerebral ischemia. Mol Neurobiol. 2001;23:1–19. doi: 10.1385/MN:23:1:01. [DOI] [PubMed] [Google Scholar]
- Ren L, Wang YK, Fang YN, Zhang AW, Li XL. Effect of electroacupuncture therapy on the expression of Na(v)1.1 and Na(v)1.6 in rat after acute cerebral ischemia. Neurol Res. 2010;32:1110–1116. doi: 10.1179/016164110X12700393823453. [DOI] [PubMed] [Google Scholar]
- Shaw J, Kirshenbaum LA. Prime time for JNK-mediated Akt reactivation in hypoxia-reoxygenation. Circ Res. 2006;98:7–9. doi: 10.1161/01.RES.0000200397.22663.b6. [DOI] [PubMed] [Google Scholar]
- Shioda N, Han F, Fukunaga K. Role of Akt and ERK signaling in the neurogenesis following brain ischemia. Int Rev Neurobiol. 2009;85:375–387. doi: 10.1016/S0074-7742(09)85026-5. [DOI] [PubMed] [Google Scholar]
- Shipp S. Structure and function of the cerebral cortex. Curr Biol. 2007;17:R443–449. doi: 10.1016/j.cub.2007.03.044. [DOI] [PubMed] [Google Scholar]
- Sun Y, Liu WZ, Liu T, Feng X, Yang N, Zhou HF. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J Recept Signal Transduct Res. 2015;35:600–604. doi: 10.3109/10799893.2015.1030412. [DOI] [PubMed] [Google Scholar]
- Vaibhav K, Shrivastava P, Khan A, Javed H, Tabassum R, Ahmed ME, Khan MB, Moshahid Khan M, Islam F, Ahmad S, Siddiqui MS, Safhi MM, Islam F. Azadirachta indica mitigates behavioral impairments, oxidative damage, histological alterations and apoptosis in focal cerebral ischemia-reperfusion model of rats. Neurol Sci. 2013;34:1321–1330. doi: 10.1007/s10072-012-1238-z. [DOI] [PubMed] [Google Scholar]
- Wang ZK, Ni GX, Liu K, Xiao ZX, Yang BW, Wang J, Wang S. Research on the changes of IL-1 receptor and TNF-alpha receptor in rats with cerebral ischemia reperfusion and the chronergy of acupuncture intervention. Zhongguo Zhen Jiu. 2012;32:1012–1018. [PubMed] [Google Scholar]
- Wu C, Wang J, Li C, Zhou G, Xu X, Zhang X, Lan X. Effect of electroacupuncture on cell apoptosis and ERK signal pathway in the hippocampus of adult rats with cerebral ischemia-reperfusion. Evid Based Complement Alternat Med. 2015;2015:414965. doi: 10.1155/2015/414965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- Xiong J, Ning LN, Bian JL, Li J, Xu JF, Zhang ZL, Guo JK, Li YD, Shi XM. Clinical effects of Xingnao Kaiqiao acupuncture on neurological impairment following cerebral infarction. Neural Regen Res. 2008;3:272–275. [Google Scholar]
- Yaidikar L, Thakur S. Punicalagin attenuated cerebral ischemia-reperfusion insult via inhibition of proinflammatory cytokines, up-regulation of Bcl-2, down-regulation of Bax, and caspase-3. Mol Cell Biochem. 2015;402:141–148. doi: 10.1007/s11010-014-2321-y. [DOI] [PubMed] [Google Scholar]
- Yang SH, Sharrocks AD, Whitmarsh AJ. Transcriptional regulation by the MAP kinase signaling cascades. Gene. 2003;320:3–21. doi: 10.1016/s0378-1119(03)00816-3. [DOI] [PubMed] [Google Scholar]
- Yang ZX, Xie JH, Liu YP, Miao GX, Wang YH, Wu SM, Li Y. Systematic review of long-term Xingnao Kaiqiao needling effcacy in ischemic stroke treatment. Neural Regen Res. 2015;10:583–588. doi: 10.4103/1673-5374.155431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Metoki N, Ishikawa A, Imaizumi T, Matsumiya T, Tanji K, Ota K, Ohyama C, Satoh K. Edaravone improves the expression of nerve growth factor in human astrocytes subjected to hypoxia/reoxygenation. Neurosci Res. 2010;66:284–289. doi: 10.1016/j.neures.2009.11.011. [DOI] [PubMed] [Google Scholar]
- Zhang L, Niu W, He Z, Zhang Q, Wu Y, Jiang C, Tang C, Hu Y, Jia J. Autophagy suppression by exercise pretreatment and p38 inhibition is neuroprotective in cerebral ischemia. Brain Res. 2014a;1587:127–132. doi: 10.1016/j.brainres.2014.08.067. [DOI] [PubMed] [Google Scholar]
- Zhang YM, Xu H, Sun H, Chen SH, Wang FM. Electroacupuncture treatment improves neurological function associated with regulation of tight junction proteins in rats with cerebral ischemia reperfusion injury. Evid Based Complement Alternat Med. 2014b;2014:989340. doi: 10.1155/2014/989340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZB, Li ZG. Cathepsin B and phospo-JNK in relation to ongoing apoptosis after transient focal cerebral ischemia in the rat. Neurochem Res. 2012;37:948–957. doi: 10.1007/s11064-011-0687-8. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Luo P, Guo Q, Li S, Zhang L, Zhao M, Xu H, Yang Y, Poon W, Fei Z. Interactions between SIRT1 and MAPK/ERK regulate neuronal apoptosis induced by traumatic brain injury in vitro and in vivo. Exp Neurol. 2012;237:489–498. doi: 10.1016/j.expneurol.2012.07.004. [DOI] [PubMed] [Google Scholar]