

Keywords: nerve regeneration, remote ischemic postconditioning, middle cerebral artery occlusion, cerebral ischemia/reperfusion, blood-brain barrier, acute cerebral ischemia, stroke, matrix metalloproteinase-9, claudin-5, neural regeneration
Abstract
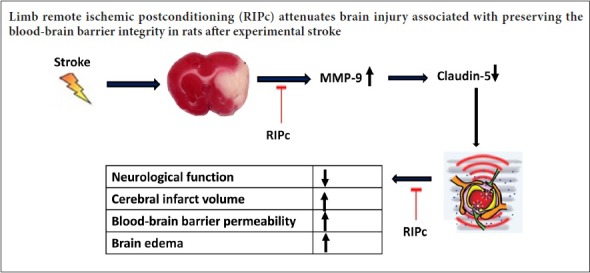
Integrity of the blood-brain barrier structure is essential for maintaining the internal environment of the brain. Development of cerebral infarction and brain edema is strongly associated with blood-brain barrier leakage. Therefore, studies have suggested that protecting the blood-brain barrier may be an effective method for treating acute stroke. To examine this possibility, stroke model rats were established by middle cerebral artery occlusion and reperfusion. Remote ischemic postconditioning was immediately induced by three cycles of 10-minute ischemia/10-minute reperfusion of bilateral hind limbs at the beginning of middle cerebral artery occlusion reperfusion. Neurological function of rat models was evaluated using Zea Longa’s method. Permeability of the blood-brain barrier was assessed by Evans blue leakage. Infarct volume and brain edema were evaluated using 2,3,5-triphenyltetrazolium chloride staining. Expression of matrix metalloproteinase-9 and claudin-5 mRNA was determined by real-time quantitative reverse transcription-polymerase chain reaction. Expression of matrix metalloproteinase-9 and claudin-5 protein was measured by western blot assay. The number of matrix metalloproteinase-9- and claudin-5-positive cells was analyzed using immunohistochemistry. Our results showed that remote ischemic postconditioning alleviated disruption of the blood-brain barrier, reduced infarct volume and edema, decreased expression of matrix metalloproteinase-9 mRNA and protein and the number of positive cells, increased expression of claudin-5 mRNA and protein and the number of positive cells, and remarkably improved neurological function. These findings confirm that by suppressing expression of matrix metalloproteinase-9 and claudin-5 induced by acute ischemia/reperfusion, remote ischemic postconditioning reduces blood-brain barrier injury, mitigates ischemic injury, and exerts protective effects on the brain.
Introduction
In recent decades, stroke has become one of the leading causes of disability and death worldwide. Despite advances in understanding of the pathogenesis of cerebral ischemia, few therapeutic options are available for acute ischemic stroke (Donnan et al., 2008). Currently, tissue plasminogen activator administered intravenously within 3 hours of symptom onset is the only Food And Drug Administration-approved therapy for rescuing brain tissue and re-establishing blood flow after acute ischemic stroke (Tilley et al., 1997). Nevertheless, mainly because of the extremely short therapeutic time window, only a small percentage of patients qualify for this treatment (Montano et al., 2013). Therefore, the current lack of clinical treatments for acute brain ischemia requires investigation of novel approaches that may eventually lead to feasible clinical application.
Recently, numerous investigators have confirmed that ischemic postconditioning (IPc) can alleviate cerebral ischemic injury in transient or permanent middle cerebral artery occlusion (MCAO) models (Zhao et al., 2006; Xing et al., 2008; Chen et al., 2016). As a new approach to induce ischemic tolerance, remote IPc (RIPc) is an endogenous protective approach, in which implementation of several brief cycles of non-infarct ischemia and reperfusion to an organ or tissue renders remote organs or tissues resistant to severe and sustained ischemia/reperfusion injury (Li et al., 2006; Lim and Hausenloy, 2012; Drunalini Perera et al., 2014). However, the underlying protective mechanisms are not well known.
Accumulating evidence demonstrates that breakdown of the blood-brain barrier (BBB) is a leading factor in many brain diseases, including ischemic stroke (Lee et al., 2013; Panahpour et al., 2014). Recently, many studies have shown that as an important member of matrix metalloproteinases (MMPs) (Chaturvedi and Kaczmarek, 2014), MMP-9 plays a dominant role in BBB disruption by further hydrolyzing tight junctions and the extracellular matrix, thereby leading to BBB breakage, brain edema, inflammatory cell exudation, and intracranial hemorrhage (Lakhan et al., 2013; Li et al., 2013; Yang et al., 2013; Jickling et al., 2014; Lv et al., 2016). As one substrate of MMP-9, claudin-5 is now widely accepted as a fundamental component of paracellular permeability and a key contributor to tight junction formation and BBB integrity (Liao et al., 2016; Lv et al., 2016).
To date, there has been little research on the effect of RIPc on structural and functional changes of the BBB induced by cerebral ischemia injury, although possible protective mechanisms of RIPc against brain injury after ischemic stroke have been described (Ren et al., 2009; Chen et al., 2014). Here, we examined the effect of RIPc on BBB disruption after ischemic stroke. Further, we also clarified whether RIPc alleviates BBB leakage by regulating critical molecules such as MMP-9 and claudin-5 in adult rats after acute ischemia reperfusion injury.
Materials and Methods
Animals
Animal use protocols were approved by the Institutional Animal Care and Use Committee at Chengdu Medical College of China (Approval No. 201401120411). The experimental procedure followed the United States National Institutes of Health Guide for the Care and Use of Laboratory Animal (NIH Publication No. 85-23, revised 1985).
A total of 81 adult female specific-pathogen-free Sprague-Dawley rats aged 15–16 weeks and weighing 250–280 g were provided by the Dashuo of Laboratory Animal Co., Ltd., Chengdu, China (License No. SYXK (Chuan) 2014-189). Rats were housed on a 12-hour dark-light cycle in specific-pathogen-free facilities at the Animal Center of Chengdu Medical College of China.
All rats were randomly assigned into three groups (n = 27 per group): sham group (sham operation), MCAO group, and RIPc group (MCAO with RIPc).
Establishment of focal cerebral ischemia/reperfusion models
All animals were allowed free access to water and food, but were fasted for 12 hours before surgery. Rats were intraperitoneally anesthetized with chloral hydrate (350 mg/kg). The transient focal MCAO model was established as previously described (Xiong et al., 2003), but with minor modifications. Briefly, the right common carotid artery, external carotid artery, and internal carotid artery were exposed in turn. A 5 cm 4-0 monofilament nylon suture with a blunt tip was inserted into the common carotid artery to block the initial segment of the MCA. The filament was softly advanced 18–20 mm along the internal carotid artery from the branch point of the carotid artery, until there was mild resistance. Sixty minutes after MCAO, reperfusion was initiated by gentle withdrawal of the monofilament. In the sham group, all surgical procedures were performed, but with no occlusion. Deep body temperature was monitored using a rectal probe and maintained at 36.2–37.2°C using light and a heating pad throughout the experiment. The skin was closed and the rat returned to its cage.
Limb RIPc
Limb RIPc was induced immediately by three cycles of 10-minute ischemia/10-minute reperfusion of bilateral hind limbs at the beginning of MCAO reperfusion, as described in previous methods (Ren et al., 2011; Cheng et al., 2014) but with slight modifications. Bilateral proximal hind limbs of each rat were encircled with rubber bands and pulse sensors placed on bilateral dorsalis pedis artery areas. Reversible ischemia of bilateral hind limbs was produced by tying the rubber bands as tight as possible until blood flow in the limbs was blocked. Blood flow of bilateral hind limbs was completely prevented as confirmed by hypothermia and cyanosis in the hind limbs and disappearance of a pulse. Limb ischemia was maintained for 10 minutes, and then reperfusion achieved by relaxing the rubber band for 10 minutes.
Assessment of neurological deficits
Ten minutes before being sacrificed (24 hours after reperfusion), rats were neurologically evaluated by three observers blinded to the animal groups. According to Zea Longa’s method (Longa et al., 1989), only rats scoring 1–2 were used for the following experimental procedure. Standard assessment was as follows: 0 point, neurological symptoms not obvious; 1 point, left forelimb cannot stretch completely; 2 points, rat rotates to the left; 3 points, rat falls to the left side; and 4 points, no spontaneous walking with a depressed level of consciousness. Final neurobehavioral scoring was performed according to a previous study (Garcia et al., 1995), with average scores finally obtained. Criteria included the following six tests: (1) spontaneous activity (0–3 points); (2) movement symmetry (0–3 points); (3) forepaw outstretching (0–3 points); (4) climbing (1–3 points); (5) body proprioception (1–3 points); and (6) vibrissae touching response (1–3 points). Minimum score was 3 and maximum score was 18.
Preparation of cerebral specimens
Deep anaesthesia was achieved, and then transcardial perfusion was performed for all rats at 24 hours after reperfusion. According to the requirements of different experimental techniques, rats were also randomly divided into groups for: 2,3,5-triphenyltetrazolium chloride (TTC) staining (n = 8 per group), real-time quantitative reverse transcription-polymerase chain reaction (qRT-PCR) (n = 5 per group), Evans blue detection (n = 5 per group), western blot assay (n = 5 per group), and histological investigation (n = 4 per group). Ischemic rats were excluded from the study if evidence of a subarachnoid hemorrhage was found during brain tissue sampling.
For morphological staining, rats were first quickly cleaned with 0.9% NaCl at 4°C, and then slowly pre-fixed with 4% paraformaldehyde (pH 7.3) at 4°C. After rapid removal of the brains, coronal brain slices were obtained from bregma −2 to 2 mm, and post-fixed in 4% paraformaldehyde for 24 hours, then embedded in paraffin and cut into 5-µm-thick coronal sections.
Evaluation of BBB permeability
To evaluate BBB permeability, Evans blue leakage into brain tissue was detected after intravenous injection. Briefly, 2% Evans blue (4 mL/kg) (Sigma–Aldrich, Steinheim, Germany) diluted in 0.9% NaCl was injected into the tail vein at 1 hour before the animal was sacrificed. Rats were transcardially perfused with normal saline to wash away remaining dye in the blood vessels. Next, each brain was rapidly removed, dissected, weighed, and ischemic hemispheres cut into fine particles and then immersed in methanamide (1 mL/100 mg) for 24 hours at 60°C. Following centrifugation at 12,000 × g for 20 minutes, absorption of supernatant was detected at 630 nm with an ultraviolet spectrophotometer (Bio-Rad, Hercules, CA, USA). Evans blue content was quantified as micrograms of Evans blue per gram of ischemic hemisphere using a standardized curve, with methanamide as negative control.
Measurement of cerebral infarct volume and brain edema
Brains were immediately collected and dissected into 2 mm coronal slices. Six continuous sections were immersed in TTC solution (0.5% TTC in PBS) at 37°C for 30 minutes, and then fixed in 4% paraformaldehyde buffer. Infarcted brain tissue retained its pale color, whereas living tissue was stained red. Sections were imaged using a digital camera (Olympus, Tokyo, Japan). The infarction zone was measured and analyzed by Image software (IPP, version 6.0, Media Cybernetics, Inc., Rockville, MD, USA). Using an indirect method (Lin et al., 1993), a corrected ratio of infarction to normal volume was calculated as follows: percentage of corrected infarct volume = contralateral hemisphere area−(ipsilateral hemisphere − infarction area measured)/contralateral hemisphere area × 100%. Cerebral edema was assessed in sections stained by TTC according to the formula: ipsilateral volume − contralateral volume/contralateral volume × 100%.
qRT-PCR
Brain tissue for each group was rapidly obtained from the right hemispheric MCA territory. Total RNA was isolated using Trizol reagent (Invitrogen, Waltham, MA, USA) according to the manufacturer’s protocol, and then reverse transcribed to generate cDNA with the Revert Aid TM First Strand cDNA Synthesis kit (TaKaRa Biotechnology, Dalian, China). cDNA was used as template in real-time PCR to analyze expression levels of target genes. The primer sequences used were: MMP-9, forward: 5′-TCC TAC TCT GCC TGC ACC A-3′; reverse: 5′-CAG TTA CAG TGA CGT CGG CT-3′; claudin-5, forward: 5′-CTG CCC ATG TGG CAG GTG A-3′; reverse: 5′-TGC ATG TGC CCG GTG CTC T-3′; and β-actin forward: 5′-GAA GAT CAA GAT CAT TGC TCC T-3′; reverse: 5′-TAC TCC TGC TTG CTG ATC CA-3′. Reverse transcription reactions were amplified using a Bio-Rad CFX96 Detection System (Bio-Rad) with the Plexor™ One-Step qRT-PCR System (Promega, Madison, WI, USA). The PCR conditions were: pre-denaturation at 95°C for 5 minutes, denaturation at 95°C for 30 seconds, annealing at 51°C for 30 seconds, and extension at 72°C for 1 minute. All operations were performed for 45 cycles. Relative expression quantities were calculated by normalization to β-actin values using the 2-ΔΔCt method.
Western blot assay
Brain meninges was carefully removed and ischemic brain tissue homogenized on ice in a lysis buffer containing 0.05 M Tris-HCl, 0.5 M ethylenediamine tetraacetic acid, 30% Triton-X-100, NaCl, 10% sodium dodecyl sulphate, and 1 mM phenylmethyl sulfonylfluoride. Homogenates were centrifuged at 12,000 r/min for 5 minutes at 4°C. The supernatant was collected and stored at −80°C for further use. Next, 20 µL samples were loaded onto each lane and electrophoresed on 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis for 2.5 hours at a constant voltage of 120 V. Proteins were transferred onto polyvinylidene difluoride membranes after electrophoresis. Membranes were blocked in 5% low fat milk in Tris-buffered saline-Tween-20, incubated at 37°C for 60 minutes with antibodies. Membranes were analyzed using Enhanced Chemiluminescence (Bio-Rad Laboratories, Philadelphia, PA, USA). The following primary antibodies were used: rabbit anti-MMP-9 (poly-antibody, 1:2000; Abcam, Cambridge, UK) and rabbit anti-claudin-5 (poly-antibody,1:1000; Abcam). The secondary antibody was goat anti-rabbit IgG (1:400; Proteintech Group, Wuhan, Hebei Province, China). Expression of target proteins was determined using β-actin protein (rabbit anti-β-actin poly-antibody, 1:8000; Abcam) as a loading control. The optical density intensity of each band was measured using BIO-RAD ChemiDoc XRS and Quantity One software (Bio-Rad). The optical density value in the sham group was used as the baseline reference value, with the ratio of each group to the sham group representing change.
Immunohistochemistry
Sections were dried, dewaxed, hydrated, and underwent high-pressure antigen retrieval. After immersion in 0.3% H2O2 at 37°C for 10 minutes, all sections were then blocked with 5% normal goat serum at 37°C for 20 minutes. Next, sections were incubated with primary anti-MMP-9 polyclonal antibody (rabbit, 1:100; Abcam) or anti-claudin-5 polyclonal antibody (rabbit, 1:200; Abcam) at 4°C overnight. After washing in PBS, sections were incubated with Polink-1 HRP DAB Detection System (goat anti-rabbit; Zhongshan, Beijing, China) at 37°C for 1 hour, visualized using 3,3′-diaminobenzidine, and counterstained with hematoxylin. Control sections were incubated in PBS instead of primary antibody. For each section, total number of positive cells in ischemic brain was counted in five different fields around the infarct area in a blinded manner using an Olympus BX-53 microscope (Olympus) at 400× magnification. Data from four rats in each group were averaged.
Double immunofluorescence
After standard histological procedures, sections were treated by high pressure antigen retrieval for 2 minutes and blocked with 5% goat serum at 37°C for 20 minutes. Afterwards, sections were incubated overnight at 4°C in mixtures of anti-MMP-9 antibody (rabbit, polyclonal, 1:50; Abcam)/anti-glial fibrillary acidic protein (GFAP) (mouse, monoclonal, 1:200; Proteintech), anti-claudin-5 antibody (rabbit, polyclonal, 1:100; Abcam)/anti-GFAP (mouse, monoclonal, 1:200; Proteintech), anti-MMP-9 antibody (rabbit, polyclonal, 1:50; Abcam)/anti-NeuN (mouse, monoclonal, 1:200; Abcam), anti-claudin-5 antibody (rabbit, polyclonal, 1:100; Abcam)/anti-NeuN (mouse, monoclonal, 1:200; Abcam), or anti-claudin-5 antibody (rabbit, polyclonal, 1:100; Abcam)/anti-MMP-9 (mouse, monoclonal, 1:50; Abcam). After washing in PBS, sections were incubated with fluorescence secondary antibody IgG (monoclonal, anti-mouse Alexa Fluor488-conjugated, green, 1:100; Proteintech)/IgG (polyclonal, anti-rabbit Alexa Fluor 594-conjugated, red, 1:100; Proteintech) for 2 hours at 37°C. After DAPI mounting, sections were imaged using a fluorescence microscope (Olympus).
Statistical analysis
All data were analyzed by an investigator blinded to treatments, and presented as mean ± SEM. Differences in quantitative data were evaluated by one-way analysis of variance followed by Student’s t-test. A value of P < 0.05 was considered statistically significant. Statistical analysis was performed with SPSS 17.0 software (SPSS, Chicago, IL, USA).
Results
Brain protection of RIPc on neurological deficits, cerebral infarct volume, edema, and BBB integrity in rats with stroke
Using the 18-point scale of Garcia et al., (1995), rats in the sham group showed nearly no neurological deficits. Neurological deficit scores in the MCAO group and RIPc group were also recorded. Scores were higher in the RIPc group than the MCAO group. These results demonstrate that neurological outcome is markedly improved after RIPc treatment (Figure 1A).
Figure 1.

Effect of RIPc on neurological function, cerebral infarction, cerebral edema, and blood-brain barrier integrity in stroke rats.
(A) Neurological scores (Garcia JH scores; Garcia et al., 1995). (B) Representative images of TTC staining in different groups. (C) Percentage of infarct size. (D) Percentage of cerebral edema. (E) Quantification of Evans blue dye. (A, C, D, G) Data are expressed as the mean ± SEM (one-way analysis of variance followed by Student’s t-test). *P < 0.05, vs. MCAO group; ##P < 0.01, vs. sham group. The experiment was performed five times. RIPc: Remote ischemic postconditioning; MCAO: middle cerebral artery occlusion; TTC: 2,3,5-triphenyltetrazolium chloride.
The protective effect of RIPc on cerebral infarction was assessed by calculating cerebral infarct volume with TTC staining. The results show that no ischemic damage was detected in the sham group. Compared with the MCAO group, infarct volume (Figure 1B, C) and edema (Figure 1D) were significantly reduced in the RIPc group (41.9 ± 6.4% and 27.6 ± 3.1% reduction, respectively) (P < 0.05). To estimate the influence of RIPc on BBB integrity after acute cerebral ischemic stroke, Evans blue leakage was detected. As expected, Evans blue content was very low in the sham group. Compared with the sham group, Evans blue transudation in the MCAO and RIPc groups markedly increased, and was mainly distributed in the ipsilateral striatum and cortex. Furthermore, compared with the MCAO group, Evans blue leakage was significantly diminished in the RIPc group (P < 0.05; Figure 1E). These results show that RIPc markedly attenuates damage of the BBB caused by transient focal cerebral ischemia.
RIPc inhibited expression of MMP-9 induced by ischemic stroke
We used qRT-PCR to detect expression of MMP-9 mRNA in the different groups. Expression of MMP-9 mRNA was very low in the sham group, but increased visibly in the MCAO and RIPc groups. Compared with the MCAO group, expression of MMP-9 mRNA was significantly decreased in the RIPc group (P < 0.05; Figure 2A).
Figure 2.

Effect of limb RIPc on transcription and expression of MMP-9 in ischemic brain.
(A) Relative expression of MMP-9 mRNA (real-time quantitative reverse transcription-polymerase chain reaction). Results are expressed as fold change over the sham group. (B) Representative western blot images of MMP-9 protein in ischemic brain of different groups. (C) Protein expression levels of MMP-9. (D) Double immunofluorescence images of MMP-9 and GFAP in the MCAO group. Nuclei were labeled with DAPI (blue). MMP-9-positive cells were labeled green by Alexa Fluor 488, while GFAP-positive cells were labeled red by Alexa Fluor 594. White arrows indicate corresponding positive cells or co-localization of MMP-9 and GFAP. Scale bar: 20 μm. (E) Double immunofluorescence images of MMP-9 and NeuN in the MCAO group. Nuclei were labeled with DAPI (blue). MMP-9-positive cells were labeled green by Alexa Fluor 488, while NeuN-positive cells were labeled red by Alexa Fluor 594. White arrows indicate corresponding positive cells or co-localization of MMP-9 and NeuN. Scale bar: 20 μm. (F) Immunohistochemistry images of MMP-9. Black arrows indicate MMP-9-positive cells. Scale bar: 20 μm. (G) Number of MMP-9-positive cells around the infarct area. (A, C, G) Data are expressed as the mean ± SEM (one-way analysis of variance followed by Student’s t-test and post hoc Fisher’s tests). *P < 0.05, vs. MCAO group; #P < 0.05, ##P < 0.01, vs. sham group. The experiment was performed five times. RIPc: Remote ischemic postconditioning; MCAO: middle cerebral artery occlusion; MMP-9: matrix metalloproteinase-9; GFAP: glial fibrillary acidic protein.
Further, levels of MMP-9 protein were examined by western blot assay. Our results show very low levels of MMP-9 protein in the sham group (Figure 2B). While after cerebral ischemia/reperfusion, levels of MMP-9 protein were significantly increased in both the RIPc and MCAO groups compared with the sham group (P < 0.01; Figure 2C). Overexpression of MMP-9 induced by stroke was markedly inhibited by RIPc treatment.
In the cortical ischemic penumbra, immunohistochemistry showed that MMP-9 was mainly expressed in the cytoplasm of neurons (MMP-9 and GFAP double-positive cells) (Figure 2E), with expression in only a few astrocytes (MMP-9 and NeuN double-positive cells) (Figure 2D). In the sham group, almost no MMP-9 labeled cells were apparent in the brain. However, MMP-9 immunostaining was significantly enhanced in the MCAO and RIPc groups (P < 0.01; Figure 2F, G). Furthermore, MMP-9-positive cell number was less in the RIPc group compared with the MCAO group (P < 0.05; Figure 2G).
RIPc inhibited the decrease of claudin-5 protein induced by acute cerebral ischemia
Expression of the gene for claudin-5 (Cldn5) was reduced in both the MCAO group and RIPc group compared with the sham group (P < 0.05; Figure 3A). While expression of claudin-5 mRNA after ischemia/reperfusion was lower in the MCAO group than RIPc group (P > 0.05). Levels of claudin-5 protein were examined by western blot assay (Figure 3B). The results showed that claudin-5 protein levels were significantly decreased in both the MCAO and RIPc groups compared with the sham group (P < 0.01). Moreover, compared with the RIPc group, reduction of claudin-5 was significant in the MCAO group (P < 0.05; Figure 3C).
Figure 3.

Effect of limb RIPc on transcription and expression of claudin-5 in the cortex.
(A) Relative expression of transcriptional levels of claudin-5 (real-time quantitative reverse transcription-polymerase chain reaction). Relative expression of claudin-5 mRNA, fold change over the sham group. (B) Representative western blot images of claudin-5 protein in the ischemic cortex. (C) Protein expression levels of claudin-5. Compared with the MCAO group, RIPc significantly decreased the infarct size induced by focal cerebral ischemia. (D) Double immunofluorescence images of claudin-5 and GFAP in the MCAO group. Nuclei were labeled with DAPI (blue). Claudin-5-positive cells were labeled green with Alexa Fluor488, while GFAP-positive cells were labeled red with Alexa Fluor 594. White arrows indicate corresponding positive cells or co-localization of claudin-5 and GFAP. Scale bar: 20 μm. (E) Double immunofluorescence images of claudin-5 and NeuN in the MCAO group. Nuclei were labeled with DAPI (blue). Claudin-5-positive cells were labeled green with Alexa Fluor 488, and NeuN-positive cells were labeled red with Alexa Fluor 594. White arrows indicate corresponding positive cells or co-localization of claudin-5 and NeuN. Scale bar: 20 μm. (F) Immunohistochemistry images of claudin-5. Some microvessel walls immunostained by claudin-5 (black arrows). Scale bar: 20 μm. (G) Number of claudin-5-positive cells around the infarct area. (A, C, G) Data are expressed as the mean ± SEM (one-way analysis of variance followed by Student’s t-test and post hoc Fisher’s tests). *P < 0.05, vs. MCAO group; ##P < 0.01, vs. sham group. The experiment was performed five times. RIPc: Remote ischemic postconditioning; MCAO: middle cerebral artery occlusion; GFAP: glial fibrillary acidic protein; DAPI: 4′,6-diamidino-2-phenylindole.
Immunohistochemistry showed that claudin-5 immunoreactivity was detected on endothelial cells of brain microvessels (claudin-5 and GFAP double-positive cells) (Figure 3D) and individual neurons (claudin-5 and NeuN double-positive cells) (Figure 3E). Indeed, there was more positive staining than the sham group (P < 0.01; Figure 3F, G). Number of claudin-5 immunopositive cells was greater in the RIPc group than the MCAO group (P < 0.05; Figure 3G).
Discussion
Many studies have confirmed that IPc is a powerful protective strategy against brain injury after stroke in rats (Du et al., 2014; Gulati and Singh, 2014; Wang et al., 2014; Zhao et al., 2014). Moreover, extensive research into the protective mechanisms underlying IPc has been performed, with multiple factors found to be involved including suppression of oxidative stress, activation of signaling pathways, decrease of apoptosis, and inhibition of endoplasmic stress (Pignataro et al., 2008; Yuan et al., 2011). Considering the potential limitations of the clinical application of IPc, it soon became evident that the concept could also be used for distant organs. As a distant organ, the limb was chosen and RIPc performed before the onset of myocardial reperfusion. This proved effective and feasible for protecting the myocardium from ischemia reperfusion (Li et al., 2006). Subsequently, Ren et al., (2009) found that RIPc remarkably decreased infarct volume following permanent focal cerebral ischemia. Given the current potential clinical role and simple operational procedure, limb RIPc has attracted widespread interest, and is generally considered a creative and promising approach for protecting important organs or tissue from injury factors of severe acute ischemia (Li et al., 2006; Lim and Hausenloy, 2012).
In most studies, RIPc was performed promptly at the start of reperfusion, and indeed a relatively narrow time window for a protective effect has been noted (Anrather and Hallenbeck, 2013). Furthermore, Xu et al., (2014) suggested that rapid RIPc can exert its maximum protective effect if the accumulative time of limb occlusion/reperfusion lasted from 40 to 60 minutes. In this study, we chose a procedure of 10-minute ischemia and 10-minute reperfusion for three cycles (therefore an overall accumulation of 60 minutes), and consistently confirmed that this dramatically reduced infarct size. Although identifying the molecular mechanisms underlying RIPc is only just beginning, to date a growing body of evidence has indicated that RIPc might share common triggers, mediators, effectors, and mechanistic signaling pathways with classical intra-organ ischemic preconditioning/postconditioning and inter-organ remote limb preconditioning (Pan et al., 2016). After repeated brief sub-irreversible injurious ischemia/reperfusion in a distant organ (limb) or tissue, lactate/pyruvate ratio due to anaerobic glycolysis accumulates within minutes, ultimately leading to acidosis (Kauffman and Albuquerque, 1970). Subsequently, a drop in tissue pH can activate sensitive sensory neurons, resulting in marked activation of afferent neural pathways (Kauffman and Albuquerque, 1970). Meanwhile, activation of endothelium, blood cells, and platelets (Strecker et al., 2005; Doeuvre et al., 2009) leads to release of protective neural, humoral, or cellular transmitters from remote organs (Anrather and Hallenbeck, 2013), reaching the target organ (brain) through neural pathways and/or blood circulation, which may activate prosurvival pathways (phosphatidylinositol-4,5-bisphosphate 3-kinase [PI3K]/Akt and extracellular signal–regulated kinase [ERK]) or suppress proinflammatory pathways (P38/ mitogen-activated protein kinase [MAPK] and toll-like receptors [TLRs]), thereby leading to remote ischemic tolerance (Noshita et al., 2002; Valen, 2009; Rohailla et al., 2014).
Anatomical structure of the BBB includes the cerebral microvascular endothelium, which together with astrocytes, pericytes, neurons, and the extracellular matrix, comprises a “neurovascular unit” that is indispensable for homeostasis and function of the central nervous system (del Zoppo, 2009; Dong et al., 2009). Since increased BBB permeability is often associated with brain edema and swelling (Wang and Shuaib, 2007; Lee et al., 2013), BBB leakage may result in central nervous system dysfunction, and even endanger patient life. Recent studies with IPc have shown that some drugs or early exercise can attenuate cerebral ischemia injury by improving BBB structure and function (Zhang et al., 2013; Liu et al., 2014b). Nevertheless, little is known about whether non-invasive RIPc can protect the brain from ischemic damage through the same mechanism. Here, we detected structural integrity of the BBB by Evans blue extravasation in a MCAO reperfusion rat model with RIPc intervention. As expected, our results show that local ischemia for 60 minutes results in marked BBB damage and cerebral edema. Further, at the early stage of ischemic stroke, infarct volume or cerebral edema enlarged correspondingly while Evans blue transudation increased, which was suppressed by RIPc treatment. This finding is consistent with previous studies (Cheng et al., 2014; Liu et al., 2014a). Therefore, we have confirmed that RIPc can exert protective effects on cerebral ischemia injury by stabilizing BBB structure and function.
Numerous animal studies and clinical data have shown that MMP-9 protein levels are visibly up-regulated in serum and brain tissue during acute ischemic stroke, which correlates with cerebral infarct volume and disease severity (Lee et al., 2009; Demir et al., 2012). Here, we show that transcription and expression of MMP-9 are up-regulated after reperfusion, and obviously inhibited by RIPc treatment. These results are consistent with previous studies (Guo et al., 2008; Liu et al., 2012). Growing evidence shows that proinflammatory pathways and downstream transcription factors (e.g., nuclear factor [NF]-κB and activator protein 1 [AP]-1) can enhance MMP-9 promoter activity and activate MMP-9 gene transcription (Yang et al., 2015). Furthermore, the MMP-9 gene promoter comprises a highly conserved motif that matches a NF-κB p65 binding component, while suppression of NF-κB inhibits MMP-9 up-regulation in ischemic brain (Cheng et al., 2006). Overall, we deduced that RIPc might inhibit ischemia-triggered MMP-9 over-regulation by suppressing activation of the NF-κB gene and corresponding pathways. It is currently accepted that increased MMP-9 is strongly associated with BBB disruption via extracellular matrix breakdown, whereas BBB damage can be attenuated by both reducing MMP-9 and deleting the MMP-9 gene (Guo et al., 2008; Liu et al., 2012; Yang et al., 2015).
It is well-known that tight junctions are continuous specialized membrane strands located at the most apical region between cerebral endothelial cells, and comprise a close complex of various proteins including transmembrane and cytoplasmic proteins. (Horstmann et al., 2003; Lv et al., 2016). As a predominant component of the BBB, claudin-5 regulates paracellular permeability of the BBB through the actin cytoskeleton via interaction with TJ proteins, including occludin, junctional adhesion molecule-1, claudin-1, zonula occludens (ZO) proteins, and caveolin-1 (CAV-1). Decreased expression of claudin-5 can open the BBB, leading to brain edema and neuronal cell death (Yang et al., 2007). Since claudin-5 is a component of the BBB that maintains stability, claudin-5 transcription and expression were detected in this study to determine whether brain protection of RIPc treatment is involved in alteration of claudin-5, at least partially. Our results indicate that RIPc visibly inhibits the decrease in claudin-5 protein. Wolburg et al., (2009) found that astrocytic end feet help regulate cerebral microvessel blood flow, which is dedicated to both maintenance and creation of the BBB microenvironment. In the present study, a few claudin-5-positive cells were co-located in astrocytes, especially around microvessels. Thus, our results indicate that structural stability of the BBB relies on subtle molecular regulation of astrocytes. However, contrary to our expectation, reduced Cldn5 transcription was not significantly suppressed by RIPc treatment. Indeed, we believe transcription of Cldn5 is earlier than subsequent protein expression.
In summary, RIPc can improve neurological function, reduce cerebral infarct volume and edema, and alleviate BBB leakage, ultimately leading to cerebral ischemia tolerance. The underlying neuroprotective mechanism induced by RIPc may involve decreased overexpression of MMP-9 and suppressed degradation of claudin-5 after transient focal ischemic stroke. Finally, our study has shortcomings. For improved and more impartial evaluation of the role of RIPc, we should further observe ultrastructural damage to the BBB after acute cerebral ischemia, and perform reperfusion for a longer time.
Additional file: Open peer review report 1 (5.6KB, pdf) .
Footnotes
Conflicts of interest: We declare that we have no conflict of interest.
Financial support: This study was supported by the National Natural Science Foundation of China, No. 30960107; the Natural Science Foundation of the Education Department of Sichuan Province of China, No. 14ZA0223. The funders had no role in study conception and design, literature retrieval, experimental studies, data acquisition and analysis, statistical analysis, paper writing and decision to submit the paper for publication.
Institutional review board statement: The study protocol was approved by the Institutional Animal Care and Use Committee at Chengdu Medical College of China (approval No. 201401120411). The experimental procedure followed the United States National Institutes of Health Guide for the Care and Use of Laboratory Animal (NIH Publication No. 85-23, revised 1985).
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Tianhao Bao, China.
Funding: This study was supported by the National Natural Science Foundation of China, No. 30960107; the Natural Science Foundation of the Education Department of Sichuan Province of China, No. 14ZA0223.
(Copyedited by James R, Maxwell R, Yu J, Li CH, Qiu Y, Song LP, Zhao M)
References
- Anrather J, Hallenbeck JM. Biological networks in ischemic tolerance - rethinking the approach to clinical conditioning. Transl Stroke Res. 2013;4:114–129. doi: 10.1007/s12975-012-0244-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi M, Kaczmarek L. Mmp-9 inhibition: a therapeutic strategy in ischemic stroke. Mol Neurobiol. 2014;49:563–573. doi: 10.1007/s12035-013-8538-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Yang J, Lu G, Guo J, Dou Y. Limb remote ischemic post-conditioning reduces brain reperfusion injury by reversing eNOS uncoupling. Indian J Exp Biol. 2014;52:597–605. [PubMed] [Google Scholar]
- Chen G, Ye X, Zhang J, Tang T, Li L, Lu P, Wu Q, Yu B, Kou J. Limb remote ischemic postconditioning reduces ischemia-reperfusion injury by inhibiting NADPH oxidase activation and MyD88-TRAF6-P38MAP-Kinase pathway of neutrophils. Int J Mol Sci. 2016;17:E1971. doi: 10.3390/ijms17121971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T, Petraglia AL, Li Z, Thiyagarajan M, Zhong Z, Wu Z, Liu D, Maggirwar SB, Deane R, Fernandez JA, LaRue B, Griffin JH, Chopp M, Zlokovic BV. Activated protein C inhibits tissue plasminogen activator-induced brain hemorrhage. Nat Med. 2006;12:1278–1285. doi: 10.1038/nm1498. [DOI] [PubMed] [Google Scholar]
- Cheng Z, Li L, Mo X, Zhang L, Xie Y, Guo Q, Wang Y. Non-invasive remote limb ischemic postconditioning protects rats against focal cerebral ischemia by upregulating STAT3 and reducing apoptosis. Int J Mol Med. 2014;34:957–966. doi: 10.3892/ijmm.2014.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Zoppo GJ. Inflammation and the neurovascular unit in the setting of focal cerebral ischemia. Neuroscience. 2009;158:972–982. doi: 10.1016/j.neuroscience.2008.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demir R, Ulvi H, Özel L, Özdemir G, Güzelcik M, Aygül R. Relationship between plasma metalloproteinase-9 levels and volume and severity of infarct in patients with acute ischemic stroke. Acta Neurol Belg. 2012;112:351–356. doi: 10.1007/s13760-012-0067-4. [DOI] [PubMed] [Google Scholar]
- Doeuvre L, Plawinski L, Toti F, Anglés-Cano E. Cell-derived microparticles: a new challenge in neuroscience. J Neurochem. 2009;110:457–468. doi: 10.1111/j.1471-4159.2009.06163.x. [DOI] [PubMed] [Google Scholar]
- Dong X, Song YN, Liu WG, Guo XL. Mmp-9, a potential target for cerebral ischemic treatment. Curr Neuropharmacol. 2009;7:269–275. doi: 10.2174/157015909790031157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnan GA, Fisher M, Macleod M, Davis SM. Stroke. Lancet. 2008;371:1612–1623. doi: 10.1016/S0140-6736(08)60694-7. [DOI] [PubMed] [Google Scholar]
- Drunalini Perera PN, Hu Q, Tang J, Li L, Barnhart M, Doycheva DM, Zhang JH, Tang J. Delayed remote ischemic postconditioning improves long term sensory motor deficits in a neonatal hypoxic ischemic rat model. PLoS One. 2014;9:e90258. doi: 10.1371/journal.pone.0090258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du GJ, Dang MR, Tan ZM, Su RQ, Shen Q, Fang JS. Limb ischemic postconditioning promotes the proliferation of endogenous neural stem cells in rats with cerebral ischemia injury. Zhongguo Zuzhi Gongcheng Yanjiu. 2014;18:1597–1602. [Google Scholar]
- Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke. 1995;26:627–635. doi: 10.1161/01.str.26.4.627. [DOI] [PubMed] [Google Scholar]
- Gulati P, Singh N. Neuroprotective mechanism of ischemic postconditioning in mice: a possible relationship between protein kinase C and nitric oxide pathways. J Surg Res. 2014;189:174–183. doi: 10.1016/j.jss.2014.02.019. [DOI] [PubMed] [Google Scholar]
- Guo M, Cox B, Mahale S, Davis W, Carranza A, Hayes K, Sprague S, Jimenez D, Ding Y. Pre-ischemic exercise reduces matrix metalloproteinase-9 expression and ameliorates blood-brain barrier dysfunction in stroke. Neuroscience. 2008;151:340–351. doi: 10.1016/j.neuroscience.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Horstmann S, Kalb P, Koziol J, Gardner H, Wagner S. Profiles of matrix metalloproteinases, their inhibitors, and laminin in stroke patients: influence of different therapies. Stroke. 2003;34:2165–2170. doi: 10.1161/01.STR.0000088062.86084.F2. [DOI] [PubMed] [Google Scholar]
- Jickling GC, Liu D, Stamova B, Ander BP, Zhan X, Lu A, Sharp FR. Hemorrhagic transformation after ischemic stroke in animals and humans. J Cereb Blood Flow Metab. 2014;34:185–199. doi: 10.1038/jcbfm.2013.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman FC, Albuquerque EX. Effect of ischemia and denervation on metabolism of fast and slow mammalian skeletal muscle. Exp Neurol. 1970;28:46–63. doi: 10.1016/0014-4886(70)90161-5. [DOI] [PubMed] [Google Scholar]
- Lakhan SE, Kirchgessner A, Tepper D, Leonard A. Matrix metalloproteinases and blood-brain barrier disruption in acute ischemic stroke. Front Neurol. 2013;4:32. doi: 10.3389/fneur.2013.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Park JW, Kim SP, Lo EH, Lee SR. Doxycycline inhibits matrix metalloproteinase-9 and laminin degradation after transient global cerebral ischemia. Neurobiol Dis. 2009;34:189–198. doi: 10.1016/j.nbd.2008.12.012. [DOI] [PubMed] [Google Scholar]
- Lee JH, Cui HS, Shin SK, Kim JM, Kim SY, Lee JE, Koo BN. Effect of propofol post-treatment on blood-brain barrier integrity and cerebral edema after transient cerebral ischemia in rats. Neurochem Res. 2013;38:2276–2286. doi: 10.1007/s11064-013-1136-7. [DOI] [PubMed] [Google Scholar]
- Li CM, Zhang XH, Ma XJ, Luo M. Limb ischemic postconditioning protects myocardium from ischemia-reperfusion injury. Scand Cardiovasc J. 2006;40:312–317. doi: 10.1080/14017430600925292. [DOI] [PubMed] [Google Scholar]
- Li DD, Song JN, Huang H, Guo XY, An JY, Zhang M, Li Y, Sun P, Pang HG, Zhao YL, Wang JF. The roles of MMP-9/TIMP-1 in cerebral edema following experimental acute cerebral infarction in rats. Neurosci Lett. 2013;550:168–172. doi: 10.1016/j.neulet.2013.06.034. [DOI] [PubMed] [Google Scholar]
- Liao Z, Yang Z, Piontek A, Eichner M, Krause G, Li L, Piontek J, Zhang J. Specific binding of a mutated fragment of Clostridium perfringens enterotoxin to endothelial claudin-5 and its modulation of cerebral vascular permeability. Neuroscience. 2016;327:53–63. doi: 10.1016/j.neuroscience.2016.04.013. [DOI] [PubMed] [Google Scholar]
- Lim SY, Hausenloy DJ. Remote ischemic conditioning: from bench to bedside. Front Physiol. 2012;3:27. doi: 10.3389/fphys.2012.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TN, He YY, Wu G, Khan M, Hsu CY. Effect of brain edema on infarct volume in a focal cerebral ischemia model in rats. Stroke. 1993;24:117–121. doi: 10.1161/01.str.24.1.117. [DOI] [PubMed] [Google Scholar]
- Liu Q, Zhou S, Wang Y, Qi F, Song Y, Long S. A feasible strategy for focal cerebral ischemia-reperfusion injury: remote ischemic postconditioning. Neural Regen Res. 2014a;9:1460–1463. doi: 10.4103/1673-5374.139463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Zhang T, Yu H, Shen H, Xia W. Adjudin protects against cerebral ischemia reperfusion injury by inhibition of neuroinflammation and blood-brain barrier disruption. J Neuroinflammation. 2014b;11:107. doi: 10.1186/1742-2094-11-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XR, Luo M, Yan F, Zhang CC, Li SJ, Zhao HP, Ji XM, Luo YM. Ischemic postconditioning diminishes matrix metalloproteinase 9 expression and attenuates loss of the extracellular matrix proteins in rats following middle cerebral artery occlusion and reperfusion. CNS Neurosci Ther. 2012;18:855–863. doi: 10.1111/j.1755-5949.2012.00366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- Lv JM, Guo XM, Chen B, Lei Q, Pan YJ, Yang Q. The noncompetitive AMPAR antagonist perampanel abrogates brain endothelial cell permeability in response to ischemia: involvement of Claudin-5. Cell Mol Neurobiol. 2016;36:745–753. doi: 10.1007/s10571-015-0257-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montano A, Staff I, McCullough LD, Fortunato G. Community implementation of intravenous thrombolysis for acute ischemic stroke in the 3- to 4, 5-hour window. Am J Emerg Med. 2013;31:1707–1709. doi: 10.1016/j.ajem.2013.08.032. [DOI] [PubMed] [Google Scholar]
- Noshita N, Sugawara T, Hayashi T, Lewen A, Omar G, Chan PH. Copper/zinc superoxide dismutase attenuates neuronal cell death by preventing extracellular signal-regulated kinase activation after transient focal cerebral ischemia in mice. J Neurosci. 2002;22:7923–7930. doi: 10.1523/JNEUROSCI.22-18-07923.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J, Li X, Peng Y. Remote ischemic conditioning for acute ischemic stroke: dawn in the darkness. Rev Neurosci. 2016;27:501–510. doi: 10.1515/revneuro-2015-0043. [DOI] [PubMed] [Google Scholar]
- Panahpour H, Nekooeian AA, Dehghani GA. Candesartan attenuates ischemic brain edema and protects the blood-brain barrier integrity from ischemia/reperfusion injury in rats. Iran Biomed J. 2014;18:232–238. doi: 10.6091/ibj.13672.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignataro G, Meller R, Inoue K, Ordonez AN, Ashley MD, Xiong Z, Gala R, Simon RP. In vivo and in vitro characterization of a novel neuroprotective strategy for stroke: ischemic postconditioning. J Cereb Blood Flow Metab. 2008;28:232–241. doi: 10.1038/sj.jcbfm.9600559. [DOI] [PubMed] [Google Scholar]
- Ren C, Yan Z, Wei D, Gao X, Chen X, Zhao H. Limb remote ischemic postconditioning protects against focal ischemia in rats. Brain Res. 2009;1288:88–94. doi: 10.1016/j.brainres.2009.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren C, Gao M, Dornbos D, 3rd, Ding Y, Zeng X, Luo Y, Ji X. Remote ischemic post-conditioning reduced brain damage in experimental ischemia/reperfusion injury. Neurol Res. 2011;33:514–519. doi: 10.1179/016164111X13007856084241. [DOI] [PubMed] [Google Scholar]
- Rohailla S, Clarizia N, Sourour M, Sourour W, Gelber N, Wei C, Li J, Redington AN. Acute, delayed and chronic remote ischemic conditioning is associated with downregulation of mTOR and enhanced autophagy signaling. PLoS One. 2014;9:e111291. doi: 10.1371/journal.pone.0111291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strecker T, Messlinger K, Weyand M, Reeh PW. Role of different proton-sensitive channels in releasing calcitonin gene-related peptide from isolated hearts of mutant mice. Cardiovasc Res. 2005;65:405–410. doi: 10.1016/j.cardiores.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Tilley BC, Lyden PD, Brott TG, Lu M, Levine SR, Welch KM. Total quality improvement method for reduction of delays between emergency department admission and treatment of acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. Arch Neurol. 1997;54:1466–1474. doi: 10.1001/archneur.1997.00550240020008. [DOI] [PubMed] [Google Scholar]
- Valen G. Extracardiac approaches to protecting the heart. Eur J Cardiothorac Surg. 2009;35:651–657. doi: 10.1016/j.ejcts.2008.12.023. [DOI] [PubMed] [Google Scholar]
- Wang CX, Shuaib A. Critical role of microvasculature basal lamina in ischemic brain injury. Prog Neurobiol. 2007;83:140–148. doi: 10.1016/j.pneurobio.2007.07.006. [DOI] [PubMed] [Google Scholar]
- Wang Y, Ge P, Yang L, Wu C, Zha H, Luo T, Zhu Y. Protection of ischemic post conditioning against transient focal ischemia-induced brain damage is associated with inhibition of neuroinflammation via modulation of TLR2 and TLR4 pathways. J Neuroinflammation. 2014;11:15. doi: 10.1186/1742-2094-11-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolburg H, Noell S, Mack A, Wolburg-Buchholz K, Fallier-Becker P. Brain endothelial cells and the glio-vascular complex. Cell Tissue Res. 2009;335:75–96. doi: 10.1007/s00441-008-0658-9. [DOI] [PubMed] [Google Scholar]
- Xing B, Chen H, Zhang M, Zhao D, Jiang R, Liu X, Zhang S. Ischemic postconditioning inhibits apoptosis after focal cerebral ischemia/reperfusion injury in the rat. Stroke. 2008;39:2362–2369. doi: 10.1161/STROKEAHA.107.507939. [DOI] [PubMed] [Google Scholar]
- Xiong L, Zheng Y, Wu M, Hou L, Zhu Z, Zhang X, Lu Z. Preconditioning with isoflurane produces dose-dependent neuroprotection via activation of adenosine triphosphate-regulated potassium channels after focal cerebral ischemia in rats. Anesth Analg. 2003;96:233–237. doi: 10.1097/00000539-200301000-00047. [DOI] [PubMed] [Google Scholar]
- Xu C, Yi C, Guo H, Liu Q, Wang L, Li H. Limb remote ischemic postconditioning is effective but also time-course-limited in protecting the brain from I/R injury. Turk J Med Sci. 2014;42:918–929. [Google Scholar]
- Yang CM, Hsieh HL, Yu PH, Lin CC, Liu SW. IL-1beta induces MMP-9-dependent brain astrocytic migration via transactivation of PDGF receptor/NADPH oxidase 2-derived reactive oxygen species signals. Mol Neurobiol. 2015;52:303–317. doi: 10.1007/s12035-014-8838-y. [DOI] [PubMed] [Google Scholar]
- Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2007;27:697–709. doi: 10.1038/sj.jcbfm.9600375. [DOI] [PubMed] [Google Scholar]
- Yang Y, Thompson JF, Taheri S, Salayandia VM, McAvoy TA, Hill JW, Yang Y, Estrada EY, Rosenberg GA. Early inhibition of MMP activity in ischemic rat brain promotes expression of tight junction proteins and angiogenesis during recovery. J Cereb Blood Flow Metab. 2013;33:1104–1114. doi: 10.1038/jcbfm.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Guo Q, Ye Z, Pingping X, Wang N, Song Z. Ischemic postconditioning protects brain from ischemia/reperfusion injury by attenuating endoplasmic reticulum stress-induced apoptosis through PI3K-Akt pathway. Brain Res. 2011;1367:85–93. doi: 10.1016/j.brainres.2010.10.017. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zhang P, Shen X, Tian S, Wu Y, Zhu Y, Jia J, Wu J, Hu Y. Early exercise protects the blood-brain barrier from ischemic brain injury via the regulation of MMP-9 and occludin in rats. Int J Mol Sci. 2013;14:11096–11112. doi: 10.3390/ijms140611096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Sapolsky RM, Steinberg GK. Interrupting reperfusion as a stroke therapy: ischemic postconditioning reduces infarct size after focal ischemia in rats. J Cereb Blood Flow Metab. 2006;26:1114–1121. doi: 10.1038/sj.jcbfm.9600348. [DOI] [PubMed] [Google Scholar]
- Zhao H, Wang R, Tao Z, Gao L, Yan F, Gao Z, Liu X, Ji X, Luo Y. Ischemic postconditioning relieves cerebral ischemia and reperfusion injury through activating T-LAK cell-originated protein kinase/protein kinase B pathway in rats. Stroke. 2014;45:2417–2424. doi: 10.1161/STROKEAHA.114.006135. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.