

Abstract
Stimulation of the autonomic parasympathetic fibers of the facial nerve system (hereafter simply “facial nerve”) rapidly dilates the cerebral arteries and increases cerebral blood flow whether that stimulation is delivered at the facial nerve trunk or at distal points such as the sphenopalatine ganglion. Facial nerve stimulation thus could be used as an emergency treatment of conditions of brain ischemia such as ischemic stroke. A rich history of scientific research has examined this property of the facial nerve, and various means of activating the facial nerve can be employed including noninvasive means. Herein, we review the anatomical and physiological research behind facial nerve stimulation and the facial nerve stimulation devices that are in development for the treatment of ischemic stroke.
Keywords: Facial nerve stimulation, future treatment, ischemic stroke
Introduction
Regulation of the cerebral arteries by the facial nerve
Our understanding of the anatomical connection of the greater superficial petrosal branch of the facial nerve to the terminal sphenopalatine ganglion (SPG) and the SPG projections to the cerebral arteries began with Vesalius’ early dissections in the 16th century. Myelinated and unmyelinated fibers carried by the SPG and distributed in plexi on either side of the vascular adventitia were more recently described[1,2] as were the origin of the preganglionic parasympathetic fibers of the facial nerve in the salivatory nucleus of the brainstem[3,4] that, when chemically stimulated with glutamate injection, produce ipsilateral increases in cerebral blood flow (CBF).[5] Those preganglionic parasympathetic neurons project through the geniculate ganglion with few, if any, synapses there[6] and run to more distal ganglia, the best studied of which is the SPG. The parasympathetic innervation carried by the facial nerve is generally distributed to the tissues derived from the branchial arch mesoderm, accounting for the facial nerve's effects on lacrimation, salivation, and blood flow to facial muscles as well as effects on the cerebral arteries. A schematic view of the facial nerve is provided in Figure 1.
Figure 1.
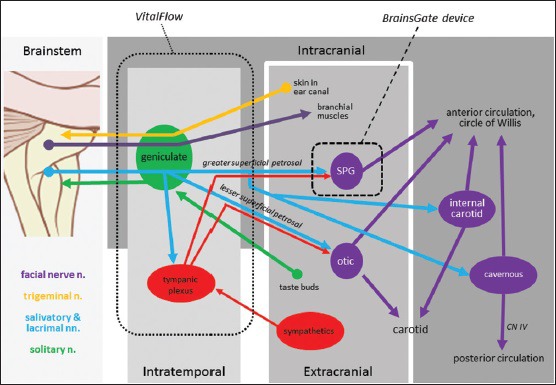
Anatomy of the facial nerve illustrating putative outflow to the various parts of the cerebral circulation, and the sites of stimulation of various devices in development for the treatment of brain ischemia: spheres and ovals = ganglia
Over the last century, many (but not all) studies of this topic have shown that electrical stimulation of the facial nerve causes dilation of the cerebral arteries and an increased CBF in normal animals.[7,8,9,10,11,12,13,14,15,16,17,18] The effect appears to critically involve nitric oxide,[13] and it may also involve neuropeptide Y and vasoactive intestinal peptide (VIP).[19] In contrast, acetylcholine - which also colocalizes with the neuropeptide markers of the parasympathetic nerve endings and which pharmacologically dilates cerebral arteries, for example, after intravenous injection - does not appear to be critically involved in the dilation of cerebral arteries after facial nerve stimulation.[7,16] Rather, if its role in the salivary glands is any indicator,[20] acetylcholine released by facial nerve terminals onto the cerebral arteries may specifically increase the permeability of the blood-brain barrier, a known effect of facial nerve stimulation.[21,22,23] In addition, acetylcholine is a critical neurotransmitter at the ganglia that project to the cerebral arteries such as the SPG.[7,8,12]
The postganglionic innervation of the facial nerve across the cerebral arterial tree is not uniform. Innervation by the VIP-expressing nerve fibers of the SPG is most dense in the proximal cerebral arteries,[24,25,26] whereas VIP-positive nerve fibers in small distal cerebral arteries may be derived from neurons of the basal forebrain[27,28] if they are present at all.[25] Similarly, the SPG is thought to innervate predominantly the anterior circulation of the brain, having little connection to the posterior circulation that nevertheless carry VIP-expressing nerve fibers at least in the rostral part of the basilar artery.[29] There is also redundancy and overlap in the innervation. In the anterior circulation, the SPG cannot be the sole source of parasympathetic innervation for the cerebral arteries since lesions of the SPG reduce VIP-expressing fiber density only by about one-third.[24] The density of VIP innervation of the anterior circulation appears to be replaced after unilateral extirpation of the SPG,[24] perhaps from the sprouting of new nerve fibers from other sources of innervation.
The SPG may be one of the several sources of parasympathetic fibers by which the facial nerve dilates the cerebral arteries. Stimulation of the main trunk of the facial nerve distal to the geniculate ganglion has no effect on cerebral artery dilation,[30] whereas stimulation of the geniculate ganglion dilates the cerebral arteries[6] (since both stimulation sites cause ipsilateral facial movements, it cannot be said that the effect on the cerebral arteries and CBF is related to facial muscle contraction). Anatomical studies also support the parasympathetic projections to the cerebral arteries leaving the facial nerve trunk at or before the geniculate ganglion, such as occurs with the greater superficial petrosal nerve that innervates the SPG. Yet, comparing across various reports that provide quantifiable data would suggest that proximal stimulation of the facial nerve trunk produces larger CBF responses than does distal stimulation of the SPG: stimulation of the facial nerve trunk produced 47–120% increase in CBF over baseline[8,9,11,14] versus a 19–45% increase over baseline after stimulation of the SPG.[7,10,12,13,31] Furthermore, extirpation of the SPG does not eliminate the ability of vagus nerve stimulation to reduce infarct size in a rat ischemic stroke model,[32] wherein the increase in CBF caused by vagus nerve stimulation in normal cat and monkey is completely blockable by transection of the facial nerve trunk[30] [Table 1]. Thus, connections for the parasympathetic facial nerve to the cerebral arteries other than the SPG likely exist, and the preganglionic projections separate from the facial nerve trunk in the region of the geniculate ganglion as the petrosal nerves.
Table 1.
Effect of electrical stimulation of the various cranial nerves on cortical artery diameter in the experiments of Cobb and Finesinger[30]
| Cranial nerve | Number of trials | Number of animals | Results (%) |
|---|---|---|---|
| III | 10 | 3 | 9 with no change (90), 1 constricted (10) |
| V* | 38 | 13 | 38 with no change (100) |
| VII | 147 | 25 | 122 dilated (83), 25 with no change (17) |
| VIII | 25 | 8 | 25 with no change (100) |
| IX | 14 | 6 | 14 with no change (100) |
| X** | 41 | 19 | 33 dilated (80), 8 with no change (20) |
| With VII transected | 20 | 5 | 20 with no change (100) |
| Trunk stimulated distal to transection | 20 | 5 | 20 with no change (100) |
| Trunk stimulated proximal to transection | 22 | 4 | 19 dilated (86), 3 with no change (14) |
| XI | 42 | 11 | 42 with no change (100) |
| XII | 17 | 5 | 17 with no change (100) |
Cats (n=25) and monkeys (n=4) were used. The stimulation parameters and use of anesthesia were not welldescribed in the manuscript. *Several studies have since shown that trigeminal stimulation causes cerebral artery vasodilation, but it appears that this effect is dependent largely upon a brainstem reflex that has as its efferent loop the facial nerve[9] **Note the relevance of the findings to reports of vagal nerve stimulator as a potential treatment for ischemic stroke, as per[87]
To that point, some animals including monkeys and man[18,33,34,35] exhibit an internal carotid ganglion in the cavernous carotid region that has fibers and neuron cell bodies within it expressing parasympathetic markers. The internal carotid ganglion – sometimes referred to as a “mini-ganglion” or an ectopic SPG – receives innervation from the greater superficial petrosal nerve and projects to the intra- and extracranial internal carotid artery; projections from the internal carotid ganglion also appear to reach into the circle of Willis and its branches in man[35] but not in rat.[36,37] Similar appearing ganglia known as cavernous ganglia also exist within the trabeculae of the cavernous sinus.[35] Furthermore, the otic ganglion, which also receives parasympathetic projections from the facial nerve (as well as glossopharyngeal nerve) sends projections to the cerebral arteries in cat[38] and rat.[34] Projections from the internal carotid, cavernous, and otic ganglia might account at least in part for the incomplete loss of VIP nerve fibers on the anterior circulation after SPG lesions,[24] but other than the otic ganglion's ability to induce vasodilation in and salivation from the parotid gland, the physiological roles for these ganglia have been untested. It is reasonable to suspect that they dilate the cerebral arteries of the anterior circulation[1] if they are physiologically analogous to the SPG, but to our knowledge, this physiological action has never been tested. At best, the otic ganglion may have been retrogradely activated in the experiments of Sato et al.[14] who stimulated the proximal cut end of the lingual nerve (which carries the parasympathetic projections of the facial nerve to the otic ganglion) and observed dilation of the common carotid artery without changes in frontal cortex CBF; since Sato et al. observed that increase in both measures of blood flow after stimulation of the distal end of the cut facial nerve trunk, it may be that the otic ganglion was responsible for the effect on common carotid artery, whereas other ganglia were responsible for the effect on the frontal cortex. SPG stimulation, by comparison, is reported to dilate the intra- and extra-cranial internal carotid artery,[12,39] without report of any effect on the upstream carotid. Though the extent of cerebral artery regulation by the facial nerve, and the territorial responsibilities of the facial nerve's various ganglia over the cerebral arterial tree, are incompletely understood, one might reasonably hypothesize if not conclude outright from these observations that the facial nerve has multiple postganglionic routes to the cerebral arteries and is not limited solely to the SPG.
The otic ganglion does not appear to innervate the posterior circulation[36,37] as some have hypothesized.[39] The parasympathetic innervation of the basilar and vertebral arteries either expresses little VIP or else is itself minimal. In man and monkey, posterior circulation innervation appeared to be derived from the cavernous ganglia and conveyed to the posterior arteries along the abducens nerve.[33,35]
Practically speaking, stimulation of the facial nerve for the purpose of causing cerebral artery dilation appears to be maximally effective around the 10 Hz frequency,[4,8,12,13] a property consistent all the way through the system to the postganglionic nerve fiber acting on ex vivo arteries.[40] The frequency dependency of stimulation may extend to the duration of the CBF response as well, with 10 Hz stimulation producing longer-lasting increases in CBF than 5 or 20 Hz stimulation.[41] Continuous stimulation appears to be more effective than does burst stimulation at increasing CBF,[16] and the CBF response to brief periods (e.g., a minute[8]) of stimulation develops within a matter of tens of seconds. CBF increases with an ipsilateral predominance after unilateral facial nerve stimulation in most animal models[8,16,31] and bilateral stimulation has never been assessed.
Regulation of the extra-cerebral cranial arteries by the facial nerve
The arteries of the dura and those that are extracranial are of potential relevance to brain ischemia because they can provide collateral circulation to the brain.[42] The facial nerve appears to innervate both. Dural arteries (e.g., the meningeal arteries) have in common with cerebral arteries innervation from the facial nerve as well as sympathetic and trigeminal nerves,[43] and dural blood flow is increased after stimulation of a facial nerve trunk that is isolated from the brainstem by transection.[44] In the extracranial tissues, the facial nerve is well-known to increase blood flow to the lacrimal and salivary glands. Other extracranial tissues (e.g. the lip, tongue, branchial musculature, gingiva, and palate) also appear to have parasympathetic innervation that originates from the SPG and otic ganglion.[14,45,46]
The extracranial and dural vasculatures are of relevance to CBF because of the presence of extra- and intra-cranial collaterals that might help supply ischemic brain with added blood flow. The clinical relevance of collaterals including those between the cerebral arteries was originally described by Charcot in the 19th century and was confirmed by more recent research with endovascular clot retrieval procedures.[47] Such collaterals may not be present in all animal species or in all individuals of a species, and they may open and close under various conditions.[48] The effect of neural regulation of collateral blood flow has not, to our knowledge, been studied directly.
Variability in the cerebral artery response to facial nerve stimulation
Cerebral artery dilation and an increase in CBF after facial nerve stimulation had not been seen in all laboratories. Reliably unresponsive CBF measures were obtained by Linder[44] who stimulated the facial nerve trunk in rabbit, and by Busija and Heistad[49] who stimulated the greater superficial petrosal nerve in a cat, although both studies detected physiological responses expected from facial nerve stimulation (e.g., lacrimation and salivation, blood flow increases to the eye and lacrimal gland). Replication of these experimental protocols, and then meticulous stepwise substitution of the animal species, anesthetic, blood flow measure, etc., would likely be illuminating.
More interestingly, the effect of facial nerve stimulation is not absolutely reliable even within a given experimental protocol and quite possibly within an individual experimental subject. One need only considers the early observations of Cobb and Finesinger[30] in cat and monkey, as summarized in Table 1, wherein frequent – but not assured – dilation of superficial cortical arteries was demonstrated after stimulation of the facial nerve trunk. A comparable response rate was observed by Forbes.[50] The variability would appear to be on the part of the facial nerve rather than the cerebral arteries since the same inconsistency is also seen with ocular blood flow measures.[51,52] More modern reports make no mention of the reliability of the effect of facial nerve stimulation on CBF: The effect of facial nerve stimulation is reported as either reliably present or reliably absent. The regularity in modern reports might suggest the resolution of some unknown variabilities in early experimental protocols or, alternatively, the advent of reporting bias.
Interindividual variability in the facial nerve's regulation of the cerebral arteries clearly exists at the anatomical level. The size and number of cavernous ganglia in man, for example, varies widely: In Suzuki and Hardebo's report,[35] three of five autopsy specimens showed only collections of 10–20 cells, whereas the other two specimens had collections of 120–150 cells. An order of magnitude difference in the number of cells in the SPG, internal carotid ganglia, and cavernous ganglia between individuals was also found in monkeys and man.[33,53,54] How anatomical differences impact physiological responses is not known, but it does raise a straightforward hypothesis to explain the interindividual variability.
Finally, intraindividual variability in facial nerve responsiveness may exist. The report of Cobb and Finesinger, and of Forbes, would allow for intraindividual variability, and indeed Cobb and Finesinger speculate on variability in cardiovascular parameters as causing unresponsiveness to facial nerve stimulation. Our own results with noninvasive facial nerve stimulation also demonstrate intraindividual variability. If the facial nerve is the efferent limb in cerebral vascular reflexes with one or more sensory afferent limbs (e.g., trigeminal), there would be no reason to expect those reflex components to be uniformly responsive, just as jaw jerk reflexes are readily modulated by various physiological inputs.[55] The evidence would, in fact, suggest the facial nerve acts in this manner, as discussed in the next section.
Purpose of facial nerve regulation of the cranial vasculature
The natural question arising at this point would be, for what purpose does the facial nerve regulate the cerebral and extracerebral cranial arteries? Some insight into that question can be provided by understanding if the facial nerve modulates CBF during mundane activities, extremis, or both.
There is some indication that the facial nerve is involved in both setting basal CBF and compensating for lost CBF although the latter is more compelling. Lesioning of the SPG or its projections to the cerebral arteries does not appear to affect basal CBF levels chronically,[17,56] whereas acute lesioning produces a drop in CBF despite a simultaneous hypercapnic reaction.[12,57] The acute reaction to denervation may be nothing more than irritation of the damaged neural structures, or it may show that the facial nerve influences resting CBF in a manner that is readily compensated by other influences given sufficient time; supporting the latter possibility is the histological observation that the parasympathetic innervation of the anterior circulation regrows after destruction of the SPG.[24] The rise in CBF caused by stimulation of the facial nerve does not affect neuronal activity or brain tissue metabolism[8,9,10] although it increases the availability of oxygen in the brain tissue[31] indicative of luxury perfusion. Goadsby and Duckworth's study[58] of the mechanism by which the trigeminal nerve increases CBF in anesthetized cat also confirms the dissociation between facial nerve activity and the routine regulation of CBF: They demonstrate that trigeminal ganglion stimulation increases CBF to the contralateral parietal lobe as would be expected because of the sensory input to the somatosensory cortex of the parietal lobe, and importantly that this increase in CBF persists after facial nerve transection that otherwise blocked the increase in CBF to other brain regions.
In extremis, facial nerve stimulation appears to increase or preserve CBF, for example, in rats with SPG lesions subject to cold stress[59] and in rats with hemorrhagic hypotension subject to facial nerve stimulation[44] although these physiological stressors inherently affect cardiovascular performance and hematologic properties in a manner that can confound CBF measures. More relevant challenges for the facial nerve might be injuries limited to the brain, such as ischemic stroke and other conditions of brain ischemia (e.g., traumatic brain injury). The former has been extensively studied (the latter, not at all) as will be described below with the effect of facial nerve stimulation reliably reversing brain ischemia in the absence of systemic changes in cardiovascular performance.
It is important to recognize that stimulation of the facial nerve in normal animals may be endogenously opposed, wherein the opposition could directly inhibit the effectiveness of the facial nerve and/or reflexively activate vasoconstrictive systems. This concept gets back to the inter- and intra-individual variability in response to facial nerve stimulation and it originates with the early experiments of Cobb and Finesinger,[30] who struggled to explain the inconsistent response to facial nerve stimulation in their cat and monkey experiments. As Salanga and Waltz[60] demonstrated in the pentobarbital-anesthetized normal cat, electrical stimulation of the facial nerve trunk increased CBF only when the connection of the trunk to the brainstem was cut proximal to the stimulation site. Without proximal transection, stimulation of the facial nerve trunk produced a decrease in CBF that Salanga and Waltz interpreted to be the summation of dilation caused by the facial nerve and a larger, countervailing constriction caused by another system. Other researchers[8,9,11] also employed proximal transection as a means to show-in-full the effect of facial nerve stimulation on CBF and cerebral artery caliber – sometimes with no success[44,49] – without understanding what brainstem reflexes or reactions they were attempting to block.
One reasonably might interpret the available data as indicative that the facial nerve projections to the cranial arteries serve to protect the brain from injury, the most common forms of which are ischemic or relatively ischemic in nature. In vivo, the facial nerve might serve as the effector end of a brainstem reflex loop in which sensory nerve endings of the trigeminal nerve are first activated in response to tissue damage. Indeed, stimulation of the trigeminal ganglion in cat increases CBF in a manner that is bilateral and blockable in each hemisphere by transection of the respective facial nerve trunk.[58] A brainstem reflex involving these components, such as we suggest here, but operating in a malfunctional manner, is postulated to underlie cluster headaches.[61]
Ultimately, understanding the purpose of why the facial nerve regulates the cerebral arteries and CBF is of lesser importance than recognizing the opportunity of the facial nerve provides to counteract brain injuries in the clinical setting, which will now be reviewed.
Clinical Development of Facial Nerve Stimulator Devices
The development programs for medical devices that stimulate the facial nerve for the treatment of conditions of brain ischemia will now be reviewed. However, before doing so, it deserves mentioning that facial nerve stimulation – specifically, at the SPG – appears to be an effective treatment for at least one vascular headache condition such as cluster headache.[62,63] Cluster headache is well-known to involve dilation of the major cerebral arteries,[64] so treating cluster headache with SPG stimulation, then, would at a glance appear to be paradoxical. Yet, SPG stimulation in cluster headache patients between headache attacks decreases flow velocities in the middle cerebral artery (MCA) as measured by transcranial Doppler ultrasonography, suggesting vasodilation, and it increases infrared spectroscopy measures of cortical perfusion.[65] It may be that the cerebral artery dilation in cluster headache is an epiphenomenon of the condition or even an inadequate reaction to the pathophysiological mechanism of cluster headache, and that dilation in response to SPG stimulation is irrelevant to its therapeutic effect. As reported by Schytz et al.,[65] SPG stimulation at all frequencies caused cerebral artery dilation and an increase in CBF, but only high-frequency stimulation (80–120 Hz) is used to treat cluster headaches whereas low-frequency stimulation (5 Hz) actually triggered cluster headache attacks. At any rate, herein, we focus on the development of facial nerve stimulator devices as treatments for brain ischemia, specifically ischemic stroke, in which restoring lost CBF directly oppose the immediate pathophysiological process and has a strong predictive value for good clinical outcomes.[66]
Invasive facial nerve stimulation as a treatment for brain ischemia
Founded in 2000, the Israeli company BrainsGate has been developing a device known at various times as the Ischemic Stroke System™, Ischemic Stroke System 500, and the NeuroPath™ IS System. The BrainsGate device stimulates the SPG a distal portion of the facial nerve and is in late-stage development as a treatment for ischemic stroke. This review of the BrainsGate program is based on the publicly available information; BrainsGate did not reply to our requests to coauthor this review.
Use of the BrainsGate device involves placement of a needle-shaped electrode into the greater palatine canal of the hard palate at the back of the throat [Figure 2][93] through which electrical current can be delivered to the SPG. The electrical current is created in the electrode by means of magnetic energy delivered through the cheek from a transmission coil. The electrode takes 20 min to implant by one report from BrainsGate[67] despite the fact that the procedure involves establishing a sterile environment and neuronavigation processes [Figure 2]. The electrode is implanted unilateral to the side of infarction and only in cases where the anterior circulation is involved. Promotional materials from BrainsGate investors[94] state the surgical procedure will be performed by neurologists although a current BrainsGate trial focused on treatment safety necessitates that only “certified implanters” use their device,[95] suggesting some need for specialized training.
Figure 2.
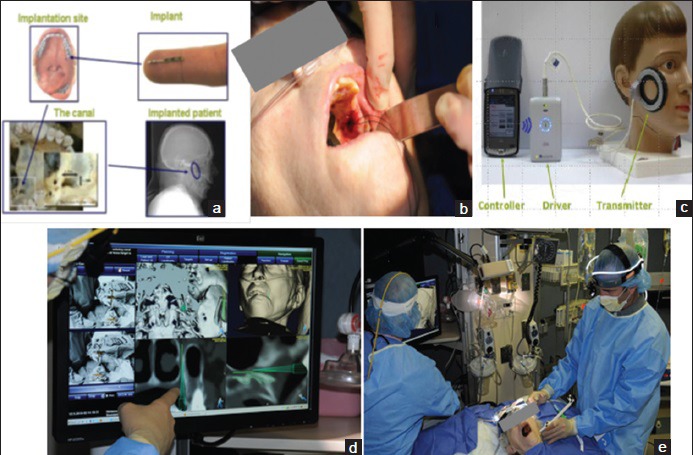
The BrainsGate device. (a) General anatomy of implantation site in the hard palate and the electrode. (b) Actual surgical implantation. (c) Transduction coil and control equipment. (d) Neuronavigation processing. (e) Surgical team including neuronavigation system in the Intensive Care Unit[93]
The concept of the BrainsGate device is, of course, strongly supported by the numerous studies described previously examining electrical stimulation of the SPG in normal animals. The concept of the BrainsGate device is further supported by the repeated observation that lesions of the SPG or its efferents to the cerebral arteries allow for larger infarct volumes in rat stroke models,[68,69,70] a finding that also supports the proposed natural protective role for the SPG against brain ischemia. BrainsGate itself sponsored preclinical studies of SPG stimulation as a treatment for ischemic stroke that are technically comparable to studies of direct electrical stimulation of the SPG by way of cranial dissection. SPG stimulation improved CBF and reduced infarct volumes in rat ischemic stroke models of transient[71] or permanent[72] MCA occlusion and of photothrombosis.[73] Improved neurological function in the rat transient[74] and permanent[75] MCA occlusion models was also reported in abstracts, and the results of a separate set of rat permanent MCA occlusion experiments published as a peer-reviewed manuscript[71] showed more rapid improvement in neurological function in SPG-stimulated rats. The stimulation parameters in the BrainsGate-sponsored rat studies appear to include 10 Hz frequency administered nearly continuously for 3 h daily over 4–7 days, with the therapeutic window ranging from immediately poststroke to 24 h poststroke.
After successful testing of SPG stimulation in its rodent models of ischemic stroke, BrainsGate pursued an additional study in the dog double-hemorrhage model of cerebral artery vasospasm.[39] Ipsilateral electrical stimulation was delivered to the SPG through a lateral temporal surgical exposure using 10 Hz pulses. The first set of three 90-s stimulation periods was delivered with each stimulation period separated by 60 s, and 30 min after completion of the first set, a second set of three stimulation periods was delivered. The assessment of the SPG stimulation on the vasospastic arteries was acute: Angiography was performed after each 90-s stimulation period and compared against prestimulation. Dilation of the vasospastic anterior and middle cerebral arteries as well as the nonvasospastic internal carotid artery as far as its extracranial portion was observed with the most pronounced effect of SPG stimulation occurred in arteries with the most severe vasospasm [Figure 3]. However, the arteries appeared to revert to the vasospastic state within 15 min of the cessation of stimulation. A comparable study was later conducted in the same laboratory using the monkey model of cerebral artery vasospasm,[76] and it confirmed a transient reduction in angiographic vasospasm severity after SPG stimulation. The monkey study was supported by Boston Scientific, which shortly thereafter invested in BrainsGate.
Figure 3.
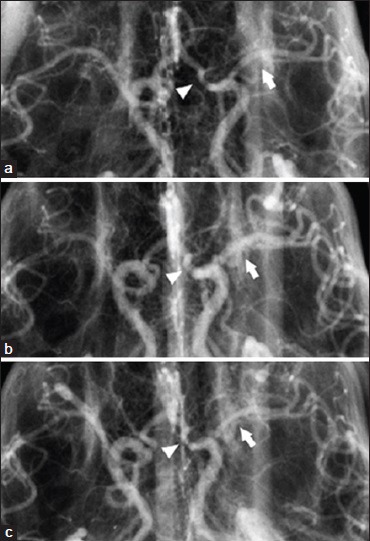
Effect of electrical stimulation of the sphenopalatine ganglion on cerebral arteries in the dog double-hemorrhage model of cerebral artery vasospasm.[39] (a) Anteroposterior cerebral angiograms from a dog before stimulation of the sphenopalatine ganglion; (b) during the second series of stimulations; and (c) 30 min after completion of the second series of stimulation. Arrow = middle cerebral artery, arrowhead = anterior cerebral artery
The subsequent clinical development of the BrainsGate device has accumulated a wealth of data on the effectiveness of facial nerve stimulation as a medical therapy for ischemic stroke, but that data have largely been undisclosed to the scientific and medical community. A safety study of the BrainsGate device, the IMPlant Augmenting Cerebral Blood Flow Trial (ImPACT)-1, was begun in 2006 and completed in 2008. While the design of the ImPACT-1 study has been published,[77] the results of the study have not been published save for an abstract of the interim analysis of the first 48 patients.[67] The abstract reports 23 of 48 patients completing the treatment and no deaths related to the treatment. The only adverse events reported were recurrent stroke with hemorrhagic transformation (n = 1), symptomatic brain edema (n = 1), wound dehiscence at the electrode implantation site (n = 2), and “malpositioning” presumably of the electrode (n = 2). In regard to treatment efficacy, the abstract reports a significant effect of treatment on “the range” of modified Rankin scale scores in a historical comparison against the 1995 NINDS rt-PA trial control group. Other efficacy data from the ImPACT-1 study were recently released in a slide deck posted online by one of the study sites[94] [Figure 4a]: In comparison with the historical control data from the NINDS rt-PA trial, NIHSS scores of neurological function appear to be positively influenced by the BrainsGate device in a group of 99 patients, presumably all of whom actually received treatment. “Positive outcome” on the NIHSS in the BrainsGate analysis is assumed to be the same as “favorable outcome” (NIHSS ≤1) in the NINDS rt-PA trial report.[78] If so, a comparison of the ImPACT-1 data against more contemporary control patient groups from trials such as ECASS-3[79] (also completed in 2008) would have suggested a smaller margin of efficacy [Figure 4b] although little is known about the poststroke treatments of the ImPACT-1 study in comparison with ECASS-3. However, in either comparison, the effect of the BrainsGate device appears promising.
Figure 4.
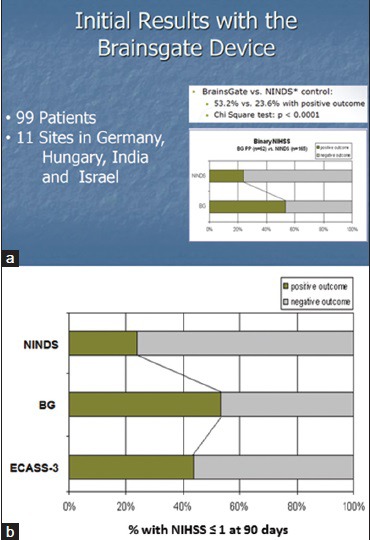
Results of the ImPACT-1 study. (a) Results comparing NIHSS scores against NINDS rt-PA trial control group. (b) Including results from a clinical trial control group (ECASS-3) concurrent with the ImPACT-1 study. BG: BrainsGate, ImPACT-1: IMPlant Augmenting Cerebral Blood Flow Trial 1
Encouraged by the ImPACT-1 study, BrainsGate pursued further clinical testing of their device although the nature of the subsequent clinical studies is not clear. A 2013 investor report stated “in 2010 BrainsGate completed a clinical trial on approximately 300 patients,” the results of which “strengthened the assessment that BrainsGate's treatment has a considerable clinical effect… stronger than the effect found in current treatments.”[89] Certainly, the analysis of scientific data by a financial investor should be considered skeptically, and it must be so considered when it is put forth in a public document to encourage further investment; yet, the ≈300 patient clinical trial was followed at the time by a Series C investment of nearly $20 M into BrainsGate, suggesting success.
The ≈300 patient clinical trial must be a free-standing trial since the 2013 investor report lists immediately under the ≈300 patient clinical trial the enrollment of “287 patients at 63 medical centers in the United States, Europe, and Asia” in “an additional clinical study… which is expected to support BrainsGate's planned PMA submission to the FDA.”[90] A 480-patient randomized controlled trial identified as “ImPACT-24” was first listed in January 2009 under the ClinicalTrials.gov identifier NCT00826059.[90] The NCT00826059 trial appears to have multiple interim analyses embedded in it, beginning with an interim analysis that was successfully completed in April 2014, when “approximately 440 patients” had been enrolled, per the SEC report of one of BrainsGate's investors[91] and the BrainsGate website.[92] Another interim analysis was planned for 600-enrolled patients, which at the time of the disclosure was expected to occur in 2015.[93] The 2015 investor report[94] confirms enrollment of the 600 patients but does not provide insight on the results of the interim analysis despite an obviously optimistic assessment of BrainsGate's ongoing clinical trial. At any rate, a clinical trial continues enrollment to this date under the original ClinicalTrials.gov identifier NCT00826059, but the trial is now unnamed.[95]
The ongoing NCT00826059 clinical trial is designed to assess the BrainsGate device in patients who had ischemic stroke limited to the anterior circulation territory and who are not candidates for rt-PA or endovascular clot retrieval catheter treatments. The side of infarction must be identified by early neuroimaging before the treatment because the BrainsGate device is applied unilaterally. The stimulation parameters employed in the clinical trial appear to be 10 Hz delivered for 4 h on each of 5 consecutive days.[94] A therapeutic window of 8-24 h poststroke is being employed, perhaps in part because treatment times <8 h operationally conflict with rt-PA and endovascular catheters procedures.
In 2013, during the ongoing conduct of the NCT00826059 clinical trial, BrainsGate initiated a new randomized controlled trial called IMPACT-24Bt (ClinicalTrials.gov identifier NCT01874093). The IMPACT-24Bt trial was designed to enroll 150 ischemic stroke patients who are treated with rt-PA (which is exclusion criteria in NCT00826059 trial), but otherwise patients are enrolled according to the inclusion and exclusion criteria of the NCT00826059 trial. The IMPACT-24Bt clinical trial is strongly focused on hemorrhagic safety concerns, namely hemorrhage from the electrode implantation site in the oral cavity and symptomatic intracranial hemorrhage. It is not clear why the IMPACT-24Bt trial was started at a time when the lead NCT00826059 trial was midway through its 800-1000 patient enrollment target. One explanation is that the IMPACT-24Bt clinical trial reflects BrainsGate's confidence in their device as a treatment for a broad range of ischemic stroke patients as would be suggested by investor materials that optimistically estimate 80% of all ischemic stroke patients to be treatable by the BrainsGate device.[94] The purpose of the IMPACT-24Bt clinical trial might then be to broaden clinical use of the BrainsGate device, and perhaps initiation of the IMPACT-24Bt trial was triggered by success at one of the interim analyses. Either sort of hemorrhage should not be surprising in a clinical trial of a surgically implanted device that treats ischemic stroke, and intracranial hemorrhage in particular might be expected in the NCT00826059 clinical trial if the time to first treatment with the BrainsGate device is comparable to that achieved in ImPACT-1 (i.e., 17.7 h[67]) because the rate of intracranial hemorrhage in ischemic stroke patients is known to increase as the time to reperfusion becomes longer.[80] Further along these lines, SPG stimulation appears to increase blood-brain barrier permeability,[21,22,23] which might promote intracranial hemorrhage in ischemic stroke patients.[81]
Noninvasive facial nerve stimulation as a treatment for brain ischemia
NeuroSpring and its for-profit research partner, Nervive, are developing a noninvasive medical device for the emergency treatment of conditions of brain ischemia known as the VitalFlow™ stimulator. Specifically, the VitalFlow stimulator is being developed as a treatment for acute ischemic stroke.
The basic concept of the VitalFlow stimulator is to position magnetic stimulation coils on the sides of the head from which magnetic fields can be generated, and to direct the magnetic fields at the portion of the facial nerve located deep to the structures of the middle ear [Figure 5]. The anatomical target of the facial nerve – the geniculate ganglion region/labyrinthine segment of the nerve - is in-line with the long axis of the ear canal. Therefore, the ear canal provides ready anatomical guidance for the orientation of the stimulation coils, and an ear plug placed on the patient facing surface of the stimulation coil is employed to take advantage of this anatomy. Other advantages of stimulating the facial nerve at the geniculate ganglion region are the larger effect the proximal stimulation has on CBF responses, and the greater reliability of activating the nerve afforded by the multiple turns the nerve takes at that point (i.e., bends in nerves are more susceptible to activation by magnetic stimulation than are straight portions of the same nerve[88]).
Figure 5.
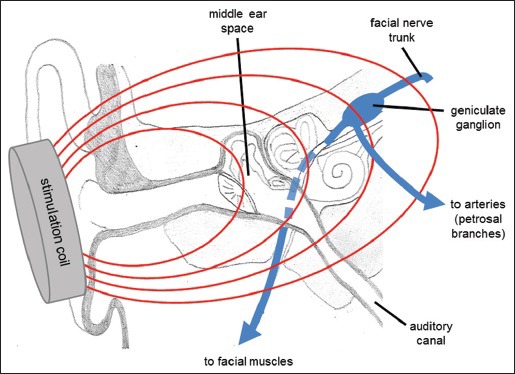
Anatomical relationship of the ear structures and the facial nerve. The ear canal provides anatomical guidance toward the branching point of the facial nerve's projections to the facial muscles and the autonomic parasympathetic projections of the petrosal branches, which separate in the region of the geniculate ganglion
In collaboration with the Sutter Research Institute in Sacramento, California, we demonstrated that stimulation of the facial nerve noninvasively with a pulsed magnetic field increases CBF and dilates the cerebral arteries. In experiments with anesthetized dogs and sheep, we positioned a prototype of the VitalFlow stimulator made from modified commercially available transcranial magnetic stimulation (TMS) equipment over the ear so that movements of the facial muscles could be reliably produced.2 Stimulation of the facial nerve for a period of 5 min was then administered, and in response increases in CBF and dilation of the cerebral arteries were observed in all 6 sheep and 4 dogs tested. These results were published in “Brain Research” in 2013.[41]
The CBF response to stimulation occurred frequently but not uniformly, and when it did occur the increase in CBF typically outlasted the 5 min period of stimulation by 1–2 min. However, in some trials, the stimulator's position resulted in an increase in CBF that outlasted the stimulation by 20-30 min. The response to facial nerve stimulation appeared to be dependent upon the position of the stimulation coil, yet a degree of the variability described by Cobb and Finesinger and by Forbes could easily be hidden therein. Examples of prolonged CBF responses and the angiographic response to magnetic stimulation of the facial nerve are shown in Figure 6. Furthermore, the size of the CBF response was related to stimulation power and frequency as has been predicted for magnetic stimulation of a physiological system,[18] with 10 Hz stimulation frequency outperforming higher or lower frequencies. More recent studies in anesthetized normal pig have shown that magnetic facial nerve stimulation for 2 min increased CBF as much as did 5 min of stimulation (around 60% over baseline) but more than did 1 min of stimulation, and that the effect of stimulation lasts about 2 h and is serially reproducible without tachyphylaxis (submitted for publication). In that study, which employed neuronavigation to ensure proper positioning of the stimulation coil, we again observed an occasional unresponsiveness to stimulation, with about 15% of stimulation trials producing less than a 20% rise in CBF. Interestingly, as supposed by the data of Cobb and Finesinger, we found that a lack of response to magnetic facial nerve stimulation in one trial did not preclude an individual pig from demonstrating a large CBF response when subject to the same stimulation parameters on another day.
Figure 6.
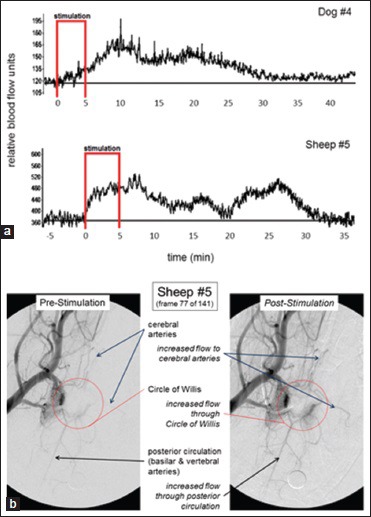
Prolonged effect of magnetic facial nerve stimulation on cerebral blood flow and cerebral arteries in normal dog and sheep. (a) Frontal cortex cerebral blood flow responses as measured by laser Doppler flowmetry. (b) Anteroposterior cerebral angiograms[41]
Our experiments in normal animals suggested the CBF response to facial nerve stimulation is rather all-or-none in nature, and that it is not going to be readily titrated to the desired level of perfusion or vasodilation. Whether or not this is an inherent restriction of the physiological system or of limited experimental resources is unclear. Considering that there are at least 5 stimulation parameters that can be readily adjusted (stimulation power, frequency, waveform shape, stimulation pattern, stimulation duration), and simplistically assuming that each parameter would have only 3 possible settings, an animal study of the various stimulation parameter combinations would need 243 groups, which at 5 animals per group would total to an astounding 1200 + animal study. That is not going to happen, particularly if the experiments have to be done in large animals because magnetic stimulation technology cannot be scaled-down proportionate to the size of rodents. A more realistic way to address this issue would be in clinical use of the VitalFlow technology as part of a long-term refinement of the VitalFlow stimulator's use in the postmarketing stage.
Based on these observations in normal (nonstroke) animals, we hypothesized that pulsed magnetic stimulation of the facial nerve would improve CBF in ischemic brain when tested in animals with ischemic stroke. We first tested the effectiveness of magnetic facial nerve stimulation in a validated dog model of ischemic stroke.[21,22] In those experiments, conducted in partnership with the National Center for Medical Imaging and Instrumentation Research (CI3M) at the Metropolitan University of Mexico City, the MCA was occluded by injection of autologous blood clots through an angiography catheter inserted into the distal internal carotid in anesthetized dogs. Occlusion of the MCA was confirmed on angiography and loss of CBF was confirmed by contrast perfusion MRI. Then, 30–90 min later, magnetic facial nerve stimulation was administered ipsilateral to the side of infarction and thereafter CBF was repeatedly measured with contrast perfusion MRI over 1 h. Neuronavigation-guided placement of the stimulation coil in those experiments, and the geniculate ganglion was employed as the target for the magnetic field. By stimulating the facial nerve trunk in this manner, CBF was restored immediately to nearly prestroke levels [Figure 7], an effect observable in 11 dogs treated with VitalFlow stimulation in comparison with 6-unstimulated dogs. The effect on CBF translated into the preservation of brain tissue: Using established tissue perfusion thresholds, volumetric analysis of the change in tissue perfusion demonstrated that VitalFlow stimulation reduced core volume in a durable manner. The improvement in CBF to the ischemic brain did not occur at the expense of surrounding brain regions or the contralateral brain, which also exhibited increased CBF versus prestimulation levels. We also did not observe any edema formation or hemorrhagic conversion in stimulated dogs. These results were published in Stroke in 2014.[82]
Figure 7.
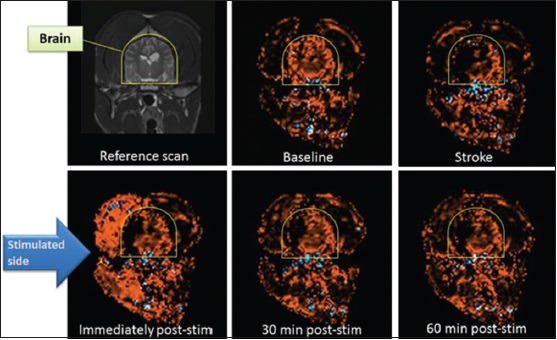
Effect of magnetic facial nerve stimulation on cerebral blood flow in dogs with right middle cerebral artery occlusion caused by an autologous blood clot. Coronal section, cerebral blood flow measured by contrast-perfusion magnetic resonance imaging. Image panels are sequential from top to bottom, left to right[82]
What is more, we have examined the effect of facial nerve stimulation on stable hematoma size after rupture of the intracranial internal carotid artery. In our model, a Tuohy needle was sterotaxically advanced through the frontal cortex to puncture the internal carotid artery; the puncture site was later confirmed by autopsy. Hematoma size was assessed using repeated MRI studies and facial nerve stimulation was administered once the hematoma was stable in size. Stimulation was administered using parameters applied in the dog ischemic stroke study described above. In 3 dogs assessed qualitatively, no enlargement of the hematoma was observed after facial nerve stimulation using parameters effective against ischemic stroke [Figure 8]. Furthermore, CBF did not obviously increase and unlike normal animals and animals with ischemic stroke, we did not see an increase in extracranial blood flow despite the expected movements of the facial musculature [compare bottom left panel of Figure 7 with bottom panel of Figure 8].
Figure 8.
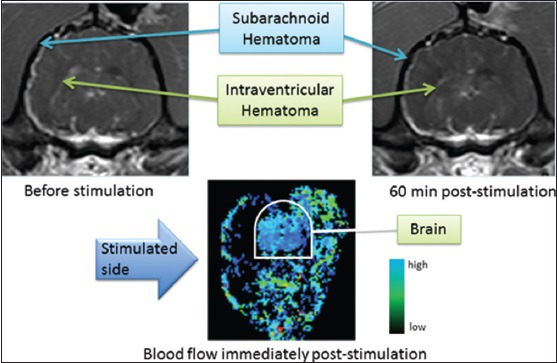
Effect of magnetic facial nerve stimulation on hematoma volume and cerebral blood flow in a dog with rupture of the intracranial internal carotid artery. Top panels, T1 magnetic resonance imaging demonstrating subarachnoid and intraventricular hemorrhage immediately before stimulation (left) and 60 min after stimulation with parameters as used in dogs with ischemic stroke, as in Figure 7[82]
The surprising observation in dogs with ruptured internal carotid arteries led us to undertake a series of safety studies, the first of which modified the dog hemorrhage model to use pigs with a ruptured MCA. A randomized, sham-stimulated control design was employed in 10 pigs (submitted for publication). The pig experiments assessed the following physiologic measures: Hematoma volume, intracranial pressure, and brain perfusion index. As in the dogs, magnetic facial nerve stimulation was delivered after hemorrhage induction in the pigs and at parameters that induce cerebral artery dilation and increase CBF in normal animals and animals with ischemic stroke. In comparison with sham stimulation, magnetic facial nerve stimulation in pigs did not further increase hematoma volume, but it did reverse a transient reduction of CBF that may have represented acute vasospasm caused by arterial irritation. Importantly, an increase in intracranial pressure of about 7 mmHg was observed immediately upon completing VitalFlow stimulation that resolved within 5 min after stimulation was completed. The increase in intracranial pressure was considerably shorter than the effect on CBF suggesting that the two are unrelated. Instead, the increase in intracranial pressure may reflect an artifact of head vibration caused by repetitive facial muscle contraction,[83] and to support that interpretation, stimulation of the SPG does not cause an increase in intracranial pressure or induce facial muscle contraction.[76] Nevertheless, an increase in intracranial pressure is concerning and may underestimate the effect since the measure was made outside of the stimulation period and with an subdural monitor.[84]
Recently, we began a study of the effect of magnetic facial nerve stimulation in rabbits with embolism-induced stroke, a model that causes ischemic stroke with a high rate of mortality and not infrequent hemorrhagic conversion and outright intracranial hemorrhage. Use of intravenous rt-PA is well-established in the rabbit model, and we included it as a treatment arm in a 2 × 2 design to assess the effectiveness of each treatment individually and in combination against an untreated control group. The importance of this ongoing study will be to confirm the finding of effectiveness of VitalFlow stimulation in a second animal species and to assess the potential interaction of VitalFlow stimulation and rt-PA in an animal model that commonly involves ‘spontaneous’ intracranial hemorrhages. After an interim analysis of 38 of the 170 total rabbits, we found no increase in intracranial hemorrhage or mortality after combined use of rt-PA and VitalFlow stimulation. Rather, a single VitalFlow stimulation improved neurological scores to a degree comparable to rt-PA,3 and the two treatments together acted synergistically [Figure 9]. To achieve definitive results, however, the rabbit study will need to progress to completion.
Figure 9.
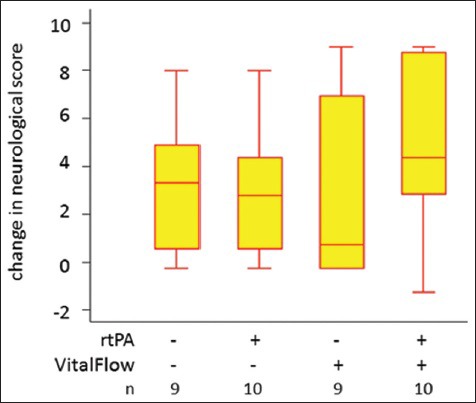
VitalFlow stimulation with and without rt-PA treatment in the rabbit ischemic stroke model. Box plots of the change in a 10-point neurological exam score[98] from poststroke/prestimulation to 2 days poststimulation, with larger numbers reflecting greater improvement. The variability in response to facial nerve stimulation can again be seen. In many animals treated with a single VitalFlow stimulation only, little improvement in neurological score was observed 2 days later, but in some animals, the treatment produced very large improvements. It may be that, in some animals, the transient vasodilation produced turbulent blood flow around the occlusive blood clot that facilitated clot breakdown and recanalization
In terms of product development of the VitalFlow for clinical use, Nervive has developed a novel magnetic stimulation coil design (the “Koala” coil) that allows for effective stimulation of the facial nerve in the temporal bone with 70% less stimulation of the overlying brain in comparison with commercially available TMS coils [Figure 10]. Reduced brain exposure to magnetic energy decreases the potential to induce adverse events in the acutely brain-injured patient although the general safety of TMS of the brain is supported by the medical literature (as comprehensively reviewed by Rossi et al.[85]) and by FDA guidance documents.[99] A particular concern with VitalFlow stimulation would be the potential to induce or increase seizure activity by nonspecific stimulation of injured brain. We addressed that concern in a safety study in rats and pigs with chemically induced status epilepticus, in which the epileptiform activity was not made worse by magnetic facial nerve stimulation, but rather could often be reduced (submitted for publication). Nevertheless, the Koala coil design should minimize the concern about nonspecific brain stimulation in clinical use of the VitalFlow.
Figure 10.
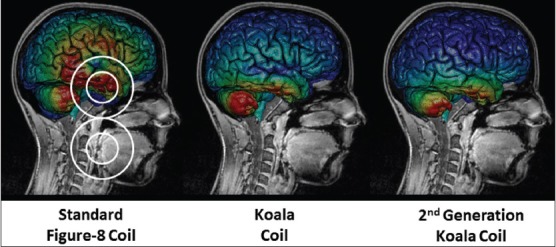
Computer modeling of induced electrical currents in the human brain caused by magnetic facial nerve stimulation using various coil designs. The magnetic field focus is directed at the geniculate ganglion portion of the facial nerve, which resides in the temporal bone underneath the brain, and the magnetic field strength at the facial nerve target it set at a constant level for all stimulation coils. Figure 8 coil outline shown; Nervive's Koala stimulation coil designs are considered proprietary and are not shown
Koala coils have been incorporated into a headrest that can be placed underneath a person's head so that the coils can be positioned over both the ears using adjustable positioning arms [Figure 11]. Though we continue to use neuronavigation for the time being, we intend that in clinical use the VitalFlow stimulation coils will be positioned in a manner based on external head anatomy. Computer modeling using high-resolution reconstructions of a human head conducted by Nervive indicates that centering the Koala coil by means of an ear plug placed into the ear canal, and rotating the coil's stimulation axis to align it with the zygomatic prominence, would optimally orient the magnetic field generated by the coil relative to the target region of the facial nerve and to the axis of the nerve as it runs through the temporal bone. The positioning process will be validated on normal subjects and patients in upcoming clinical studies in comparison to neuronavigation-assisted coil placement.
Figure 11.
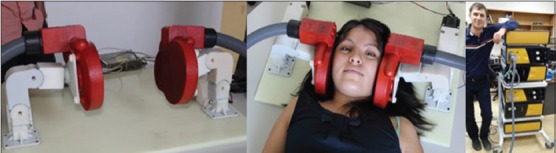
The clinical prototype VitalFlow stimulator. Koala stimulation coils (red) are positioned against the sides of the head of a subject under neuronavigation guidance in the current use of the device
The clinical prototype VitalFlow has been successfully employed in early clinical studies. The clinical prototype VitalFlow stimulates the facial nerve bilaterally under the assumption that two is better than one, and that the VitalFlow will be used to treat ischemic stroke patients in a very rapid manner that would not allow for the side of infarction to be determined in advance by neuroimaging. An initial study in 35 normal subjects with the clinical prototype VitalFlow demonstrated safety, tolerability, and efficacy at increasing CBF.[97] Stimulation power was gradually increased with the permission of the subject up to a power that could be tolerated by the subject for a 3-min period, and with gentle encouragement the subjects could readily tolerate stimulation powers of 80–90% of the maximum device output (corresponding to 1.6–1.8 Tesla magnetic field strength at coil surface). The most common side effects of stimulation were jaw pain or soreness (n = 14), sweating (n = 12), metallic taste sensation (n = 4), neck pain or soreness (n = 3), visual flashes (n = 3), and nausea (n = 2). All side effects were transient and limited to the 3-min period of stimulation. No injury to auditory or vestibular function was found on electrophysiological testing nor did intraocular pressure increase after VitalFlow stimulation. CBF measured 10 min after completion of stimulation at 80–90% power was 32 ± 6% (mean ± standard error of the mean) above baseline.
Since the clinical prototype VitalFlow appeared safe and tolerable in normal subjects, a study in patients suffering from delayed cerebral artery vasospasm (“vasospasm”) after subarachnoid hemorrhage was initiated at the National Institute of Neurology and Neurosurgery in Mexico City. The purpose of the pilot vasospasm study is to obtain proof of concept that the VitalFlow can cause vasodilation in a stroke-like condition of brain ischemia. At the time of this publication, 3 SAH patients with angiography confirmed cerebral artery vasospasm have been stimulated with the VitalFlow using 10 Hz pulses delivered for 3 min at 80% power. Patients with Fisher Grade 3 SAH are being enrolled on the 3rd or 4th day posthemorrhage, at a time after the aneurysm is surgically controlled. Before VitalFlow stimulation, all patients routinely received vasospasm prophylaxis including maintenance of euvolemia and systemic nimodipine. The development of vasospasm is monitored with daily transcranial Doppler ultrasonography, and when flow velocities achieved velocities consistent with probably vasospasm, the patient is evaluated with catheter angiography using a Zeego angiography system (Siemens).
While on the angiography table, and after confirmation of vasospasm defined by >50% constriction of one or more cerebral arteries, VitalFlow stimulation is delivered as described above. After VitalFlow stimulation, no adverse events have been reported by the 3 patients stimulated to date, all of whom were sedated per the routine of the angiography procedure. All three patients demonstrated dilation of the vasospastic cerebral arteries [Figure 12] on average by 32% from the prestimulation diameter. Moreover, aside from the relief of the vasospastic arteries, VitalFlow stimulation diffusely dilated unaffected cerebral arteries, increasing their diameters by an average of 13%. Similarly, the time to peak for contrast signal was reduced by 0.5 s across 18 measured arteries. Such results appear to us as quite promising for the VitalFlow technology as a treatment for conditions of brain ischemia.
Figure 12.
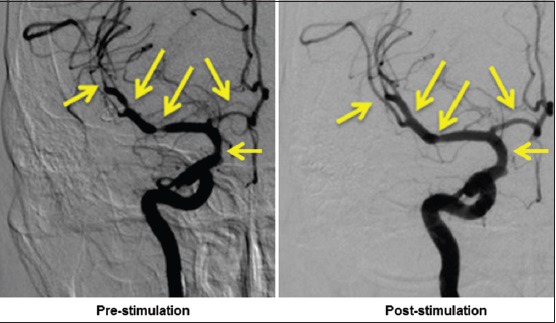
The clinical prototype VitalFlow causes cerebral artery dilation in a subarachnoid hemorrhage patient who developed vasospasm. Anteroposterior view. Left: Prestimulation; right: Poststimulation. Automated pressure-and volume-controlled contrast injection
Summary and Future Directions
Facial nerve stimulation increases CBF in a manner that appears to reverse brain ischemia and improve neurological function. This is well-documented in the preclinical setting, and can be inferred from information available on BrainsGate's invasive facial nerve stimulation device that is in late-stage clinical testing as a treatment for ischemic stroke patients. Successful development of a facial nerve stimulator would represent the first fundamentally new therapy for ischemic stroke in 20 years.
Noninvasive facial nerve stimulation as an emergency treatment for ischemic stroke may offer considerable benefit to patients if it can be rapidly applied without much need for diagnostic testing or the involvement of specialized healthcare practitioners. Our preclinical findings with the VitalFlow stimulator suggest that acute facial nerve stimulation is safe in the context of intravenous rt-PA treatment and even ruptured cerebral arteries. A rapid use of facial nerve stimulation close to the time of onset of the ischemic stroke may not have intracranial hemorrhage as a limiting concern, then. In addition, misapplication of facial nerve stimulation to stroke mimics including active seizure may not prove to be a safety concern in the clinical setting, as has even been shown for rt-PA[86] despite its dangerous hemorrhagic complications.[78] If these preclinical findings were to prove true in the clinical setting, and with a simple and straightforward design, a noninvasive facial nerve stimulator such as the VitalFlow might be used for stroke treatment at the earliest opportunity.
Financial support and sponsorship
Nil.
Conflicts of interest
Drs. Borsody and Sacristan are founders of Nervive, Inc., a for-profit company that is developing a noninvasive facial nerve stimulation device.
References
- 1.Dowgjallo N. Contributions to the theory of the innervation of the peripheral vascular system. J Anat. 1932;97:9–54. [Google Scholar]
- 2.Stohr P. On the innervation of the pia mater and the choroid plexus of man. J Anat. 1922;63:562–607. [Google Scholar]
- 3.Mitchell J, Templeton D. The origin of the preganglionic parasympathetic fibres to the mandibular and sublingual salivary glands in the rat: A horseradish peroxidase study. J Anat. 1981;132(Pt 4):513–8. [PMC free article] [PubMed] [Google Scholar]
- 4.Suzuki N, Hardebo JE, Skagerberg G, Owman C. Central origins of preganglionic fibers to the sphenopalatine ganglion in the rat. A fluorescent retrograde tracer study with special reference to its relation to central catecholaminergic systems. J Auton Nerv Syst. 1990;30:101–9. doi: 10.1016/0165-1838(90)90133-4. [DOI] [PubMed] [Google Scholar]
- 5.Nakai M, Tamaki K, Ogata J, Matsui Y, Maeda M. Parasympathetic cerebrovasodilator center of the facial nerve. Circ Res. 1993;72:470–5. doi: 10.1161/01.res.72.2.470. [DOI] [PubMed] [Google Scholar]
- 6.Forbes HS, Nason GI, Cobb S, Wortman RC. Cerebral circulation. XLV. Vasodilation in the pia following stimulation of the geniculate ganglion. Arch Neurol Psychiatry Chic. 1937;37:776–81. [Google Scholar]
- 7.D’Alecy LG, Rose CJ. Parasympathetic cholinergic control of cerebral blood flow in dogs. Circ Res. 1977;41:324–31. doi: 10.1161/01.res.41.3.324. [DOI] [PubMed] [Google Scholar]
- 8.Goadsby PJ. Characteristics of facial nerve-elicited cerebral vasodilatation determined using laser Doppler flowmetry. Am J Physiol. 1991;260(1 Pt 2):R255–62. doi: 10.1152/ajpregu.1991.260.1.R255. [DOI] [PubMed] [Google Scholar]
- 9.Goadsby PJ. Effect of stimulation of facial nerve on regional cerebral blood flow and glucose utilization in cats. Am J Physiol. 1989;257(3 Pt 2):R517–21. doi: 10.1152/ajpregu.1989.257.3.R517. [DOI] [PubMed] [Google Scholar]
- 10.Goadsby PJ. Sphenopalatine ganglion stimulation increases regional cerebral blood flow independent of glucose utilization in the cat. Brain Res. 1990;506:145–8. doi: 10.1016/0006-8993(90)91211-x. [DOI] [PubMed] [Google Scholar]
- 11.Goadsby PJ, Hoskin KL. Cerebral blood flow is not coupled to neuronal activity during stimulation of the facial nerve vasodilator system. Brain Res. 1994;647:192–8. doi: 10.1016/0006-8993(94)91317-x. [DOI] [PubMed] [Google Scholar]
- 12.Toda N, Ayajiki K, Tanaka T, Okamura T. Preganglionic and postganglionic neurons responsible for cerebral vasodilation mediated by nitric oxide in anesthetized dogs. J Cereb Blood Flow Metab. 2000;20:700–8. doi: 10.1097/00004647-200004000-00007. [DOI] [PubMed] [Google Scholar]
- 13.Toda N, Tanaka T, Ayajiki K, Okamura T. Cerebral vasodilatation induced by stimulation of the pterygopalatine ganglion and greater petrosal nerve in anesthetized monkeys. Neuroscience. 2000;96:393–8. doi: 10.1016/s0306-4522(99)00557-6. [DOI] [PubMed] [Google Scholar]
- 14.Sato M, Izumi H, Karita K, Iwatsuki N. Comparative effects of lingual and facial nerve stimulation on intracranial and extracranial vasomotor responses in anesthetized cats. Tohoku J Exp Med. 1997;182:103–13. doi: 10.1620/tjem.182.103. [DOI] [PubMed] [Google Scholar]
- 15.Forbes HS, Schmidt CF, Nason GI. Evidence of vasodilator innervation in the parietal cortex of the cat. Am J Physiol. 1939;125:216–9. [Google Scholar]
- 16.Suzuki N, Hardebo JE, Kåhrström J, Owman C. Selective electrical stimulation of postganglionic cerebrovascular parasympathetic nerve fibers originating from the sphenopalatine ganglion enhances cortical blood flow in the rat. J Cereb Blood Flow Metab. 1990;10:383–91. doi: 10.1038/jcbfm.1990.68. [DOI] [PubMed] [Google Scholar]
- 17.Suzuki N, Gotoh F, Gotoh J, Koto A. Evidence for in vivo cerebrovascular neurogenic vasodilatation in the rat. Clin Auton Res. 1991;1:23–6. doi: 10.1007/BF01826054. [DOI] [PubMed] [Google Scholar]
- 18.Chorobski J, Penfield W. Cerebral vasodilator nerves and their pathway from the medulla oblongata. Arch Neurol Psychiatry. 1932;28:1257–89. [Google Scholar]
- 19.Suzuki N, Hardebo JE, Kåhrström J, Owman C. Neuropeptide Y co-exists with vasoactive intestinal polypeptide and acetylcholine in parasympathetic cerebrovascular nerves originating in the sphenopalatine, otic, and internal carotid ganglia of the rat. Neuroscience. 1990;36:507–19. doi: 10.1016/0306-4522(90)90001-7. [DOI] [PubMed] [Google Scholar]
- 20.Hilton SM, Lewis GP. The cause of the vasodilatation accompanying activity in the submandibular salivary gland. J Physiol. 1955;128:235–48. doi: 10.1113/jphysiol.1955.sp005302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yarnitsky D, Gross Y, Lorian A, Shalev A, Lamensdorf I, Bornstein R, et al. Blood-brain barrier opened by stimulation of the parasympathetic sphenopalatine ganglion: A new method for macromolecule delivery to the brain. J Neurosurg. 2004;101:303–9. doi: 10.3171/jns.2004.101.2.0303. [DOI] [PubMed] [Google Scholar]
- 22.Yarnitsky D, Gross Y, Lorian A, Shalev A, Shorer S, Tanaka T, et al. Increased BBB permeability by parasympathetic sphenopalatine ganglion stimulation in dogs. Brain Res. 2004;1018:236–40. doi: 10.1016/j.brainres.2004.05.103. [DOI] [PubMed] [Google Scholar]
- 23.Delépine L, Aubineau P. Plasma protein extravasation induced in the rat dura mater by stimulation of the parasympathetic sphenopalatine ganglion. Exp Neurol. 1997;147:389–400. doi: 10.1006/exnr.1997.6614. [DOI] [PubMed] [Google Scholar]
- 24.Hara H, Jansen I, Ekman R, Hamel E, MacKenzie ET, Uddman R, et al. Acetylcholine and vasoactive intestinal peptide in cerebral blood vessels: Effect of extirpation of the sphenopalatine ganglion. J Cereb Blood Flow Metab. 1989;9:204–11. doi: 10.1038/jcbfm.1989.30. [DOI] [PubMed] [Google Scholar]
- 25.Gibbins IL, Brayden JE, Bevan JA. Perivascular nerves with immunoreactivity to vasoactive intestinal polypeptide in cephalic arteries of the cat: Distribution, possible origins and functional implications. Neuroscience. 1984;13:1327–46. doi: 10.1016/0306-4522(84)90301-4. [DOI] [PubMed] [Google Scholar]
- 26.Hara H, Hamill GS, Jacobowitz DM. Origin of cholinergic nerves to the rat major cerebral arteries: Coexistence with vasoactive intestinal polypeptide. Brain Res Bull. 1985;14:179–88. doi: 10.1016/0361-9230(85)90077-2. [DOI] [PubMed] [Google Scholar]
- 27.Edvinsson L, Ekman R. Distribution and dilatory effect of vasoactive intestinal polypeptide (VIP) in human cerebral arteries. Peptides. 1984;5:329–31. doi: 10.1016/0196-9781(84)90229-8. [DOI] [PubMed] [Google Scholar]
- 28.Lorén I, Emson PC, Fahrenkrug J, Björklund A, Alumets J, Håkanson R, et al. Distribution of vasoactive intestinal polypeptide in the rat and mouse brain. Neuroscience. 1979;4:1953–76. doi: 10.1016/0306-4522(79)90068-x. [DOI] [PubMed] [Google Scholar]
- 29.Hara H, Weir B. Different distributions of substance P and vasoactive intestinal polypeptide in the cerebral arterial innervation in rat and guinea pig. Anat Anz. 1987;163:19–23. [PubMed] [Google Scholar]
- 30.Cobb S, Finesinger JE. Cerebral circulation. XIX. The vagal pathway of the vasodilator impulses. Arch Neurol Psychiatry. 1932;28:1243–89. [Google Scholar]
- 31.Seylaz J, Hara H, Pinard E, Mraovitch S, MacKenzie ET, Edvinsson L. Effect of stimulation of the sphenopalatine ganglion on cortical blood flow in the rat. J Cereb Blood Flow Metab. 1988;8:875–8. doi: 10.1038/jcbfm.1988.145. [DOI] [PubMed] [Google Scholar]
- 32.Ay I, Ay H. Ablation of the sphenopalatine ganglion does not attenuate the infarct reducing effect of vagus nerve stimulation. Auton Neurosci. 2013;174:31–5. doi: 10.1016/j.autneu.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hardebo JE, Arbab M, Suzuki N, Svendgaard NA. Pathways of parasympathetic and sensory cerebrovascular nerves in monkeys. Stroke. 1991;22:331–42. doi: 10.1161/01.str.22.3.331. [DOI] [PubMed] [Google Scholar]
- 34.Suzuki N, Hardebo JE, Owman C. Origins and pathways of choline acetyltransferase-positive parasympathetic nerve fibers to cerebral vessels in rat. J Cereb Blood Flow Metab. 1990;10:399–408. doi: 10.1038/jcbfm.1990.70. [DOI] [PubMed] [Google Scholar]
- 35.Suzuki N, Hardebo JE. Anatomical basis for a parasympathetic and sensory innervation of the intracranial segment of the internal carotid artery in man. Possible implication for vascular headache. J Neurol Sci. 1991;104:19–31. doi: 10.1016/0022-510x(91)90211-o. [DOI] [PubMed] [Google Scholar]
- 36.Suzuki N, Hardebo JE, Owman C. Origins and pathways of cerebrovascular vasoactive intestinal polypeptide-positive nerves in rat. J Cereb Blood Flow Metab. 1988;8:697–712. doi: 10.1038/jcbfm.1988.117. [DOI] [PubMed] [Google Scholar]
- 37.Shimizu T. Distribution and pathway of the cerebrovascular nerve fibers from the otic ganglion in the rat: Anterograde tracing study. J Auton Nerv Syst. 1994;49:47–54. doi: 10.1016/0165-1838(94)90019-1. [DOI] [PubMed] [Google Scholar]
- 38.Walters BB, Gillespie SA, Moskowitz MA. Cerebrovascular projections from the sphenopalatine and otic ganglia to the middle cerebral artery of the cat. Stroke. 1986;17:488–94. doi: 10.1161/01.str.17.3.488. [DOI] [PubMed] [Google Scholar]
- 39.Yarnitsky D, Lorian A, Shalev A, Zhang ZD, Takahashi M, Agbaje-Williams M, et al. Reversal of cerebral vasospasm by sphenopalatine ganglion stimulation in a dog model of subarachnoid hemorrhage. Surg Neurol. 2005;64:5–11. doi: 10.1016/j.surneu.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 40.Ignacio CS, Curling PE, Childres WF, Bryan RM., Jr Nitric oxide-synthesizing perivascular nerves in the rat middle cerebral artery. Am J Physiol. 1997;273(2 Pt 2):R661–8. doi: 10.1152/ajpregu.1997.273.2.R661. [DOI] [PubMed] [Google Scholar]
- 41.Borsody MK, Yamada C, Bielawski D, Heaton T, Lyeth B, Garcia A, et al. Effect of pulsed magnetic stimulation of the facial nerve on cerebral blood flow. Brain Res. 2013;1528:58–67. doi: 10.1016/j.brainres.2013.06.022. [DOI] [PubMed] [Google Scholar]
- 42.Bozzao L, Fantozzi LM, Bastianello S, Bozzao A, Fieschi C. Early collateral blood supply and late parenchymal brain damage in patients with middle cerebral artery occlusion. Stroke. 1989;20:735–40. doi: 10.1161/01.str.20.6.735. [DOI] [PubMed] [Google Scholar]
- 43.Uddman R, Hara H, Edvinsson L. Neuronal pathways to the rat middle meningeal artery revealed by retrograde tracing and immunocytochemistry. J Auton Nerv Syst. 1989;26:69–75. doi: 10.1016/0165-1838(89)90109-4. [DOI] [PubMed] [Google Scholar]
- 44.Linder J. Effects of facial nerve section and stimulation on cerebral and ocular blood flow in hemorrhagic hypotension. Acta Physiol Scand. 1981;112:185–93. doi: 10.1111/j.1748-1716.1981.tb06803.x. [DOI] [PubMed] [Google Scholar]
- 45.Sudo E, Ishii H, Niioka T, Hirai T, Izumi H. Parasympathetic vasodilator fibers in rat digastric muscle. Brain Res. 2009;1302:125–31. doi: 10.1016/j.brainres.2009.09.035. [DOI] [PubMed] [Google Scholar]
- 46.Ishii H, Niioka T, Sudo E, Izumi H. Evidence for parasympathetic vasodilator fibres in the rat masseter muscle. J Physiol. 2005;569(Pt 2):617–29. doi: 10.1113/jphysiol.2005.087643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pham M, Bendszus M. Facing time in ischemic stroke: An Alternative hypothesis for collateral failure. Clin Neuroradiol. 2016;26:141–51. doi: 10.1007/s00062-016-0507-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liebeskind DS. Collateral circulation. Stroke. 2003;34:2279–84. doi: 10.1161/01.STR.0000086465.41263.06. [DOI] [PubMed] [Google Scholar]
- 49.Busija DW, Heistad DD. Effects of cholinergic nerves on cerebral blood flow in cats. Circ Res. 1981;48:62–9. doi: 10.1161/01.res.48.1.62. [DOI] [PubMed] [Google Scholar]
- 50.Forbes H. The cerebral circulation: Observations and measurements of pial vessels. Arch Neurol Psychiatry. 1928;19:751–61. [Google Scholar]
- 51.Gloster J. Influence of the facial nerve on intra-ocular pressure. Br J Ophthalmol. 1961;45:259–78. doi: 10.1136/bjo.45.4.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nilsson SF, Linder J, Bill A. Characteristics of uveal vasodilation produced by facial nerve stimulation in monkeys, cats and rabbits. Exp Eye Res. 1985;40:841–52. doi: 10.1016/0014-4835(85)90129-0. [DOI] [PubMed] [Google Scholar]
- 53.Ruskell GL. The orbital branches of the pterygopalatine ganglion and their relationship with internal carotid nerve branches in primates. J Anat. 1970;106(Pt 2):323–39. [PMC free article] [PubMed] [Google Scholar]
- 54.Gellért A. Ganglia of the internal carotid plexus. J Anat. 1934;68(Pt 3):318–22. [PMC free article] [PubMed] [Google Scholar]
- 55.Cadden SW. Modulation of human jaw reflexes: Heterotopic stimuli and stress. Arch Oral Biol. 2007;52:370–3. doi: 10.1016/j.archoralbio.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 56.Tanaka K, Fukuuchi Y, Shirai T, Nogawa S, Nozaki H, Nagata E, et al. Chronic transection of post-ganglionic parasympathetic and nasociliary nerves does not affect local cerebral blood flow in the rat. J Auton Nerv Syst. 1995;53:95–102. doi: 10.1016/0165-1838(94)00167-i. [DOI] [PubMed] [Google Scholar]
- 57.Boysen NC, Dragon DN, Talman WT. Parasympathetic tonic dilatory influences on cerebral vessels. Auton Neurosci. 2009;147:101–4. doi: 10.1016/j.autneu.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goadsby PJ, Duckworth JW. Effect of stimulation of trigeminal ganglion on regional cerebral blood flow in cats. Am J Physiol. 1987;253(2 Pt 2):R270–4. doi: 10.1152/ajpregu.1987.253.2.R270. [DOI] [PubMed] [Google Scholar]
- 59.Wang YC, Kuo JS, Lin SZ. The effect of sphenopalatine postganglionic neurotomy on the alteration of local cerebral blood flow of normotensive and hypertensive rats in acute cold stress. Proc Natl Sci Counc Repub China B. 1998;22:122–8. [PubMed] [Google Scholar]
- 60.Salanga VD, Waltz AG. Regional cerebral blood flow during stimulation of seventh cranial nerve. Stroke. 1973;4:213–7. doi: 10.1161/01.str.4.2.213. [DOI] [PubMed] [Google Scholar]
- 61.Láinez MJ, Puche M, Garcia A, Gascón F. Sphenopalatine ganglion stimulation for the treatment of cluster headache. Ther Adv Neurol Disord. 2014;7:162–8. doi: 10.1177/1756285613510961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sanders M, Zuurmond WW. Efficacy of sphenopalatine ganglion blockade in 66 patients suffering from cluster headache: A 12-to 70-month follow-up evaluation. J Neurosurg. 1997;87:876–80. doi: 10.3171/jns.1997.87.6.0876. [DOI] [PubMed] [Google Scholar]
- 63.Schoenen J, Jensen RH, Lantéri-Minet M, Láinez MJ, Gaul C, Goodman AM, et al. Stimulation of the sphenopalatine ganglion (SPG) for cluster headache treatment. Pathway CH-1: A randomized, sham-controlled study. Cephalalgia. 2013;33:816–30. doi: 10.1177/0333102412473667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.May A, Bahra A, Büchel C, Frackowiak RS, Goadsby PJ. PET and MRA findings in cluster headache and MRA in experimental pain. Neurology. 2000;55:1328–35. doi: 10.1212/wnl.55.9.1328. [DOI] [PubMed] [Google Scholar]
- 65.Schytz HW, Barløse M, Guo S, Selb J, Caparso A, Jensen R, et al. Experimental activation of the sphenopalatine ganglion provokes cluster-like attacks in humans. Cephalalgia. 2013;33:831–41. doi: 10.1177/0333102413476370. [DOI] [PubMed] [Google Scholar]
- 66.Khatri P, Abruzzo T, Yeatts SD, Nichols C, Broderick JP, Tomsick TA IMS I and II Investigators. Good clinical outcome after ischemic stroke with successful revascularization is time-dependent. Neurology. 2009;73:1066–72. doi: 10.1212/WNL.0b013e3181b9c847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kaul S, Khurana D, Csani A, Ichaporia N, Schneider D, Lichy C, Weiss S, et al. Implant for augmentation of cerebral blood flow clinical trial – (ImpACT-1). An interim analysis of safety and effectiveness of the Neuropath IS system in the treatment of acute ischemic stroke, in World Stroke Congress, Vienna. 2008 [Google Scholar]
- 68.Koketsu N, Moskowitz MA, Kontos HA, Yokota M, Shimizu T. Chronic parasympathetic sectioning decreases regional cerebral blood flow during hemorrhagic hypotension and increases infarct size after middle cerebral artery occlusion in spontaneously hypertensive rats. J Cereb Blood Flow Metab. 1992;12:613–20. doi: 10.1038/jcbfm.1992.85. [DOI] [PubMed] [Google Scholar]
- 69.Diansan S, Shifen Z, Zhen G, Heming W, Xiangrui W. Resection of the nerves bundle from the sphenopalatine ganglia tend to increase the infarction volume following middle cerebral artery occlusion. Neurol Sci. 2010;31:431–5. doi: 10.1007/s10072-010-0238-0. [DOI] [PubMed] [Google Scholar]
- 70.Kano M, Moskowitz MA, Yokota M. Parasympathetic denervation of rat pial vessels significantly increases infarction volume following middle cerebral artery occlusion. J Cereb Blood Flow Metab. 1991;11:628–37. doi: 10.1038/jcbfm.1991.114. [DOI] [PubMed] [Google Scholar]
- 71.Bar-Shir A, Shemesh N, Nossin-Manor R, Cohen Y. Late stimulation of the sphenopalatine-ganglion in ischemic rats: Improvement in N-acetyl-aspartate levels and diffusion weighted imaging characteristics as seen by MR. J Magn Reson Imaging. 2010;31:1355–63. doi: 10.1002/jmri.22110. [DOI] [PubMed] [Google Scholar]
- 72.Henninger N, Fisher M. Stimulating circle of Willis nerve fibers preserves the diffusion-perfusion mismatch in experimental stroke. Stroke. 2007;38:2779–86. doi: 10.1161/STROKEAHA.107.485581. [DOI] [PubMed] [Google Scholar]
- 73.Levi H, Schoknecht K, Prager O, Chassidim Y, Weissberg I, Serlin Y, et al. Stimulation of the sphenopalatine ganglion induces reperfusion and blood-brain barrier protection in the photothrombotic stroke model. PLoS One. 2012;7:e39636. doi: 10.1371/journal.pone.0039636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Solberg Y, Yarnitsky D, Borenstein N, Tanne D, Dayan A, Weiss S, Fisher M. Effectiveness of sphenopalatine ganglion stimulation therapy in focal ischemic stroke models: A 24-hour post-stroke window of treatment, in International Stroke Conference, Honolulu, Hawaii. 2008 [Google Scholar]
- 75.Yarnitsky D, Lorian A, Dayan A, Avnon Y, Krakovsky M, Lamensdorf I. Orlando, FL: International Stroke Conference; 2006. Sphenopalatine ganglion (SPG) stimulation in acute stroke model: A novel method for neuroprotection. [Google Scholar]
- 76.Takahashi M, Zhang ZD, Macdonald RL. Sphenopalatine ganglion stimulation for vasospasm after experimental subarachnoid hemorrhage. J Neurosurg. 2011;114:1104–9. doi: 10.3171/2010.6.JNS091525. [DOI] [PubMed] [Google Scholar]
- 77.Khurana D, Kaul S, Bornstein NM ImpACT-Study Group. Implant for augmentation of cerebral blood flow trial 1: A pilot study evaluating the safety and effectiveness of the Ischaemic Stroke System for treatment of acute ischaemic stroke. Int J Stroke. 2009;4:480–5. doi: 10.1111/j.1747-4949.2009.00385.x. [DOI] [PubMed] [Google Scholar]
- 78.Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. N Engl J Med. 1995;333:1581–7. doi: 10.1056/NEJM199512143332401. [DOI] [PubMed] [Google Scholar]
- 79.Hacke W, Kaste M, Bluhmki E, Brozman M, Dávalos A, Guidetti D, et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med. 2008;359:1317–29. doi: 10.1056/NEJMoa0804656. [DOI] [PubMed] [Google Scholar]
- 80.Trouillas P, von Kummer R. Classification and pathogenesis of cerebral hemorrhages after thrombolysis in ischemic stroke. Stroke. 2006;37:556–61. doi: 10.1161/01.STR.0000196942.84707.71. [DOI] [PubMed] [Google Scholar]
- 81.Leigh R, Jen SS, Hillis AE, Krakauer JW, Barker PB STIR and VISTA Imaging Investigators. Pretreatment blood-brain barrier damage and post-treatment intracranial hemorrhage in patients receiving intravenous tissue-type plasminogen activator. Stroke. 2014;45:2030–5. doi: 10.1161/STROKEAHA.114.005249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Borsody MK, Yamada C, Bielawski D, Heaton T, Castro Prado F, Garcia A, et al. Effects of noninvasive facial nerve stimulation in the dog middle cerebral artery occlusion model of ischemic stroke. Stroke. 2014;45:1102–7. doi: 10.1161/STROKEAHA.113.003243. [DOI] [PubMed] [Google Scholar]
- 83.Anderson WR, Boster RA, Willems GC. Empirical model of intracranial pressure and head motion resulting from a vibrating seated rhesus. Aviat Space Environ Med. 1978;49(1 Pt 2):240–52. [PubMed] [Google Scholar]
- 84.Sundbärg G, Nordström CH, Messeter K, Söderström S. A comparison of intraparenchymatous and intraventricular pressure recording in clinical practice. J Neurosurg. 1987;67:841–5. doi: 10.3171/jns.1987.67.6.0841. [DOI] [PubMed] [Google Scholar]
- 85.Rossi S, Hallett M, Rossini PM, Pascual-Leone A Safety of TMS Consensus Group. Safety, ethical considerations, and application guidelines for the use of transcranial magnetic stimulation in clinical practice and research. Clin Neurophysiol. 2009;120:2008–39. doi: 10.1016/j.clinph.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chernyshev OY, Martin-Schild S, Albright KC, Barreto A, Misra V, Acosta I, et al. Safety of tPA in stroke mimics and neuroimaging-negative cerebral ischemia. Neurology. 2010;74:1340–5. doi: 10.1212/WNL.0b013e3181dad5a6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cai PY, Bodhit A, Derequito R, Ansari S, Abukhalil F, Thenkabail S, et al. Vagus nerve stimulation in ischemic stroke: Old wine in a new bottle. Front Neurol. 2014;5:107. doi: 10.3389/fneur.2014.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rotem A, Moses E. Magnetic stimulation of curved nerves. IEEE Trans Biomed Eng. 2006;53:414–20. doi: 10.1109/TBME.2005.869770. [DOI] [PubMed] [Google Scholar]
- 89.Elron Electronic Industries Ltd., Investor Presentation, May. 2013 [Google Scholar]
- 90.BrainsGate, “ImpACT24 Interim Results.” July. 2014. [Last accessed on 2016 Oct 05]. Available from: http://www.brainsgate.com/eng/pr.php?actions=detailsand id=50 .
- 91.Elron Electronic Industries Ltd., Form 6. K Securities and Exchange Commission filing, May. 2014 [Google Scholar]
- 92.BrainsGate, Clinical Data. [Last accessed on 2016 Oct 05]. Available from: http://www.brainsgate.com/eng/page.php?id=14 and instance_id=9 .
- 93.Erlanger Hospital, the University of Tennessee College of Medicine - Chattanooga, and BrainsGate. “High Technology Stroke Update,” April. 2010. [Last accessed on 2016 Oct 05]. Available from: http://www.slideshare.net/erlangerhealth/brainsgate-press-conf-dr-devlin .
- 94.Elron Electronics Industries, LTD. “Discover. Innovation.” Investor presentation, November 2015, copy available from corresponding author on request [Google Scholar]
- 95.U.S. NIH, Clinical Trials. [Last accessed on 2016 Oct 05]. Available from: https://www.clinicaltrials.gov/archive/NCT00826059 .
- 96.Snell RP. Clinical Anatomy for Medical Students. 5th ed. Boston: Little Brown and Co; 1995. [Google Scholar]
- 97.Garcia A, Castro-Prado F, Perez M, Lara-Estrada R, Sanchez O, Ramirez M, et al. Facial Nerve Stimulation in Healthy Human Subjects. IEEE abstract; Annual Meeting, Orlando FL. 2016 [Google Scholar]
- 98.Vernau KM, Osofsky A, LeCouteur RA. The neurological examination and lesion localization in the companion rabbit (Oryctolagus cuniculus) Vet Clin North Am Exot Anim Pract. 2007;10:731–58. doi: 10.1016/j.cvex.2007.05.003. v. [DOI] [PubMed] [Google Scholar]
- 99.U.S. Food and Drug Administration, Class II Special Controls Guidance Document: Repetitive Transcranial Magnetic Stimulation (rTMS) Systems. [Last accessed on 2016 Sep 08]. Available from: http://www.fda.gov/RegulatoryInformation/Guidances/ucm265269.htm .