

Abstract
Microbiota and its induced inflammation in colorectal mucosa have been considered risk factors for the development of colorectal carcinogenesis. Previous studies demonstrated that the coexisting elements of microbiota in the gut, such as short chain fatty acids (SCFAs) and lipopolysaccharides (LPS), which exhibited regulatory effects on the intestinal epithelial cells individually. Unfortunately, the association between butyrate and the toll-like receptor (TLR) signaling pathway in the development of colon cancer is not fully elucidated. In the present study, by culturing human colon cancer SW480 cells or mouse colon cancer CT26 cells with butyrate and/or TLR4 ligand LPS in vitro, it was identified that butyrate suppressed the growth and promoted apoptosis of these cancer cells. Notably, the expression levels of TLR4 and CD14 were markedly increased on these butyrate-treated cells, but not on LPS-alone treated cells. Additionally, butyrate treatment induced the phosphorylation of extracellular signal-regulated kinase, tumor protein 38, c-Jun NH2-terminal kinase and nuclear factor-κB (NF-κB) p65, and then promoted the pro-inflammatory cytokine tumor necrosis factor-α, but not interleukin 6 secretion in SW480 and CT26 cells. Therefore, butyrate treatment regulates the expression of TLR4, mitogen-activated protein kinase and NF-κB signal pathway activation and pro-inflammatory response in vitro. Although the exact mechanisms have not been fully explored, these results suggested that butyrate and LPS-TLR4 signaling mediated innate immunity in colon cancer cells through two distinct but inter-regulated pathways. Thus, butyrate can further initiate innate immunity against tumor cells by upregulating the TLR4 expression and activation to preserve intestinal homeostasis.
Keywords: butyrate, apoptosis, inflammatory, colon cancer cells, toll-like receptor
Introduction
Colorectal cancer (CRC) is the third most common cancer type in the world and is cancer with the second-highest mortality rate, and its incidence rates continue to increase in a number of countries, such as Brazil and Russia (1,2). Numerous factors, including a high-fat diet, which may induce chronic inflammation in the intestine, clearly increase the risk for CRC (3–5). Accumulating evidence suggests that cancer incidence in the colon is increased compared with that of the small intestine (6,7). With advances in metagenomics technology and use of germ-free mice, it has been demonstrated that the intestinal microbiota serves a vital role in CRC initiation and development (8,9). Indeed, the microbiota and mucosal gene expression profiles in the gut of patients with CRC differ from those in healthy subjects (10). Although a number of studies have documented a critical association between the microbiota microenvironment and development of CRC (11–13), the roles of toll-like receptors (TLRs) and the underlying mechanisms have not been investigated as thoroughly.
TLRs are germline-encoded type I transmembrane receptors that serve as pathogen recognition receptors (PRRs) to identify microbe-associated molecular patterns. TLR4 is one of the characterized PRRs on a number of intestinal immune and non-immune cell types, which recognizes lipopolysaccharide (LPS) from Gram staining-negative bacteria in the gut. Generally, TLR4 is expressed at low concentrations on intestinal mucosa under steady-state conditions to maintain tolerance to commensal bacteria (14). Activation of TLR4 on colonic epithelial cells induces a tumor-promoting microenvironment to drive the tumorigenesis of colitis-associated cancer (15–17). In addition, TLR4 innate immune signaling alters the colorectal cancer chemotherapeutic response and radio-sensitivity through regulating the autophagy pathway (18,19). The administration of a TLR4 inhibitor or immune-modulator significantly regulates the TLR4 signal pathway and decreases tumor burden (20,21). Thus, gut microbiota are associated with carcinogenesis via modulating TLR, particularly the TLR4 signal pathway in the colon epithelial cells.
Generally, the metabolites from microbiota in the gut also alter host intestinal cancer risk using different mechanisms, including affecting host cell growth or turnover, regulating the local immune microenvironment and metabolizing ingested and host-derived products (22). Previous evidence revealed that short-chain fatty acids (SCFA), primarily acetate, propionate and butyrate, are major metabolites of microbiota with pleiotropic effects on the epithelial and immune cells (23). SCFA are catabolic end products of the microbial fermentation of dietary carbohydrates in the gut and primarily resistant starches and dietary fiber (24). As a C4 fatty acid, butyrate functions as the principle energy source for colonocytes. It also exerts functions in the differentiation, maturation and apoptosis of a number of different cell types, and may decrease the risk of gastrointestinal inflammation, colon cancer and cardiovascular disease (25). Numerous studies have indicated that the different activities of butyrate and its concomitant outcomes should be considered with the metabolism of target cells (26–29). Indeed, in contrast to normal colonocytes, cancerous colonocytes prefer to use aerobic glycolysis for glucose metabolism, and butyrate acts as a histone deacetylase (HDAC) inhibitor to suppress cell growth and to induce apoptosis by altering gene expression profiles (30).
Therefore, as the coexisting elements of microbiota in the gut, SCFAs and LPS have demonstrated the regulatory effect on normal intestinal epithelial cells or colon cancer cells. However, knowledge of the association between SCFAs and the TLR4 signaling pathway in the development of colon cancer remains incomplete. The present study, by culturing human colon cancer SW480 cells or mouse colon cancer CT26 cells with butyrate in vitro, identified that butyrate suppressed cell growth and proliferation, and increased the numbers of apoptotic cells. Notably, the expression levels of TLR4 and cluster of differentiation 14 (CD14) on these cells were clearly increased. In addition, butyrate treatment induced the phosphorylation of extracellular signal-regulated kinase (ERK), tumor protein 38 (p38), c-Jun NH2-terminal kinase (JNK) and nuclear factor-κB (NF-κB) p65, and then promoted pro-inflammatory cytokine tumor necrosis factor-α (TNF-α) secretion in SW480 and CT26 cells. These results suggested that butyrate had the ability to regulate innate immunity in colon cancer cells via promoting TLR4 expression and the phosphorylation of mitogen activated protein kinases (MAPKs) and NF-κB.
Materials and methods
Reagents and antibodies
RPMI-1640 medium and fetal calf serum was purchased from PAN Biotech (PAN-Biotech GmbH, Aidenbach, Germany). The CellTrace™ carboxyfluorescein succinimidyl ester (CFSE) Cell Proliferation kit and the Pierce™ BCA protein assay kit were purchased from Thermo Fisher Scientific, Inc. (Waltham, MA, USA). Butyrate, LPS, bovine serum albumin (BSA) and 2-mercaptoethanol were purchased from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). The 7-aminoactinomycin D (7-AAD), anti-human CD14-fluorescein isothiocyanate (FITC; cat no. 11-0149-42), anti-mouse CD14-FITC (cat no. 11-0141-82), anti-human TLR4-Alexa Fluor 488 (cat no. 53-9917-42), anti-mouse TLR4-Alexa Fluor 488 (cat no. 53-9917-42), anti-ERK1/2 (cat no. LF-MA0178) and anti-phosphorylated (phosphor)-ERK1/2 (cat no. PA5-36776) antibodies, interleukin (IL)-6 and TNF-α ELISA kits (human and mouse) and Annexin V Apoptosis Detection kit were purchased from the Thermo Fisher Scientific, Inc. Radioimmunoprecipitation assay (RIPA) cell lysis buffer and phenylmethane sulfonyl fluoride (PMSF) were purchased from the Beyotime Institute of Biotechnology (Haimen, China). The Cell Cycle Staining kit and goat anti-mouse IgG-horseradish peroxidase (HRP) antibody were purchased from Hangzhou MultiSciences (Lianke) Biotech Co., Ltd. (Hangzhou, China). Antibodies against p38, phospho-p38, JNK, phospho-JNK, NF-κB p65, NF-κB phospho-p65 and cleaved caspase-3 were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). Goat anti-rabbit IgG-HRP and HRP-conjugated-anti-actin antibodies were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
Colon cancer cell lines
The human colon cancer SW480 cell line and mouse colon cancer CT26 cell line were obtained from the Chinese Academy of Sciences (Shanghai, China). Cells were grown in RPMI-1640 medium supplemented with 10% heat-inactivated fetal calf serum, 100 U/ml penicillin and 100 mg/ml streptomycin in a humidified incubator at 37°C and 5% CO2. In certain experiments, 5 mM butyrate and/or 1 µg/ml LPS were added into the culture wells.
Flow cytometric analysis
Cells were treated with trypsin for 2 min at room temperature and then terminated with RPMI-1640 medium containing 10% fetal calf serum. Cells were dispersed with pipette and washed with PBS twice and passed through 70 µm cell strainers (FalconTM, Corning Incorporated, Corning, NY, USA). Then, the single cell suspension was incubated with 2% rabbit serum (Stemcell Technologies, Vancouver, BC, Canada) for 30 min at room temperature to block Fc receptors on the cell membrane, followed by staining with anti-TLR4-FITC or anti-CD14-FITC (both from Thermo Fisher Scientific, Inc.) at 4°C for 30 min. Following washing with PBS 3 times, cells were re-suspended with PBS (1 mM EDTA) and analyzed with a FACS Calibur flow cytometer (BD Bioscience, Franklin Lakes, NJ, USA). All data were analyzed using CellQuest (version 6.1; BD Biosciences) or FlowJo (version 8.7; Tree Star, Inc., Ashland, OR, USA).
Cell proliferation and apoptosis assays
For the cell proliferation assays, SW480 and CT26 cells were harvested with trypsin and the density of the cell suspension was adjusted to 2×106/ml with PBS. An equal volume of 4 µM carboxyfluorescein succinimidyl ester (CFSE) labeling buffer (Invitrogen; Thermo Fisher Scientific, Inc.) was added to the cell suspension, and the cells were gently re-suspended. The cell suspension was then incubated in the dark at 37°C for 20 min. Next, RPMI-1640 complete medium was added to terminate CFSE labeling and the cell suspension was then washed twice with PBS. Cells were resuspended with fresh RPMI-1640 complete medium and cultured in an incubator at 37°C and 5% CO2 for 24 h with or without 5 mM butyrate treatment. Cells were digested with trypsin as previously described and pelleted by centrifugation for 5 min in 150 × g at 4°C, and then stained with 7-AAD (1:100) for 2 min at room temperature to remove dead cells, and were then immediately analyzed with a flow cytometer (BD FACSCalibur; BD Biosciences).
For cell apoptosis detection, cells were prepared and harvested as aforementioned in the cell proliferation experimental protocol, and then stained with the Annexin V Apoptosis Detection kit according to the manufacturer's protocol. Cells were additionally analyzed with a flow cytometer (BD FACSCalibur; BD Biosciences), and the data were analyzed using FlowJo (version 8.7; Tree Star, Inc., Ashland, OR, USA).
ELISA
The cell supernatant was collected following centrifugation at 300 × g for 10 min at 4°C. The protein levels of TNF-α and IL-6 in the supernatant were measured using ELISA kits (Thermo Fisher Scientific, Inc.) following the manufacturer's protocols.
Western blot analysis assay
Cells were prepared as aforementioned and washed twice with cold PBS. Then, the cells were lysed with RIPA cell lysis buffer and mixed with PMSF (100:1) and 1X phosphatase inhibitor cocktail (Bimake, Houston, USA) for 20 min on ice. Lysed cells were scraped into the 1.5 ml tube and centrifuged at 12,000 × g for 10 min at 4°C. The supernatants of the cell lysates were harvested and the protein concentrations were quantified with the Pierce™ BCA Protein Assay kit (Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol. The lysate was mixed with the 5X loading buffer, to which 5% 2-mercaptoethanol was added and boiled at 100°C for 5 min. All lysate samples (30 µg per lane) were subjected to SDS-PAGE for protein separation and then electrophoretically transferred onto polyvinylidene difluoride (PVDF) membranes (Merck KGaA). Depending on the molecular weight of the target protein, 10% SDS-PAGE gels were used. Subsequent to being blocked with TBS containing 5% BSA and 0.1% Tween-20 for 2 h at room temperature, the PVDF membranes were incubated with HRP-conjugated anti-actin (cat no., sc-47778; 1:10,000), anti-ERK1/2 mAb (cat no., 14-9108-82; 1:1,000), anti-phospho-ERK1/2 mAb (cat no., 14-9109-82; MILAN8R; 1:1,000), anti-p38 MAPK mAb (cat no., 2387; 1:1,000), anti-phospho-p38MAPKThr180/Tyr182 mAb (cat no., 5175; 1:1,000), anti-SAPK/JNK mAb (cat no., 9252; 1:1,000), anti-phospho-SAPK/JNKThr183/Tyr185 (cat no., 4668; 1:1,000), anti-NF-κB P65 mAb (cat no., 8242; 1:1,000), anti-phospho-NF-kB P65Ser536 (cat no., 3033; 1:1,000) and anti-cleaved caspase-3 (cat no., 9664; 1:1,000) for at least 12 h at 4°C. Then, the membranes were incubated with HRP-conjugated secondary antibodies, including mouse anti-rabbit IgG-HRP (cat no., sc-2357; 1:2,000) and goat anti-mouse IgG-HRP (cat no., 70-GAM0072; 1:10,000) for 2 h at room temperature. Following washing three times with TBS, the PVDF membranes were visualized using the Immobilon™ Western Chemiluminescent HRP Substrate (Merck KGaA) and quantified by Fusion Solo 6 S Chemiluminescence (Vilber Lourmat Deutschland GmbH, Eberhardzell, Germany).
Cell cycle assay
For cell cycle detection, cells were prepared and harvested as aforementioned in the cell proliferation and apoptosis experiments. Then, cells were labeled with Cell cycle staining kit following the manufacture's protocols. The DNA content of cells was detected using flow cytometry (BD FACSCalibur; BD Biosciences), and the data were analyzed with Modfit LT 5.0 (American Verity Software House, Topsham ME, USA).
Statistical analysis
Data were statistically analyzed using the two-tailed unpaired Student's t-test or post-hoc test (Scheffe) for two-way analysis of variance (ANOVA) on GraphPad Prism version 5.0 (GraphPad Software, Inc., La Jolla, CA, USA). All data are presented as the mean ± standard deviation. P<0.05 was considered to indicate a statistically significant difference. The representative data presented are from more than 3 independent experiments.
Results
Butyrate suppresses the proliferation of colon cancer cells
The effects of butyrate on the growth of colon cancer cells were first examined. By treating human colon cancer SW480 cells and mouse colon cancer CT26 cells with butyrate, it was identified that the growth of colon cancer cells was evidently inhibited and able to be directly observed under the microscope (Fig. 1A). These results were additionally confirmed by the data of the cell numbers of colon cancer cells (Fig. 1B), and CFSE tracing cell proliferation experiments (Fig. 1C and D). After 24 h culturing, the proliferation of the SW480 and CT26 cells were significantly suppressed by butyrate treatment (Fig. 1C and D). By analyzing the cell cycle with a flow cytometer, an increased number of cells in S phase were identified in the NC group compared with the Bu group, and butyrate treatment increased the numbers of cells in G1 and G2 phases compared with the NC group (Fig. 2). Therefore, the growth and the proliferation of colon cancer cells are significantly inhibited by butyrate.
Figure 1.

Butyrate suppresses the proliferation of colon cancer cells. Human colon cancer SW480 cells and mouse colon cancer CT26 cells were cultured with or without 5 mM butyrate treatment for 24 h. (A) Representative cell images were captured under a microscope (magnification, ×40). (B) Total cell numbers were quantified. (C and D) SW480 and CT26 cells were firstly labeled with CFSE and cultured for 24 h. Then the dilutions of CFSE in 7-AAD-negative live cells were analyzed using a flow cytometer (C), and the q percentages of CFSE-labeled dividing cancer cells (CFSElow) in different groups were quantitatively analyzed. Three replicate wells in each group and all experiments were repeated ≥3 times, and representative graphs or data are presented. Data in (B and D) are presented as the mean ± standard deviation. *P<0.05 and **P<0.01. Numbers in graphs indicate the percentage of cells. Bu, butyrate; NC, negative control; SSC, side scatter; CFSE, carboxyfluorescein succinimidyl ester; 7-ADD, 7-aminoactinomycin D.
Figure 2.

Butyrate regulates the cell cycles of colon cancer cells. The cell cycles of SW480 and CT26 were (A) analyzed by flow cytometry, and then (B) quantitatively analyzed. All experiments were repeated ≥3 times, and representative graphs or data are presented. *P<0.05 and **P<0.01; NS, not significant; Bu, butyrate; NC, negative control.
Butyrate promotes apoptosis in colon cancer cells in vitro
Next, the effects of butyrate on apoptosis induction in colon cancer cells were investigated. As demonstrated in Fig. 3A and B, the percentages of Annexin V-positive apoptotic SW480 and CT26 cells were significantly increased in butyrate treatment groups compared with the NC groups. As caspases and their cleaved substrates contribute to cell apoptosis (31), the levels of cleaved caspase-3 protein (17 and 19 kD) in SW480 and CT26 cells was additionally detected using western blot analysis. The data in Fig. 3C indicates that butyrate treatment evidently increased the protein levels of cleaved caspase-3 in the SW480 and CT26 cells. Therefore, butyrate treatment induces apoptosis in colon cancer cells in vitro.
Figure 3.

Butyrate promotes the apoptosis of colon cancer cells in vitro. SW480 and CT26 cells were cultured and treated with butyrate and the apoptotic cells were (A) analyzed with Annexin-V-fluorescein isothiocyanate/propidium iodide using a flow cytometer, and then (B) quantitative analyzed. (C) The cleaved caspase 3 proteins in colon cancer cells were detected using a western blot analysis assay. All experiments were repeated ≥3 times. **P<0.01; ***P<0.001. PI, propidium iodide; Bu, butyrate; NC, negative control.
Butyrate upregulates the expression of TLR4 and CD14 on colon cancer cells
Butyrate is produced in the colonic lumen, and a high concentration of LPS is detected in the gut microenvironment (32). Butyrate has been demonstrated to exert immune modulatory effects on colonic epithelial cells (32), and to possess a regulatory function on colon cancer cells (33). TLR4 and CD14 function as the receptor for LPS recognition (34,35). Next, the effects of butyrate on the expression of TLR4 and CD14 in the SW480 and CT26 cells were detected. As demonstrated in Fig. 4, there was a difference in the baseline expression levels of TLR4 and CD14 in the human colon cancer SW460 cells and mouse colon cancer CT26 cells. Markedly low expression levels of TLR4 and CD14 were detected in the SW480 cells, but moderate expression of these molecules in CT26 cells (Fig. 4). Notably, regardless of what the baseline expression levels of TLR4 and CD14 on the cell membrane were, no marked effects on the expression of these molecules were observed in the two types of colon cancer cells following LPS stimulation. However, if SW480 and CT26 cells were treated with butyrate itself or butyrate + LPS, the expression levels of TLR4 and CD14 were significantly increased in the two cell types (Fig. 4). Therefore, these results suggested that butyrate upregulated the expression of TLR4 and CD14 in SW480 and CT26 cells, and that the effects of butyrate were species-independent.
Figure 4.

Butyrate upregulates the levels of TLR4 and CD14 on colon cancer cells. The expression levels of TLR4 and CD14 on the membrane of SW480 cells and CT26 cells treated by butyrate and/or LPS were (A) analyzed using a flow cytometer, and (B) the MFI values of TLR4 and CD14 were quantitative analyzed. These experiments were repeated ≥3 times, and representative graphs are presented. Compared with the NC group, *P<0.05, **P<0.01 and ***P<0.001. NS, not significant; MFI, mean fluorescence index; Bu, butyrate; NC, negative control; LPS, lipopolysaccharide; TLR4, toll-like receptor 4; CD14, cluster of differentiation 14.
Butyrate induces the phosphorylation of ERK, p38, JNK and NF-κB p65 in colon cancer cells
Stimulation of the TLR4 signaling pathway leads to the activation of three distinct MAPKs, ERK, JNK, p38 and the activation of NF-κB p65 (36). The present study therefore elucidated the roles of the TLR4 signaling pathway in the cellular response to butyrate and LPS in colon cancer cells. The phosphorylation status of ERK, p38, JNK and NF-κB p65 were analyzed using western blot analysis. In SW480 cells, as indicated in Fig. 5, the amount of p-ERK, p38, JNK and NF-κB p65 were not altered by LPS stimulation alone but were evidently increased in the butyrate and butyrate + LPS groups. Different from SW480, LPS-alone simulation increased the phosphorylation of P-38 and JNK, but not the phosphorylation of ERK and NF-κB p65 in CT26 cells. This difference may depend on the different baseline expression levels of TLR4 and CD14 on SW480 and CT26 cells (Fig. 4), and indicated the species-specific LPS response in these colon cancer cells. Notably, butyrate itself and butyrate +LPS stimulation increased the phosphorylation of ERK, JNK, p38, NF-κB p65 protein in SW480 and CT26 cells (Fig. 5). These data suggested that butyrate treatment resulted in the activation of certain MAPKs (ERK, JNK and p38) and the NF-κB signaling pathway in human and mouse colon cancer cells, and that the butyrate-mediated activation of these signaling pathways in these cancer cells was species-independent.
Figure 5.

Butyrate modifies the phosphorylation of ERK, p38, JNK and NF-κB p65 in colon cancer cells. The phosphorylation of ERK, p38, JNK and NF-κB p65 proteins in SW480 and CT26 cells treated with butyrate and/or LPS were detected using a western blot analysis assay. (A) Representative western blot analysis images of P-ERK/ERK, P-p38/p38 and P-JNK/JNK are presented. (B) The ratios of P-ERK/ERK, P-p38/p38 and P-JNK/JNK protein intensities were calculated. (C) Representative western blot analysis images of P-NF-κB p65/NF-κB p65 protein expression in cells are presented. (D) P-NF-κB p65/p65 protein intensities were calculated. β-actin was used as the control. Experiments were performed in triplicate and data are expressed as the mean ± standard deviation. Compared with the NC group, *P<0.05, **P<0.01 and ***P<0.001. These experiments were repeated ≥3 times, and representative images are presented. P, phosphorylated; ERK, extracellular signal-regulated kinase; p38, tumor protein 38; JNK, c-Jun NH2-terminal kinase; NS, not significant.
Butyrate induces TNF-α secretion in colon cancer cells
The inflammatory cytokine secretion of SW480 and CT26 cells prior to and following butyrate treatment with or without LPS was additionally analyzed. The data in Fig. 6A demonstrate that LPS alone was unable to increase TNF-α production in SW40 cells, but that butyrate or butyrate + LPS treatment significantly increased the secretion of TNF-α (Fig. 6). In the case of CT26 cells, butyrate and LPS individually stimulated these cells to produce increased levels of TNF-α (Fig. 6B). Notably, the production of IL-6 was not promoted by butyrate and LPS in SW480 cells. Concurrently, in the CT26 cells, LPS itself and butyrate + LPS induced high levels of IL-6, but butyrate alone did not increase IL-6 production. These data suggested that butyrate promoted TNF-α secretion, but not IL-6 production, in colon cancer cells.
Figure 6.
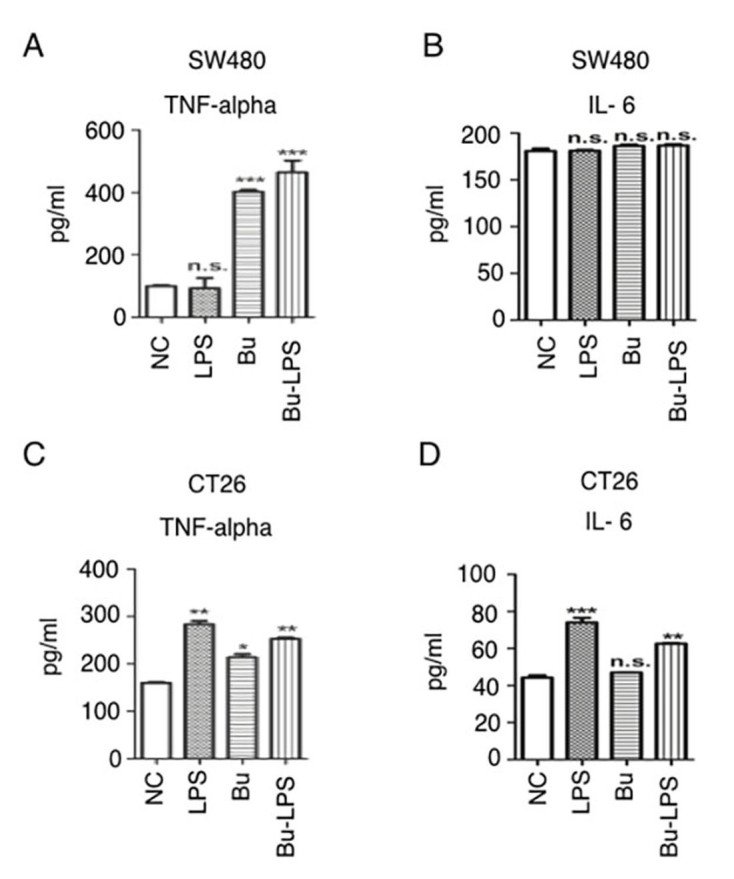
Butyrate induces inflammatory cytokine secretion in colon cancer cells. (A) SW480 cells were cultured and treated with/without butyrate and/or LPS for 24 h, and the supernatants were collected for inflammatory cytokine detection using a TNF-α ELIA kit. (B) SW480 cells were cultured and treated with/without butyrate and/or LPS for 24 h, and the supernatants were collected for inflammatory cytokine detection using an IL-6 ELISA kit. (C) CT26 cells were cultured and treated with/without butyrate and/or LPS for 24 h, and the supernatants were collected for inflammatory cytokine detection using a TNF-α ELISA kit. (D) CT26 cells were cultured and treated with or without butyrate and/or LPS for 24 h, and the supernatants were collected for inflammatory cytokine detection using an IL-6 ELISA kit. The protein levels in the different groups were statistically analyzed. These experiments were repeated ≥3 times, and representative data are presented as the mean ± standard deviation. *P<0.05, **P<0.01 and ***P<0.001. TNF-α, tumor necrosis factor α; IL-6, interleukin 6; Bu, butyrate; NC, negative control; LPS, lipopolysaccharide; NS, not significant.
Discussion
Microbiota and its induced inflammation in colorectal mucosa are considered risk factors for the development of colorectal carcinogenesis (8,37,38), but the potential molecular mechanisms are yet to be elucidated. Previous evidence suggests that microbiota-derived molecular mediators, including SCFAs, regulate the metabolic and immune homeostasis in the colon (39). In the present study, by using culture cells in vitro, it was identified that butyrate not only induced apoptotic cancer cell production, but also promoted TNF-α secretion and enhanced the TLR4 signaling pathway in SW480 and CT26 cells. So, in the pathological condition in colon cancer, butyrate exhibits a function in upregulating the TLR4 sensors on cancer cells, and then initiating bacterial mediated innate immunity to maintain intestinal homeostasis.
Previous studies have suggested that butyrate exerts different anti-proliferative properties in normal colonocytes and colon cancer cells by altering cellular metabolism (40,41). For normal colonic epithelia cells, butyrate is the primary energy source and promotes the turnover of the colonic epithelium to maintain normal homeostasis of the gut (42). However, cancerous colonocytes undergo the Warburg effect and the preferred energy source for these cells is glucose, not butyrate (42). Therefore, accumulated butyrate functions as an HDAC inhibitor to promote cell arrest and induce apoptosis (40,41). This butyrate-mediated growth inhibition in colon cancer cells is associated with the direct silencing of HDAC3 expression, the repression of Cyclin B or the upregulation of cyclin-dependent kinase inhibitor 1 (43,44). Furthermore, the present study demonstrated that the activation of caspase-3 contributed to butyrate-induced apoptosis in SW480 and CT26 colon cancer cells. In accordance with the results in the present study, Zhang et al (45) previously demonstrated that butyrate effectively induces human colon cancer apoptosis through JNK MAPK activation, characterized by an increased B-cell lymphoma 2 (Bcl-2)-associated X protein (Bax)-to-Bcl-2 expression ratio and caspase cascade activation. An additional study indicated that microRNA transcription regulation or autophagy induction also contributed to butyrate-mediated colon cancer cell arrest and apoptosis (46). Therefore, the inhibitory effects of butyrate on the growth and apoptosis of colon cancer cells may depend on multiple molecular mechanisms.
In the gut, the expression, localization and signaling of TLR4 on colonic epithelia are developmentally regulated in a compartmentalized manner (47). Compared with the high expression on postnatal day 1, decreased levels of TLR4 were detected in crypts in the mature colon of humans and mice (48). These data indicate that the expression of TLR4 on intestinal epithelial cells is closely associated with the flora community in the intestine. Although data concerning the expression levels of TLR4 on colon cells are contradictory, they indicate an association between the TLR4 signaling pathway and the development of colon cancer (49–51). The present study identified that the expression levels of TLR4 on human colon cancer SW480 cells and mouse colon cancer CT26 cells were low (Fig. 3), but were significantly increased following butyrate treatment. Collectively, these data indicated that butyrate modified the immune microenvironment in the intestinal tract by regulating the expression of TLR4 and its signaling pathway in the colon epithelial or colorectal cancer cells.
The myeloid differentiation primary response gene 88 (MyD88)-dependent and MyD88-independent pathways are initiated in the TLR4 signaling pathway (52). LPS treatment alone is not able to induce activation of the TLR4 signaling pathway, and no differences in the phosphorylation of different MAPKs (ERK, p38 and JNK) and NF-κB p65 were identified between the control group and LPS-treated SW480 cells (Fig. 4). However, the LPS treatment alone significantly induced the phosphorylation of p38 and JNK in CT26 cells, although no difference was observed in ERK and p65 in the control and LPS-treated groups. These different responses to treatment with LPS alone may be caused by the different original expression levels of TLR4 on SW480 and CT26 cells.
There are conflicting data about the association between SCFAs and TLRs in the literature. Certain studies have suggested that butyrate exhibits anti-inflammatory effects by downregulating the TLR4-dependent signaling cascade and the secretion of inflammatory cytokines, but others demonstrated its function in promoting inflammatory cytokines (53,54). The present study demonstrated that if butyrate or butyrate + LPS were used, the expression levels of TLR4 on SW480 and CT26 cells were significantly upregulated, and the protein levels of phosphorylated ERK, p38, JNK and NF-κB p65 were increased. Similarly, Alva-Murillo et al (55) identified that butyrate induced p38 phosphorylation and improved antimicrobial defense in bovine mammary epithelial cells. Butyrate also increased TLR4 expression and the NF-κB response to TLR ligand stimulation in human L-cells, HEK293 or HeLa epithelial cells (56,57). In the present study, although butyrate upregulated TLR4 expression, MAPKs and NF-κB phosphorylation in SW480 and CT26 cells, only an increase in TNF-α production was induced and no change of IL-6 expression was detected (Fig. 5). Similarly, this specific effect of butyrate on the expression of different inflammatory cytokine genes was also identified in bovine mammary epithelial cells (55). The regulatory effects of butyrate on the production and functions of TNF-α were previously explored in colonocytes (58,59). Through interacting with TNF-α, butyrate induced higher NF-κB activities in adenocarcinoma HT-29 and fetal FHC human colon cells in vitro (59). Even in the same types of cells, butyrate exhibited counter-regulatory effects on TNF-α-induced complement and inflammatory protein production, which were associated with the modulation of transcription factor activation (58,59). Therefore, combined with the data in the present study, it is suggested that the butyrate mediated pro-inflammatory response may involve multiple mechanisms of action.
In summary, butyrate inhibited the growth of colon cancer cells, but also promoted the TLR4 expression, MAPKs and NF-κB phosphorylation in these cells, and then induced microbe mediated innate immunity to maintain microenvironment homeostasis in the gut. Although, the potential mechanisms of butyrate in intestinal tolerance and colon cancer development remain to be elucidated, the findings of the present study indicated the potential usage of butyrate in the treatment of colorectal cancer.
Acknowledgements
Thanks for all members in Shao Lab and Zhou Lab of School of Medicine, Jiangsu University (Zhenjiang, China) for helpful discussion and technical assistance. Thanks to the technician in the Clinical Lab of the affiliated hospital of Jiangsu University for the flow cytometric analysis.
Glossary
Abbreviations
- Bu
butyrate
- CRC
colorectal cancer
- ERK
extracellular signal-regulated kinase
- HDACs
histone deacetylases
- IL
interleukin
- JNK
c-Jun NH2-terminal kinase
- LPS
lipopolysaccharide
- MAPK
mitogen-activated protein kinase
- NF-κB
nuclear factor-κB
- SCFA
short-chain fatty acids
- TLR
toll-like receptor
- PRRs
pathogen recognition receptors
- TNF
tumor necrosis factor
Funding
The present study was funded by grants from the National Natural Science Foundation of China (grant nos., 31570879 and 31428006) and the Key Research and Development Program of Jiangsu Province, China (grant no., BE2017696).
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors' contributions
TX and SW performed the majority of experiments and analyzed the data. CY and CZ were responsible for the culture of cell lines. NY and JX performed the flow cytometric analysis for the phenotype and the apoptosis of the cells. HJ, CL and YW conducted the ELISA analysis and the western blotting. SX and QS designed the experiments. SX analyzed the data and wrote the manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Center MM, Jemal A, Ward E. International trends in colorectal cancer incidence rates. Cancer Epidemiol Biomarkers Prev. 2009;18:1688–1694. doi: 10.1158/1055-9965.EPI-09-0090. [DOI] [PubMed] [Google Scholar]
- 2.Center MM, Jemal A, Smith RA, Ward E. Worldwide variations in colorectal cancer. CA Cancer J Clin. 2009;59:366–378. doi: 10.3322/caac.20038. [DOI] [PubMed] [Google Scholar]
- 3.Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: A meta-analysis. Gut. 2001;48:526–535. doi: 10.1136/gut.48.4.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hagemann T, Balkwill F, Lawrence T. Inflammation and cancer: A double-edged sword. Cancer Cell. 2007;12:300–301. doi: 10.1016/j.ccr.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wei EK, Giovannucci E, Wu K, Rosner B, Fuchs CS, Willett WC, Colditz GA. Comparison of risk factors for colon and rectal cancer. Int J Cancer. 2004;108:433–442. doi: 10.1002/ijc.11540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tomkovich S, Yang Y, Winglee K, Gauthier J, Mühlbauer M, Sun X, Mohamadzadeh M, Liu X, Martin P, Wang GP, et al. Locoregional effects of microbiota in a preclinical model of colon carcinogenesis. Cancer Res. 2017;77:2620–2632. doi: 10.1158/0008-5472.CAN-16-3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tjalsma H, Boleij A, Marchesi JR, Dutilh BE. A bacterial driver-passenger model for colorectal cancer: Beyond the usual suspects. Nat Rev Microbiol. 2012;10:575–582. doi: 10.1038/nrmicro2819. [DOI] [PubMed] [Google Scholar]
- 8.Wong SH, Zhao L, Zhang X, Nakatsu G, Han J, Xu W, Xiao X, Kwong TNY, Tsoi H, Wu WKK, et al. Gavage of fecal samples from patients with colorectal cancer promotes intestinal carcinogenesis in germ-free and conventional mice. Gastroenterology. 2017;153(1621–1633):e6. doi: 10.1053/j.gastro.2017.08.022. [DOI] [PubMed] [Google Scholar]
- 9.Hold GL, Garrett WS. Gut microbiota. Microbiota organization-a key to understanding CRC development. Nat Rev Gastroenterol Hepatol. 2015;12:128–129. doi: 10.1038/nrgastro.2015.25. [DOI] [PubMed] [Google Scholar]
- 10.Flemer B, Lynch DB, Brown JM, Jeffery IB, Ryan FJ, Claesson MJ, O'Riordain M, Shanahan F, O'Toole PW. Tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut. 2017;66:633–643. doi: 10.1136/gutjnl-2015-309595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zou S, Fang L, Lee MH. Dysbiosis of gut microbiota in promoting the development of colorectal cancer. Gastroenterol Rep (Oxf) 2018;6:1–12. doi: 10.1093/gastro/gox031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ray D, Kidane D. Gut microbiota imbalance and base excision repair dynamics in colon cancer. J Cancer. 2016;7:1421–1430. doi: 10.7150/jca.15480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meng C, Bai C, Brown TD, Hood LE, Tian Q. Human gut microbiota and gastrointestinal cancer. Genomics Proteomics Bioinformatics. 2018;16:33–49. doi: 10.1016/j.gpb.2017.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frosali S, Pagliari D, Gambassi G, Landolfi R, Pandolfi F, Cianci R. How the intricate interaction among toll-like receptors, microbiota, and intestinal immunity can influence gastrointestinal pathology. J Immunol Res. 2015;2015:489821. doi: 10.1155/2015/489821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fukata M, Hernandez Y, Conduah D, Cohen J, Chen A, Breglio K, Goo T, Hsu D, Xu R, Abreu MT. Innate immune signaling by toll-like receptor-4 (TLR4) shapes the inflammatory microenvironment in colitis-associated tumors. Inflamm Bowel Dis. 2009;15:997–1006. doi: 10.1002/ibd.20880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fukata M, Shang L, Santaolalla R, Sotolongo J, Pastorini C, España C, Ungaro R, Harpaz N, Cooper HS, Elson G, et al. Constitutive activation of epithelial TLR4 augments inflammatory responses to mucosal injury and drives colitis-associated tumorigenesis. Inflamm Bowel Dis. 2011;17:1464–1473. doi: 10.1002/ibd.21527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pimentel-Nunes P, Gonçalves N, Boal-Carvalho I, Afonso L, Lopes P, Roncon-Albuquerque R, Jr, Soares JB, Cardoso E, Henrique R, Moreira-Dias L, et al. Decreased Toll-interacting protein and peroxisome proliferator-activated receptor γ are associated with increased expression of Toll-like receptors in colon carcinogenesis. J Clin Pathol. 2012;65:302–308. doi: 10.1136/jclinpath-2011-200567. [DOI] [PubMed] [Google Scholar]
- 18.Yu T, Guo F, Yu Y, Sun T, Ma D, Han J, Qian Y, Kryczek I, Sun D, Nagarsheth N, et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell. 2017;170(548–563):e16. doi: 10.1016/j.cell.2017.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chung YH, Kim D. Enhanced TLR4 expression on colon cancer cells after chemotherapy promotes cell survival and epithelial-mesenchymal transition through phosphorylation of GSK3β. Anticancer Res. 2016;36:3383–3394. [PubMed] [Google Scholar]
- 20.Kuo WT, Lee TC, Yu LC. Eritoran suppresses colon cancer by altering a functional balance in toll-like receptors that bind lipopolysaccharide. Cancer Res. 2016;76:4684–4695. doi: 10.1158/0008-5472.CAN-16-0172. [DOI] [PubMed] [Google Scholar]
- 21.Rafa H, Benkhelifa S, AitYounes S, Saoula H, Belhadef S, Belkhelfa M, Boukercha A, Toumi R, Soufli I, Moralès O, et al. All-trans retinoic acid modulates TLR4/NF-κB signaling pathway targeting TNF-α and nitric oxide synthase 2 expression in colonic mucosa during Ulcerative colitis and colitis associated cancer. Mediators Inflamm. 2017;2017:7353252. doi: 10.1155/2017/7353252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Louis P, Hold GL, Flint HJ. The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol. 2014;12:661–672. doi: 10.1038/nrmicro3344. [DOI] [PubMed] [Google Scholar]
- 23.Koh A, De Vadder F, Kovatcheva-Datchary P, Bäckhed F. From dietary fiber to host physiology: Short-chain fatty acids as key bacterial metabolites. Cell. 2016;165:1332–1345. doi: 10.1016/j.cell.2016.05.041. [DOI] [PubMed] [Google Scholar]
- 24.Cummings JH, Pomare EW, Branch WJ, Naylor CP, Macfarlane GT. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut. 1987;28:1221–1227. doi: 10.1136/gut.28.10.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wong JM, de Souza R, Kendall CW, Emam A, Jenkins DJ. Colonic health: Fermentation and short chain fatty acids. J Clin Gastroenterol. 2006;40:235–243. doi: 10.1097/00004836-200603000-00015. [DOI] [PubMed] [Google Scholar]
- 26.Chisolm DA, Weinmann AS. Connections between metabolism and epigenetics in programming cellular differentiation. Annu Rev Immunol. 2018;36:221–246. doi: 10.1146/annurev-immunol-042617-053127. [DOI] [PubMed] [Google Scholar]
- 27.Zheng L, Kelly CJ, Battista KD, Schaefer R, Lanis JM, Alexeev EE, Wang RX, Onyiah JC, Kominsky DJ, Colgan SP. Microbial-derived butyrate promotes epithelial barrier function through IL-10 receptor-dependent repression of claudin-2. J Immunol. 2017;199:2976–2984. doi: 10.4049/jimmunol.1700105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mollica MP, Raso Mattace G, Cavaliere G, Trinchese G, De Filippo C, Aceto S, Prisco M, Pirozzi C, Di Guida F, Lama A, et al. Butyrate regulates liver mitochondrial function, efficiency, and dynamics in insulin-resistant obese mice. Diabetes. 2017;66:1405–1418. doi: 10.2337/db16-0924. [DOI] [PubMed] [Google Scholar]
- 29.Astakhova L, Ngara M, Babich O, Prosekov A, Asyakina L, Dyshlyuk L, Midtvedt T, Zhou X, Ernberg I, Matskova L. Short chain fatty acids (SCFA) reprogram gene expression in human malignant epithelial and lymphoid cells. PLoS One. 2016;11:e0154102. doi: 10.1371/journal.pone.0154102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fellows R, Denizot J, Stellato C, Cuomo A, Jain P, Stoyanova E, Balázsi S, Hajnády Z, Liebert A, Kazakevych J, et al. Microbiota derived short chain fatty acids promote histone crotonylation in the colon through histone deacetylases. Nat Commun. 2018;9:105. doi: 10.1038/s41467-017-02651-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Julien O, Wells JA. Caspases and their substrates. Cell Death Differ. 2017;24:1380–1389. doi: 10.1038/cdd.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Inan MS, Rasoulpour RJ, Yin L, Hubbard AK, Rosenberg DW, Giardina C. The luminal short-chain fatty acid butyrate modulates NF-kappaB activity in a human colonic epithelial cell line. Gastroenterology. 2000;118:724–734. doi: 10.1016/S0016-5085(00)70142-9. [DOI] [PubMed] [Google Scholar]
- 33.Katzenmaier EM, André S, Kopitz J, Gabius HJ. Impact of sodium butyrate on the network of adhesion/growth-regulatory galectins in human colon cancer in vitro. Anticancer Res. 2014;34:5429–5438. [PubMed] [Google Scholar]
- 34.Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: A cell biological perspective. Annu Rev Immunol. 2015;33:257–290. doi: 10.1146/annurev-immunol-032414-112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kitchens RL. Role of CD14 in cellular recognition of bacterial lipopolysaccharides. Chem Immunol. 2000;74:61–82. doi: 10.1159/000058750. [DOI] [PubMed] [Google Scholar]
- 36.Yang H, Young DW, Gusovsky F, Chow JC. Cellular events mediated by lipopolysaccharide-stimulated toll-like receptor 4. MD-2 is required for activation of mitogen-activated protein kinases and Elk-1. J Biol Chem. 2000;275:20861–20866. doi: 10.1074/jbc.M002896200. [DOI] [PubMed] [Google Scholar]
- 37.Mariani F, Sena P, Roncucci L. Inflammatory pathways in the early steps of colorectal cancer development. World J Gastroenterol. 2014;20:9716–9731. doi: 10.3748/wjg.v20.i29.9716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pesic M, Greten FR. Inflammation and cancer: Tissue regeneration gone awry. Curr Opin Cell Biol. 2016;43:55–61. doi: 10.1016/j.ceb.2016.07.010. [DOI] [PubMed] [Google Scholar]
- 39.Sivaprakasam S, Bhutia YD, Ramachandran S, Ganapathy V. Cell-surface and nuclear receptors in the colon as targets for bacterial metabolites and its relevance to colon health. Nutrients. 2017;9 doi: 10.3390/nu9080856. pii: E856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blouin JM, Penot G, Collinet M, Nacfer M, Forest C, Laurent-Puig P, Coumoul X, Barouki R, Benelli C, Bortoli S. Butyrate elicits a metabolic switch in human colon cancer cells by targeting the pyruvate dehydrogenase complex. Int J Cancer. 2011;128:2591–2601. doi: 10.1002/ijc.25599. [DOI] [PubMed] [Google Scholar]
- 41.Bultman SJ. Molecular pathways: Gene-environment interactions regulating dietary fiber induction of proliferation and apoptosis via butyrate for cancer prevention. Clin Cancer Res. 2014;20:799–803. doi: 10.1158/1078-0432.CCR-13-2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hague A, Butt AJ, Paraskeva C. The role of butyrate in human colonic epithelial cells: An energy source or inducer of differentiation and apoptosis? Proc Nutr Soc. 1996;55:937–943. doi: 10.1079/PNS19960090. [DOI] [PubMed] [Google Scholar]
- 43.Archer SY, Johnson J, Kim HJ, Ma Q, Mou H, Daesety V, Meng S, Hodin RA. The histone deacetylase inhibitor butyrate downregulates cyclin B1 gene expression via a p21/WAF-1-dependent mechanism in human colon cancer cells. Am J Physiol Gastrointest Liver Physiol. 2005;289:G696–G703. doi: 10.1152/ajpgi.00575.2004. [DOI] [PubMed] [Google Scholar]
- 44.Wilson AJ, Byun DS, Popova N, Murray LB, L'Italien K, Sowa Y, Arango D, Velcich A, Augenlicht LH, Mariadason JM. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J Biol Chem. 2006;281:13548–13558. doi: 10.1074/jbc.M510023200. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y, Zhou L, Bao YL, Wu Y, Yu CL, Huang YX, Sun Y, Zheng LH, Li YX. Butyrate induces cell apoptosis through activation of JNK MAP kinase pathway in human colon cancer RKO cells. Chem Biol Interact. 2010;185:174–181. doi: 10.1016/j.cbi.2010.03.035. [DOI] [PubMed] [Google Scholar]
- 46.Hu S, Liu L, Chang EB, Wang JY, Raufman JP. Butyrate inhibits pro-proliferative miR-92a by diminishing c-Myc-induced miR-17-92a cluster transcription in human colon cancer cells. Mol Cancer. 2015;14:180. doi: 10.1186/s12943-015-0450-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ortega-Cava CF, Ishihara S, Rumi MA, Kawashima K, Ishimura N, Kazumori H, Udagawa J, Kadowaki Y, Kinoshita Y. Strategic compartmentalization of Toll-like receptor 4 in the mouse gut. J Immunol. 2003;170:3977–3985. doi: 10.4049/jimmunol.170.8.3977. [DOI] [PubMed] [Google Scholar]
- 48.Meng D, Zhu W, Shi HN, Lu L, Wijendran V, Xu W, Walker WA. Toll-like receptor-4 in human and mouse colonic epithelium is developmentally regulated: A possible role in necrotizing enterocolitis. Pediatr Res. 2015;77:416–424. doi: 10.1038/pr.2014.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang EL, Qian ZR, Nakasono M, Tanahashi T, Yoshimoto K, Bando Y, Kudo E, Shimada M, Sano T. High expression of Toll-like receptor 4/myeloid differentiation factor 88 signals correlates with poor prognosis in colorectal cancer. Br J Cancer. 2010;102:908–915. doi: 10.1038/sj.bjc.6605558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lu CC, Kuo HC, Wang FS, Jou MH, Lee KC, Chuang JH. Upregulation of TLRs and IL-6 as a marker in human colorectal cancer. Int J Mol Sci. 2014;16:159–177. doi: 10.3390/ijms16010159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paarnio K, Väyrynen S, Klintrup K, Ohtonen P, Mäkinen MJ, Mäkelä J, Karttunen TJ. Divergent expression of bacterial wall sensing Toll-like receptors 2 and 4 in colorectal cancer. World J Gastroenterol. 2017;23:4831–4838. doi: 10.3748/wjg.v23.i26.4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rosadini CV, Kagan JC. Early innate immune responses to bacterial LPS. Curr Opin Immunol. 2017;44:14–19. doi: 10.1016/j.coi.2016.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu JL, Zou JY, Hu ED, Chen DZ, Chen L, Lu FB, Xu LM, Zheng MH, Li H, Huang Y, et al. Sodium butyrate ameliorates S100/FCA-induced autoimmune hepatitis through regulation of intestinal tight junction and toll-like receptor 4 signaling pathway. Immunol Lett. 2017;190:169–176. doi: 10.1016/j.imlet.2017.08.005. [DOI] [PubMed] [Google Scholar]
- 54.Iraporda C, Errea A, Romanin DE, Cayet D, Pereyra E, Pignataro O, Sirard JC, Garrote GL, Abraham AG, Rumbo M. Lactate and short chain fatty acids produced by microbial fermentation downregulate proinflammatory responses in intestinal epithelial cells and myeloid cells. Immunobiology. 2015;220:1161–1169. doi: 10.1016/j.imbio.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 55.Alva-Murillo N, Medina-Estrada I, Báez-Magaña M, Ochoa-Zarzosa A, López-Meza JE. The activation of the TLR2/p38 pathway by sodium butyrate in bovine mammary epithelial cells is involved in the reduction of Staphylococcus aureus internalization. Mol Immunol. 2015;68:445–455. doi: 10.1016/j.molimm.2015.09.025. (2 pt B) [DOI] [PubMed] [Google Scholar]
- 56.Lin MY, de Zoete MR, van Putten JP, Strijbis K. Redirection of epithelial immune responses by short-chain fatty acids through inhibition of histone deacetylases. Front Immunol. 2015;6:554. doi: 10.3389/fimmu.2015.00554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Larraufie P, Doré J, Lapaque N, Blottière HM. TLR ligands and butyrate increase Pyy expression through two distinct but inter-regulated pathways. Cell Microbiol. 2017;19 doi: 10.1111/cmi.12648. [DOI] [PubMed] [Google Scholar]
- 58.Andoh A, Fujiyama Y, Hata K, Araki Y, Takaya H, Shimada M, Bamba T. Counter-regulatory effect of sodium butyrate on tumour necrosis factor-alpha (TNF-alpha)-induced complement C3 and factor B biosynthesis in human intestinal epithelial cells. Clin Exp Immunol. 1999;118:23–29. doi: 10.1046/j.1365-2249.1999.01038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hýzd'alová M, Hofmanová J, Pacherník J, Vaculová A, Kozubík A. The interaction of butyrate with TNF-alpha during differentiation and apoptosis of colon epithelial cells: Role of NF-kappaB activation. Cytokine. 2008;44:33–43. doi: 10.1016/j.cyto.2008.06.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.