

Abstract
Previous studies identified ebselen as a potent in vitro and in vivo inhibitor of the Mycobacterium tuberculosis (Mtb) antigen 85 (Ag85) complex, comprising three homologous enzymes required for the biosynthesis of the mycobacterial cell wall. In this study, the Mtb Ag85C enzyme was cocrystallized with azido and adamantyl ebselen derivatives, resulting in two crystallographic structures of 2.01 and 1.30 Å resolution, respectively. Both structures displayed the anticipated covalent modification of the solvent accessible, noncatalytic Cys209 residue forming a selenenylsulfide bond. Continuous difference density for both thiol modifiers allowed for the assessment of interactions that influence ebselen binding and inhibitor orientation that were unobserved in previous Ag85C ebselen structures. The kinact/KI values for ebselen, adamantyl ebselen, and azido ebselen support the importance of observed constructive chemical interactions with Arg239 for increased in vitro efficacy toward Ag85C. To better understand the in vitro kinetic properties of these ebselen derivatives, the energetics of specific protein−inhibitor interactions and relative reaction free energies were calculated for ebselen and both derivatives using density functional theory. These studies further support the different in vitro properties of ebselen and two select ebselen derivatives from our previously published ebselen library with respect to kinetics and protein−inhibitor interactions. In both structures, the α9 helix was displaced farther from the enzyme active site than the previous Ag85C ebselen structure, resulting in the restructuring of a connecting loop and imparting a conformational change to residues believed to play a role in substrate binding specific to Ag85C. These notable structural changes directly affect protein stability, reducing the overall melting temperature by up to 14.5 °C, resulting in the unfolding of protein at physiological temperatures. Additionally, this structural rearrangement due to covalent allosteric modification creates a sizable solvent network that encompasses the active site and extends to the modified Cys209 residue. In all, this study outlines factors that influence enzyme inhibition by ebselen and its derivatives while further highlighting the effects of the covalent modification of Cys209 by said inhibitors on the structure and stability of Ag85C. Furthermore, the results suggest a strategy for developing new classes of Ag85 inhibitors with increased specificity and potency.
Keywords: Mycobacterium tuberculosis, antigen 85 complex, allosteric covalent inhibition, ebselen
An old world infectious disease, Tuberculosis, is causing modern day problems.1 Despite being considered curable, current treatment requires a multidrug regimen over the course of months. As a result of failed adherence to treatment plans, modern strains of the disease have developed multidrug and total drug resistance to current antibiotics.2 As a result, there has been a significant push to discover and develop new antibiotics that target Mycobacterium tuberculosis (Mtb). One such effort led to the recent in vitro discovery of ebselen as a potent inhibitor of the three essential, homologous Mtb proteins of the antigen 85 complex (Ag85A, Ag85B, and Ag85C).3 The Ag85 complex is part of a cell wall biosynthetic pathway unique to Mtb and similar bacteria, and is responsible for the transesterification reactions that mycolate trehalose monomycolate to form trehalose dimycolate (TDM) and arabinogalactan to form mycolylarabinogalactan.4,5 Single mutant knockouts of any of the fbp genes encoding Ag85A, B, or C have been successfully cultured; however, the fbp genes appear to be synthetically lethal since the creation of a viable fbp double knockout was attempted but was unsuccessful.6,7 As a result, a drug targeting the Ag85 complex must successfully inhibit at least two of the three homologues. The inhibitory activity of ebselen was shown to do so, having an MIC of 20 µg/mL against the Mtb mc26206 strain and directly affecting the production of TDM.3
Ebselen (2-phenyl-1,2-benzisoselenazol-3(2H)-one), a selenium containing compound, was discovered by screening Ag85C activity against the NIH Clinical Collection (Figure 1a).3 Inhibition of the Ag85s by ebselen proceeds through an unexpected mechanism. Ebselen was shown to covalently modify the only cysteine, Cys209, in Ag85C, which is conserved, noncatalytic, and solvent-accessible through the reversible oxidation of the γ-sulfur of Cys209 by the selenium of ebselen.3,8 The resulting modification of Cys209 leads to the relaxation of the native, kinked α9 helix, displacing the catalytically relevant Glu228 residue from the active site, and thereby rendering the enzyme inactive through covalent allosteric inhibition (Figure 2).9
Figure 1.

Ebselen and its derivatives; the primary phenyl ring (left side of each molecule) is on the core moiety, while the secondary phenyl ring or adamantyl is amide linked. (a) Ebselen; (b) p-azido ebselen; (c) p-amino ebselen; (d) adamantyl ebselen.
Figure 2.

Inhibiting Ag85C through covalent allosteric inhibition. Native Ag85C (green; PDB, 1DQZ) superimposed with Ag85C covalently modified at Cys209 by ebselen, resulting in a structural shift in α9 helix that displaces the catalytically relevant Glu228 (gray; PDB, 4QDU).
The crystal structure of Mtb Ag85C covalently modified by ebselen was solved at 1.4 A (PDB 4QDU). However, due to the partial occupancy for the covalent modifier, complete electron density was lacking for the secondary phenyl ring of ebselen.9 As a result, a full assessment of protein—inhibitor interactions was not possible, resulting in a cursory assignment of a π—π interaction between the aromatic side chain of Phe254 and the primary phenyl ring of ebselen, and a hypothesized cation—n interaction between the guanidinium moiety of Arg239 and the carbonyl of the amide of ebselen.9–11
In an effort to optimize the original ebselen lead, a library of ebselen derivatives has been synthesized and tested for the inhibition of Ag85C in vitro and for in vivo inhibition of Mtb growth.12 Upon inspection of the growth inhibitory data (MIC50) for the whole library of ebselen derivatives, a noticeable trend was not initially apparent. The MIC50 values varied between 12.5 μg/mL and 100 μg/mL. In an attempt to better compare the various derivatives, a set concentration and incubation time were used to determine the IC50 values for those compounds against Ag85C. These data ranged between 0.5 μM and >100 μM and correlated with the determined MIC50 values but still did not fit a clear structure—activity relationship.12 To better understand the in vitro inhibition data with respect to ebselen-binding interactions, orientation, and the structural effects imparted on Ag85C by the covalent modification of Cys209, we selected two chemically dissimilar derivatives for X-ray crystallographic studies. One of these derivatives has an azido group placed on the secondary phenyl ring para to the amide linkage (Figure 1b). The second derivative has an adamantyl group in place of the secondary phenyl group (Figure 1d). Mtb Ag85C was successfully cocrystallized in the presence of these two ebselen derivatives, yielding two X-ray crystal structures of 2.01 and 1.30 A resolution. These structures and resulting interaction and reaction energies computed with density functional theory reveal factors that influence initial drug recognition and reactivity. To compliment our crystallographic and computed thermodynamic analysis, kinact/KI values were determined for ebselen and the two derivatives to provide further insight on the kinetics of inactivation. On the basis of these findings, we have established a better understanding of the in vitro properties with regard to the inhibition of Mtb Ag85C by ebselen and our library of recently published derivatives.12 Additionally, observed structural changes to Ag85C imparted by the covalent modification of Cys209 by the two ebselen derivatives help explain the decrease in protein thermal stability and also provide further insight into future drug design efforts.
■ RESULTS AND DISCUSSION
In Vitro Kinetic Properties of Selected Inhibitors.
As stated, enzymatic inhibition is dependent upon the covalent modification of Cys209, which lies outside the active site; therefore, inhibition by ebselen and its derivatives is both concentration and time dependent. To assess in vitro inhibition with respect to time and concentration, kinact/KI values for the respective derivatives were determined by monitoring the transesterase activity of Ag85C in the presence of varying concentrations of inhibitor as a function of time. We observe the expected decay in initial velocity to a plateau of terminal velocity as a result of covalent inhibition over time.13 By fitting the progress curves as a one-phase association event (equation and data fitting in methods), kobs were determined for each enzymatic reaction in the presence of specific inhibitor concentration (Figure 3a—c). Plotting the respective kobs as a function of inhibitor concentration, the kinact/KI was determined as the slope of each line (Figure 3d—f).13 The determined kinact/KI values for the inhibitors are ebselen = 0.3057 ± 0.0140 μM−1 Min−1, adamantyl ebselen = 0.0065 ± 0.0003 μM−1 Min−1, and azido ebselen = 0.1845 ± 0.0094 μM−1 Min−1. Therefore, inhibition of Ag85C by ebselen proceeds slightly faster than that of azido ebselen and almost 50-fold faster than adamantyl ebselen.
Figure 3.

kinact/KI determination: (a−c) progress curves of the enzymatic reactions in the presence of specific inhibitor at specific concentrations as a function of increasing reaction product (resorufin, μM) over time (min). The average of the triplicate reads is plotted and fitted with a one-phase association equation. (d−f) Resulting kinact/KI plots for ebselen, adamantyl ebselen, and azido ebselen.
Covalent inhibition can proceed through three schemes: nonspecific, quiescent, and mechanism-based.13 Nonspecific is a result of multiple accessible covalent sites, that readily undergo modification, yet each site may not lead to inhibition; therefore, a saturation/full inhibition point may never be obtained with respect to inhibitor concentration. Quiescent and mechanism-based modification rely on the noncovalent binding affinity of an inhibitor as well as the rate of covalent modification. While inhibition of Ag85C by ebselen and its derivatives is most likely represented by quiescent inhibition, as full inhibition is certainly obtainable and there must be an initial short-lived noncovalent binding event, we analyzed the data as nonspecific for two major reasons. Complete inhibition by ebselen and azido ebselen at higher concentrations (2 and 3 μM) is obtained within 2 to 4 min, drastically decreasing in time as concentration increases. This becomes problematic as the transition from initial rate to terminal rate is simply lost since the time-resolution of our experimental setup is not sufficiently high; therefore, accurately determining kobs is challenging. The second issue is the high concentration of enzyme required, 500 nM, for the assay, which limits our ability to use lower inhibitor concentrations as we are already near a 1 to 1 stoichiometric equivalent of inhibitor to enzyme. For these reasons, kinact/KI was determined using nonspecific inhibition analysis as a reasonable compromise.
Structure Determination of Ag85C Covalently Modified by Ebselen Derivatives.
The Ag85C-azido ebselen and Ag85C-adamantyl ebselen structures were solved at 2.01 and 1.30 A, respectively. Complete data collection and refinement statistics are presented in Table 1. The generated 2fo-fc map for the azido ebselen structure displayed electron density up to the second phenyl ring of the modifier, where partial density was observed when contouring the map at 1.5σ. Difference density was not observed for the linear azide moiety. As a result, further refinement of this portion of the inhibitor involved modeling it as a terminal amine (Figure 1c). Analysis of the azido ebselen compound after purification indicated an intact azide on the ebselen derivative that was used for crystallographic and inhibition studies.12 We believe the terminal azide is structurally dynamic, thus explaining the lack of observable electron density.
Table 1.
Crystallographic and Refinement Statistics a
| Ag85C–azido ebselen |
Ag85C–adamantyl ebselen |
|
|---|---|---|
| PDB ID | 5KWJ | 5KWI |
| Data Collection | ||
| diffraction source | APS LS-CAT F | APS LS-CAT G |
| wavelength (Å) | 0.97872 | 0.97857 |
| space group | C2221 | P43212 |
| a, b, c (Å) | 88.474, 88.479, 161.950 |
63.405, 63.405, 160.210 |
| α, β, γ (deg) | 90, 90, 90 | 90, 90, 90 |
| mosaicity (deg) | 0.605 | 0.318 |
| resolution range (Å) | 49.51–2.01 | 40.85–1.30 |
| no. of unique reflections | 42348 (3917) | 81206 (7968) |
| completeness (%) | 99.32 (93.13) | 99.96 (99.67) |
| redundancy | 14.9 (15.0) | 13.0 (6.9) |
| ⟨I/σ(I)⟩ | 20.91 (5.50) | 20.00 (2.69) |
| Rmeas | 0.104 (0.563) | 0.071 (0.578) |
| overall B factor from Wilson plot (Å2) |
24.59 | 14.90 |
| Refinement | ||
| resolution range (Å) | 49.51–2.01 | 40.85–1.3 |
| completeness (%) | 99.32 (93.13) | 99.94 (99.67) |
| Rwork/Rfree (%) | 16.23/19.68 | 16.60/17.63 |
| total no. of atoms | 4781 | 2533 |
| protein | 4452 | 2237 |
| solvent | 328 | 296 |
| RM.S.Deviations | ||
| bonds (Å) | 0.008 | 0.006 |
| angles (deg) | 1.090 | 1.550 |
| average B factors (Å2) | 24.59 | 14.88 |
| protein | 25.80 | 15.80 |
| solvent | 34.00 | 25.20 |
| Ramachandran Plot | ||
| most favored (%) | 96.81 | 97.53 |
| allowed (%) | 3.19 | 2.47 |
Parentheses indicate the values for the highest resolution shell.
The calculated, likelihood-weighted fo-fc omit maps for both structures were generated by omitting the modeled covalent modifier as well as the γ-sulfur and β-carbon atoms of Cys209 (Figure 4). Strong difference density for the covalent linkage between the Cys209 sulfur atom of Ag85C and the selenium of the ebselen derivatives is present in both structures. This observable covalent modification of Cys209 of Ag85C is consistent with previous studies.3,9 It is worth noting, however, that in both the azido and adamantyl ebselen structures the selenium atoms have much higher occupancy values than the previous Ag85C-ebselen structure: 0.87 and 0.94 compared to 0.53, respectively.9 Also, in contrast to the Ag85C-ebselen structure, difference density for the entire adamantyl ebselen molecule is observed. With regard to azido ebselen, difference density for the primary and secondary phenyl rings is observed, yet not the azide moiety itself, further supporting the truncated p-amino model.
Figure 4.
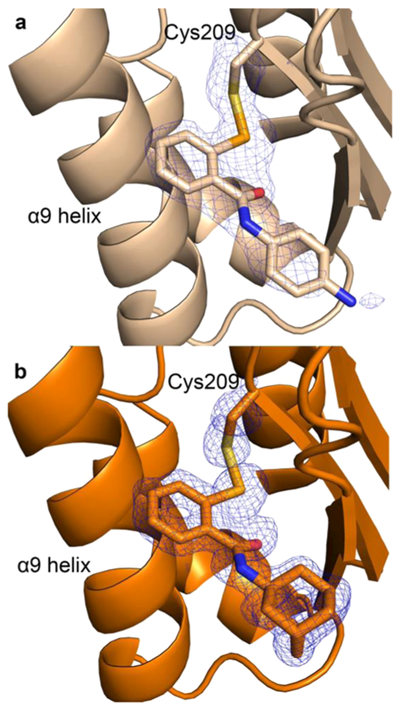
Calculated Fo−Fc omit maps for the modified Cys209 of Ag85C and respective ebselen derivatives. Both maps are contoured at 3σ in blue mesh. Atom color: carbon is beige in panel a, orange in panel b, nitrogen blue, oxygen red, sulfur yellow, and selenium light orange. (a) Ag85C covalently modified by azido ebselen and (b) Ag85C covalently modified by adamantyl ebselen.
Ag85C-Ebselen Derivative Interactions.
In both structures, the derivatized end (p-amino and adamantyl) of the covalent modifier is pointing toward solvent, with the carbonyl of the amide linkage oriented toward Cβ of Phe252. The solvent-exposed binding site therefore reduces the number of potential protein—inhibitor interactions, limiting the potential effects of steric bulk on inhibitor binding. In agreement with the previous Ag85C-ebselen structure, the primary phenyl ring of the ebselen derivative forms a centered, yet tilted π—π stacking interaction with the aromatic side chain of Phe254 in both the azido and adamantyl structures (Figure 5a and d).9 Because of the lack of complete electron density for the ebselen molecule in 4QDU, proper orientation and interactions with the secondary phenyl ring of ebselen were not resolved.9 As a result, the orientation of the amide linkage remains ambiguous in the original Ag85C ebselen structure. In contrast, complete density is observable for the secondary phenyl ring in the azido ebselen-modified structure, allowing for further characterization. Notably, a favorable cation—π interaction may be split between the parallel orientation of the guanidinium moiety of Arg239 to the sp2-hybridized nitrogen of the amide linkage and to the secondary sp2-hybridized phenyl ring of the modifier (Figure 5a). Conversely, in the adamantyl ebselen structure this secondary phenyl ring of ebselen is omitted and replaced with a bulky hydrophobic, sp3-hybridized adamantyl group. As a result, the observable cation—π interaction is no longer split and solely resides between Arg239 and the nitrogen of the amide linkage with the guanidinium moiety once again parallel to the inhibitor (Figure 5d). However, the once favorable cation—π interaction with the secondary benzylic ring now results in a van der Waals interaction between Arg239 and the hydrophobic adamantyl group, increasing the distance of the Arg239—amide interaction when compared to that of the azido derivative (Figure 5d). The distance between the central carbon of the Arg239 guanidinium and the nitrogen of the amide linkage increases from 3.6 to 4.4 Å between the azido and adamantyl modifiers (Figure 5a and d).
Figure 5.

Crystallographic interactions of ebselen derivatives with Ag85C and calculated energetics of interacting monomers after QM geometry optimization. Atom color assignment is identical to that of Figure 2. QM optimized geometries are given in Supplemental Figure 1. (a) Interactions observed in the crystal structure of Ag85C−azido ebselen are similar to those observed in the Ag85C-adamantyl ebselen structure (d). (b, c, e, and f) Interaction energetic curves for respective residue derivative interaction.
Computed Interaction Energies Using DFT.
Because of the unique mode of inhibition by allosteric covalent modification, we sought to better characterize the observed crystallographic Ag85C—inhibitor interactions with respect to the azido and adamantyl ebselen derivatives. Isothermal titration calorimetry is commonly used to measure enthalpic and entropic contributions to drug binding.14 However, because the target site is solvent-exposed, results in covalent binding, and displaces a secondary structure element in the protein, obtaining interpretable thermodynamic data is challenging due to the various sources of heat generation. Thus, to estimate the magnitude of the initial interactions that drive preliminary binding prior to covalent modification, we instead used dispersion-corrected DFT to compute non-covalent interaction energies between allosteric site residues Phe254 and Arg239 with ebselen and the two ebselen derivatives. Another contribution to the inhibition of Ag85C is the chemical reactivity of the ebselen derivatives. Therefore, we computed reaction free energies for adduct formation to investigate potential differences in electronic effects in the respective derivatives to gain insight into how reactivity of the ebselen derivatives affects inhibition in vitro.
DFT calculations were performed with a polarizable continuum solvent (SMD) to approximate the effects of the local protein and solvent environment of the allosteric site.15 Because the inhibitor binding site is located at the interface of a partially hydrophobic cleft and the solvent-exposed surface of the protein, the choice of an appropriate dielectric constant ε is not obvious. Thus, we first optimized the geometries of each model (see Methods) using three different dielectric constants, ε =10, ε = 20, and ε = 80, and compared them to the corresponding X-ray crystal structures of Ag85C-adamantyl ebselen and Ag85C-azido ebselen (Figure S3). The atomic RMS displacements for heavy atoms between the optimized models and the corresponding X-ray crystal structures were similar for the different dielectric values. Therefore, we also analyzed key interaction distances and relative residue conformations (see Supporting Information methods section for details). For the Ag85C-adamantyl ebselen model with ε = 10, the interaction distances are 3.90 A between the closest carbons of the phenyl ring of Phe254 and primary phenyl group of adamantyl ebselen, and 5.00 A between the nitrogen of the amide linkage and the central carbon of the guanidinium side chain of Arg239, compared to 3.6 and 4.4 A in the crystal structure, respectively (Figure S3F). In the Ag85C-azido ebselen ε = 10 model, the interaction distances for the same atoms are 3.85 A for Phe254-inhibitor and 3.78 A for Arg239-inhibitor compared to 3.6 and 3.7 A in the crystal structure, respectively (Figure S3B). When the dielectric constant was set to higher values (ε = 20 or ε = 80), the interaction distances between Arg239 and adamantyl ebselen increased to 5.33 A (ε = 20) and 5.97 A (ε = 80), respectively. For the Ag85C-azido ebselen model, as the dielectric was increased the interaction distances between Arg239 and azido ebselen became shorter (3.48 A in the ε = 80 model). For the higher dielectric models, the interatomic distances are in worse agreement with the X-ray structure: therefore, the quantum cluster models with ε =10 (Figure S3) were used in all further interaction energy calculations. A similar comparison was not possible for Ag85C-ebselen due to the lack of a complete model for the ebselen molecule, but ε = 10 was used on the basis of the results for the two ebselen derivatives. Hereafter, we refer to the ε =10 models as condensed phase models.
To quantify the interactions between the ebselen derivatives and the closely interacting side chains from Ag85C, we extracted pairs of moieties from the optimized cluster models. The resulting dimers, namely, Phe254-azido ebselen, Arg239-azido ebselen, Phe254-adamantyl ebselen, Arg239-adamantyl ebselen, Phe254-ebselen, and Arg239-ebselen were then used to compute noncovalent interaction energy curves in the condensed phase. In these calculations, Phe254 was modeled as benzene, and Arg239 was modeled as a guanidinium cation to reduce computation time. We computed benzene—benzene and ammonium (cation)—benzene noncovalent interaction energies to validate the DFT-D3BJ method used in this study. Further details are given in the Supplemental Methods section.
At the equilibrium geometry (intermonomer distance of 3.90 A), the noncovalent interaction energy between Phe254 and adamantyl ebselen was computed to be −8.0 kcal/mol in the condensed phase. The computed interaction energy minimum for the Phe254 and azido ebselen pair is −10.2 kcal/mol at an intermonomer separation of 3.86 A and −8.9 kcal/mol at 3.55 A for Phe254 and ebselen. The complete interaction energy curves for both derivatives with Phe254 are given in Figure 5b and e.15 For comparison, the interaction energies computed here are approximately three to four times more favorable than the benchmark CCSD(T) value for the parallel displaced benzene—benzene dimer (−2.62 kcal/mol).16 Various substituents on aromatic rings can greatly influence interaction energies.17,20 As the primary phenyl ring of ebselen and both ebselen derivatives contain selenium and a carbonyl substituent, the more favorable interactions are not surprising and can be attributed to dispersive and other interactions that are not further characterized here. Thus, both ebselen derivatives are capable of forming favorable stacking interactions with Phe254, which is expected to contribute to initial drug recognition and binding.
Of greater intrigue to us are the interaction energetics between the cationic guanidinium side chain of Arg239 and respective ebselen molecules since the observed noncovalent interaction differs between the two derivatives. The calculated minimum interaction energy for Arg239 and adamantyl ebselen is -4.1 kcal/mol for the condensed phase model at 5.04 A. Conversely, the interaction energy for Arg239 and azido ebselen is -7.0 kcal/mol for the condensed phase model at 3.78 A, approximately double that computed for the adamantyl ebselen system. The complete interaction energy curves are given in Figure 5c and f. The Arg239 interaction with ebselen at an equilibrium distance of 3.76 A is -7.4 kcal/mol. The comparable values for the ebselen and azido ebselen-Arg239 interactions are consistent with a constructive split cation—π interaction, whereas the higher interaction energetics for the Arg-adamantyl ebselen support much weaker cation—sp3 dispersive interactions. This interaction increases the distance between the guanidinium moiety and the amide nitrogen by 1.3 A when compared to ebselen and azido ebselen.20 This increase in distance may be the cause for the reduced interaction energies calculated for the Arg239-adamantyl ebselen model. The calculated interaction energies for Arg239-ebselen suggest more favorable interactions than guanidinium-benzene.21 Again, the more favorable interactions can be attributed to additional electrostatic, dispersion, and inductive interactions from other chemical moieties within ebselen but are not characterized here. Considering Arg-benzene (i.e., cation—π) interactions specifically, our computed values are closer to those of the parallel Arg-benzene orientation than those for the perpendicular orientation, which are −8.6 and −14.9 kcal/mol, respectively.21,22 The parallel interaction orientation has been previously shown to be favorably formed in solution.23
Comparing both sets of complete interaction energy curves for ebselen and the two derivatives, we see that each molecule shares similar interaction energies with Phe254 (Figure 6a). However, the interactions with Arg239 and the site of derivatization display obvious differences (Figure 6a). These observations suggest that interactions between the inhibitor and Arg239 may facilitate initial noncovalent drug binding to Ag85C and may partially explain the differences in the observed in vitro kinetic inhibition properties.
Figure 6.

Protein−inhibitor interactions and relative reaction free energies for the covalent modification of cysteine by ebselen and its derivatives. Abbreviations are as follows: AdaEbs = adamantyl ebselen, AziEbs = azido ebselen, and Ebs = ebselen. (a) Both inhibitors have similar energetics between Phe254 and the primary benzylic ring. However, the cation-sp3-hybridized adamantyl van der Waals interaction results in a much weaker calculated value. (b) Difference in calculated RXN energies between ebselen and the derivatives suggests that respective derivatives can influence the reactivity of selenium toward the sulfur of Cys209.
Reaction Free Energies.
The chemical moieties of the various ebselen derivatives could affect the reactive nature of the selenium with regard to its ability to covalently modify the sulfur of Cys209. Thus, to estimate the thermodynamics of each respective reaction we computed relative reaction free energies (AAGr) for the oxidation of cysteine by the selenium of the respective ebselen derivative using Cys209-ebselen as a reference according to the reactions shown in Figure 6b. The AAGr value for the formation of the Cys209-adamantyl ebselen adduct compared to the Cys209-ebselen adduct is 0.1 kcal/mol. Similarly, the AAGr value for the formation of the Cys209-azido ebselen compared to the Cys209-ebselen adduct is 0.5 kcal/mol. The negligible difference in the computed reaction free energies for azido and adamantyl ebselen compared to ebselen suggests that differences in the reactivity of the selenium in each inhibitor toward Cys209 are not major contributors to the observed inhibition differences in vitro kinetics (Figure 3). Instead, this finding provides additional evidence that the inhibition of Ag85C is governed by initial noncovalent binding of the ebselen derivatives.
Dynamic Nature of the α9 Helix and the Connecting Loop.
Consistent with the previously published Ag85C-ebselen structure, a relaxation of the α9 helix and restructuring of a connective loop (Gly210—Pro223) is observed as a result of covalent modification of Cys209.9 However, there is a noticeable difference in the observed movement of the α9 helix when the azido and adamantyl ebselen structures are superimposed with the previous Ag85C-ebselen structure (RMSD = 0.220 and 0.201 A2, respectively, compared with PDB entry 4QDU). The movement of the α9 helix shifts residue Phe226 by 1.8 and 1.6 A in the azido and adamantyl structures, respectively. Figure 7c highlights the movement of the α9 helix between the original Ag85C-ebselen structure and the two new Ag85C-ebselen derivative structures relative to native Ag85C. Helical movement from native to modified with respect to the azido ebselen is 9.5 A when measured from the amide backbone nitrogen of Lys225. In the new structures, helical movement directly results in a nearly identical conformational change to Trp157, rotating χ2 by +110° with respect to the previous Ag85C-ebselen structure. This side chain rotation reorients Trp157 perpendicular to the α9 helix compared to the previous parallel orientation observed in 4QDU (Figure 7a and b). An additional consequence of the larger shift in the α9 helix is the elongation and restructuring of the 14-residue loop connecting the β7 strand to the α9 helix (Gly210—Pro223). A key difference in the restructuring of this relatively dynamic loop between the azido and adamantyl structures is the conformational change in Leu217; this hydrophobic side chain now points toward the active site and the identified trehalose binding site.24,25 This orientation of Leu217 is similar to that observed in the Ag85C structure covalently modified by diethyl phosphonate, in which Leu217 is directed toward diethyl phosphate (PDB: 1DQY).26 This region, in addition to residues Phe226 and Trp157, was previously investigated by the Barry group with the potential to establish substrate specify as a result of sequence differences between the Ag85 homologues.27 Both new structures further highlight the dynamic nature of this region.
Figure 7.

Further observed structural shift of α9 helix away from the active site, restructuring the connecting loop, and resulting in the loss of thermal stability. (a) Alignment of the Ag85C ebselen structure (gray; PDB, 4QDU) with the Ag85C azido ebselen structure (beige). (b) Alignment of the Ag85C ebselen structure (gray; PDB, 4QDU) with the Ag85C adamantyl ebselen structure (orange). (c) Overlay of α-helix 9 between native enzyme (green; PDB, 1DQZ), ebselen modified enzyme (gray; PDB, 4QDU), azido ebselen modified enzyme (beige), and adamantyl ebselen modified enzyme. (d) Ag85C modified with ebselen or either of the derivatives displays a 14 °C thermal shift from native enzyme.
Destabilizing Ag85C through Covalent Modification of Cys209 by Ebselen and Its Derivatives.
We have shown that covalent modification of Cys209 by ebselen and its derivatives inhibits Ag85C enzymatic activity through the displacement of the α9 helix. We were curious to determine whether or not the resulting movement of a major secondary structural element had any significant impact on the overall thermal stability of the enzyme. To answer this question, native and covalently modified Ag85C were subjected to thermal shift assays using differential scanning fluorimetry (DSF). DSF is becoming a routine approach to investigate the thermal stability of proteins for the optimization of buffers and ligands for crystallization experiments as well as for high-throughput screening of drug and fragment libraries.28,31 Native Ag85C with no drug present displayed a melt temperature of 55.0 ± 0 °C, whereas Ag85C modified with ebselen and azido ebselen displayed melt temperatures of 40.7 ± 0.3 °C and 41.0 ± 0 °C, respectively. As expected, Ag85C modified with adamantyl ebselen resulted in a biphasic melt curve, yielding two melt temperatures of 41.0 ± 0 °C and 54.5 ± 0 °C (Figure 7d). The biphasic nature is easily explained from the in vitro studies as adamantyl ebselen displays approximately half the inhibitory ability (covalently modified enzyme) of azido ebselen when set at equal concentrations relative to protein concentration.12 This lack of reactivity therefore yields two melt peaks, one reflecting the modified and the other the unmodified enzyme.
Upon modification by ebselen and its derivatives, the thermal stability of Ag85C decreases by 14 °C. The significant loss in thermal stability is not surprising because the covalent modification of Cys209 results in the movement of a major secondary structure and connecting loop. Previous studies examining protein stability of thermophiles concluded that proteins with relatively dynamic secondary structures tend to be less thermal stable.32 As a result, various hydrogen bonds are disrupted, as highlighted in our previous paper investigating various covalent modifiers of Cys209.9 Additionally, factors associated with higher protein thermal stability include buried hydrophobic patches, the presence of numerous salt bridges, and a lack of solvent channels through the protein core structure. Each of these stabilizing characteristics is lost in Ag85C upon covalent modification by ebselen and its derivatives (Figure 8).33,34 The decrease in thermal stability lowers the protein melt temperature to near physiological temperature (37 °C), suggesting that the modified enzyme will not only be inhibited enzymatically but will begin to denature in the host environment during active and dormant stages of bacterial growth.35,36
Figure 8.

Active site region differs in each structure. Atom color assignment is consistent with Figure 3, and water molecules are depicted by red spheres. (a) Residues that line the region of intrigue present in the azido ebselen structure are shown. The path from the Ser124 to the modified Cys209 is filled by two water molecules with distances shown. (b) Void volume depicted through a surface rendering of the cavity pocket found in the azido structure. (c) The void region observed in the adamantyl structure is lined with similar residues found in the azide structure, with the addition of Leu217. Again, two water molecules span the distance from Ser124 and Cys209, with distances nearly identical to a. (d) Void volume depicted through a surface rendering of the cavity pocket found in the adamantyl structure.
Another Path for Future Drug Design.
Previous studies have shown the potential benefit of targeting noncatalytic cysteines for drug development. For example, targeting a noncatalytic cysteine near the active site of kinases resulted in selectively increasing drug residence times and reduced drug resistance.37,38 These studies further strengthen the rationale for the development of covalent inhibitors targeting the conserved Cys209 of Mtb Ag85s, such as we have with ebselen and its derivatives. However, due to the lack of conserved residues on the α9 helix and the overall lack of specific protein interactions with the solvent-exposed ebselen derivative, which is inherently reactive toward exposed cysteines, we propose another way to target all Ag85s covalently but with greater specificity.
Upon further inspection of the azido and adamantyl structures, we identified a significant solvent channel spanning from the active site region to the area near the modified Cys209. This common region begins at the restructured loop (Gly210-Pro223) and encompasses the substrate-binding site, nucleophilic Ser124, and follows the α9 helix to the modified Cys209. Cys209 is approximately 14 A from the Ser124 nucleophile; however, the channel adopts a slight bend to accommodate a connective loop (Leu147-Trp157). In each new structure, two water molecules occupy this channel formed by residues Ser148, Gly149, Phe150, Thr231, Asn235, the catalytic His260, and the backbone carbonyl of Gly210 (Figure 8a and c). The channel of interest is approximately 8 A across at its widest, tapering down to 2 A near Cys209. Various linkers could potentially be used to form favorable interactions with the residues that line this channel, allowing us to target both the active site and Cys209. Differing between the two structures is the flipped-down side chain of Leu217 present in the adamantyl structure, which greatly reduces the volume of this region. This pocket is further detailed in Figure 8b and d. As stated previously, this region of the active site extends to the trehalose binding site and can be exploited in future drug design efforts aimed at improving drug specificity. This follows our previous work where the trehalose binding site was successfully targeted with an arabinose moiety to increase the specificity and inhibitory activity of thiophene compounds targeting Ag85s.39 Therefore, our knowledge of carbohydrate site targeting and the dynamic region detailed in these novel Ag85C-inhibitor structures illustrates an intriguing area for future drug development efforts targeting Mtb Ag85s.
METHODS
Molecular Cloning.
Cloning of Mtb Ag85C was carried out as previously described.3 Briefly, the amplified fbpC gene encoding Ag85C was inserted into a pET-29 plasmid (EMD Biosciences) using Ndel and XhoI (New England Biolabs) restriction sites, producing a noncleavable C-terminal, poly histidine tagged, recombinant Ag85C protein.
Overexpression and Purification.
Expression and purification of Mtb Ag85C were carried out as previously published.3 In short, T7 Express Escherichia col cells (New England Biolabs) were transformed with the Ag85C pET-29 construct. Cell cultures were grown to a density of 0.6 O.D.600 nm at 37 °C in Luria-Bertani broth (Research Products International). Protein expression was induced through the addition of 1 mM isopropyl β-D-1-thiogalactopyranoside. Induced cultures were allowed to incubate for 24 to 36 h at 16 °C. Induced cell cultures were harvested by centrifugation at 4,000 rpm (3,724g). The cell pellet was resuspended in lysis buffer (20 mM Tris pH 8.0 buffer containing 5 mM β-mercaptoethanol). Resuspended cells were lysed via incubation with lysozyme (Hampton Research) and DNase I (Roche Applied Sciences) on ice for a half hour, followed by sonication (Sonicator 3000, Misonix). The lysate was clarified by centrifugation at 12,000 rpm (18,514g) for 40 min. Clarified lysate was loaded onto a 5 mL HiTrap Talon Crude cobalt column (GE Healthcare) equilibrated with lysis buffer. Following washing, protein was eluted from the column via a 0 to 150 mM imidazole gradient of 15 column volumes. Eluted fractions were directly loaded onto a 5 mL HiTrap FF Q (GE Healthcare) anion exchange column equilibrated with a 20 mM Tris pH 8.0 buffer containing 1 mM EDTA and 0.3 mM TCEP. Following washing, protein was eluted with a 0 to 1 M NaCl gradient of 15 column volumes. Pooled elution fractions were subjected to ammonium sulfate precipitation (2.6 M), pelleted at 12,000 rpm (18,514g) for 50 min, and the pellet resuspended in either a 10 mM Tris pH 7.5 buffer containing 2 mM EDTA for crystallization experiments or a 50 mM sodium phosphate buffer at pH 7.5 for inhibition assays. Purified protein was dialyzed overnight at 4 °C against the same buffer to remove residual ammonium sulfate. Purified Ag85C was concentrated for crystallographic and inhibitory studies using a 3K Amicon Ultra centrifugal concentrator (Millipore).
kinact/KI Determination.
Ag85C activity in the presence of ebselen and its derivatives was assessed using a previously established fluorometric assay that monitors the acyl transfer rate of butyrate from resorufin butyrate (RfB) to trehalose, producing a resorufin fluorescent signal.3 Briefly, each reaction comprised 500 nM of Ag85C, 4 mM trehalose, and was initiated by addition of 100 μM RfB (10 mM DMSO stock). For inhibition studies, Ag85C in the presence of trehalose was quickly titrated with ebselen or the corresponding ebselen derivative (10 mM DMSO stocks, the inhibitor was serial diluted for the desired concentrations) and an equivalent v/v % DMSO for the uninhibited reaction immediately prior to RfB addition and the subsequent fluorometric measurement. Reactions were performed in triplicate at 37 °C in a 50 mM sodium phosphate buffer at pH 7.5 (triplicate data in Figure(S1) Kinetic data were acquired every 30 s on a Synergy H4 plate reader (Biotek), using Aex = 500 nm with Aem monitored at 590 nm. After subtracting the rate of background fluorescence production and converting from relative fluorescent units to product concentration (resorufin standard curve given in Figure(S2) kobs was calculated by fitting the triplicate kinetic data in Prism 7 with a one-phase association equation, Y = Y0 + (plateau -Y0)(1-exp(-kx)). This is equivalent to the commonly used [product] = vi where Y =[product], (Y0 + (plateau - Y0) = , k = kobs, and x = t or time.13 To obtain the best fit, time points 0 to 5 min were used for 3 and 2 μM ebselen, while 0 to 10 min were used for 1.5, and 1 μM ebselen reactions. All data for azido ebselen were fitted using 0 to 5 min, while 0 to 40 min were used for adamantyl ebselen. kinact/KI was determined by plotting kobs vs inhibitor concentration and fitting the data to a line through the origin.
Crystallization and Data Collection.
Crystals for the Ag85C azido ebselen complex were achieved through cocrystallization using the hanging drop vapor diffusion method. Purified Ag85C at 4.2 mg/mL with azido ebselen (10 mM DMSO stock) added in a 1:1.1 molar ratio, respectively, and was allowed to incubate on ice for 15 min prior to drop set up. The Ag85C-azido ebselen complex crystallized in 1:1 ratio of protein to well solution after a week of incubation at 16 °C. The well solution was composed of 0.1 M ammonium acetate, 0.05 M HEPES pH 7.5, and 12.5% w/v polyethylene glycol 3,350. Crystals were cryoprotected through the addition of 0.25 μL of glycerol to the drop immediately prior to looping and flash cooling in liquid nitrogen. X-ray diffraction data were collected at the Advanced Photon Source (APS) at Argonne National Laboratory, on LS-CAT beamline F.
Crystals for the Ag85C adamantyl ebselen complex were also obtained via cocrystallization using the hanging drop vapor diffusion method. Adamantyl ebselen (40 mM DMSO stock) was added to purified Ag85C at 4.0 mg/mL in a 1:1.1 protein to inhibitor molar ratio and allowed to incubate on ice for 15 min prior to drop setup. Crystals formed in a 1:1 ratio of protein to well solution, comprising 0.1 Bis-Tris at pH 5.5, and 25% w/v polyethylene glycol 3,350 after 4 days of incubation at 16 °C. Crystals were cryoprotected through the addition of 0.25 μL of glycerol to the drops immediately prior to looping and flash cooling in liquid nitrogen. X-ray diffraction data were collected at the APS LS-CAT beamline G.
Structure Determination and Refinement.
Both data sets were indexed, integrated, and scaled using HKL2000.40 The X-ray crystal structures of Mtb Ag85C covalently modified at residue Cys209 by azido ebselen and adamantyl ebselen were solved at 2.01 and 1.3 A resolution, respectively. The Ag85C-azido ebselen crystals had a C2221 space group with the phase solution coming from molecular replacement using Phaser MR with the previous Ag85C ebselen structure with both ebselen and the ebselen modified Cys209 omitted from the search model (PDB: 4QDU).41 Two molecules were determined to be in the asymmetric unit and confirmed by the molecular replacement solution. The Ag85C-adamantyl ebselen crystals had a P43212 space group, and the phase solution was also solved by molecular replacement with the same search model as described above (PDB: 4QDU) and supported one molecule per asymmetric unit. Each structure was subjected to a rigid body and simulated annealing refinement to remove model bias followed by rounds of XYZ coordinate, real-space, occupancy, and B-factor refinements (Phenix Refine) between manual modeling with COOT.42,43 Following rounds of model refinement and manual building, final Rwork/Rfree values of 0.1623/0.1968 and 0.1660/0.1763 were obtained for the azido and adamantyl structures, respectively. Restraints for both azido and adamantyl ligands covalently linked to C209 were generated using eLBOW (electronic Ligand Building and Optimization Workbench).44 Ligand geometries were further optimized using REEL 44.
DFT Calculations.
All DFT calculations were performed with the program Gaussian 09, revision E.01.45 The interaction energies were computed at the SMD/BLYP-D3BJ/Def2-QZVP level of theory with ε = 10. To compute the free energy of the oxidation reaction that occurs with Cys209 and the covalent modifier, the SMD/M06–2X/Def2-TZVP level of theory with ε = 10 was used for both geometry optimization and vibrational frequency calculations. Additional details are provided in the Supporting Information.
Thermal Shift Assay.
Relative protein stability was determined by differential scanning fluorimetry using a BioRad CFX96 Real-Time PCR Detection System. Purified Ag85C in the crystallization buffer was concentrated to 36 μM (1.2 mg/mL) and incubated with 100 μM of either ebselen, ebselen derivatives, or an equal volume of DMSO (1% v/v) for 15 min. Ebselen and both derivative stocks were at 10 mM in DMSO. SYPRO Orange was added to a final concentration of 2.5X (5,000X DMSO stock, Life Technologies, diluted to 200X stock with buffer), after 25 μL of the respective master stock reaction solution was aliquoted into the 96 well PCR plate and sealed (Bio-Rad). The plate was centrifuged at 700 rpm (651g) for 2 min to remove any air bubbles. The melt curve protocol is as follows: reactions were initially incubated at 25 °C for 3 min followed by a 0.5 degree increase, being held for 3 s, followed by a fluorescent read using the FRET setting, until a final temperature of 95 °C was achieved. Data were analyzed using the Bio-Rad CFX Manager 3.1 software. Triplicate data sets were averaged in Excel and plotted in Prism 7 to yield the final melt peaks. Triplicate data in the form of the raw melt curves and resulting melt peaks plotted with PRISM 7 are shown in Figure S5.
Supplementary Material
■ ACKNOWLEDGMENTS
Funding for this research project was provided by NIH grant AI105084. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfec-dis.7b00003.
Methods and raw data pertaining to kinact/KI determination, DFT calculations, and DSF melt curves (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).World Health Organization (2015) Global Tuberculosis Report 2015, World Health Organization, Geneva, Switzerland. [Google Scholar]
- (2).Loewenberg S (2012) India reports cases of totally drug-resistant tuberculosis. Lancet 379, 205. [DOI] [PubMed] [Google Scholar]
- (3).Favrot L, Grzegorzewicz AE, Lajiness DH, Marvin RK, Boucau J, Isailovic D, Jackson M, and Ronning DR (2013) Mechanism of inhibition of Mycobacterium tuberculosis antigen 85 by ebselen. Nat. Commun. 4, 2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Belisle JT, Vissa VD, Sievert T, Takayama K, Brennan PJ, and Besra GS (1997) Role of the Major Antigen of Mycobacterium tuberculosis in Cell Wall Biogenesis. Science 276, 1420–1422. [DOI] [PubMed] [Google Scholar]
- (5).Sanki AK, Boucau J, Ronning DR, and Sucheck SJ (2009) Antigen 85C-mediated acyl-transfer between synthetic acyl donors and fragments of the arabinan. Glycoconjugate J. 26, 589–596. [DOI] [PubMed] [Google Scholar]
- (6).Armitige LY, Jagannath C, Wanger AR, and Norris SJ (2000) Disruption of the Genes Encoding Antigen 85A and Antigen 85B of Mycobacterium tuberculosis H37Rv: Effect on Growth in Culture and in Macrophages. Infect. Immun. 68 (2), 767–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Jackson M, Raynaud C, Lane elle M, Guilhot C, Laurent-Winter C, Ensergueix D, Gicquel B, and Daffe M (1991) Inactivation of the antigen 85C gene profoundly affects the mycolate content and alters the permeability of the Mycobacterium tuberculosis cell envelope. Mol. Microbiol. 31 (5), 1537–1587. [DOI] [PubMed] [Google Scholar]
- (8).Xu K, Zhang Y, Tang B, Laskin J, Roach PJ, and Chen H (2010) Study of Highly Selective and Efficient Thiol Derivatization Using Selenium Reagents by Mass Spectrometry. Anal. Chem. 82, 6926–6932. [DOI] [PubMed] [Google Scholar]
- (9).Favrot L, Lajiness DH, and Ronning DR (2014) Inactivation of the Mycobacterium tuberculosis antigen 85 complex by covalent, allosteric inhibitors. J. Biol. Chem. 289 (36), 25031–25040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Gallivan JP, and Dougherty DA (1999) Cation-π interactions in structural biology. Proc. Natl. Acad. Sci. U. S. A. 96, 9459–9464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ma JC, and Dougherty DA (1997) The Cation-π Interaction. Chem. Rev. 97, 1303–1324. [DOI] [PubMed] [Google Scholar]
- (12).Thanna S, Goins CM, Knudson SE, Slayden RA, Ronning DR, and Sucheck SJ (2017) Thermal and photoinduced copper-promoted C-Se bond formation: Synthesis of 2-alkyl-1,2- benzisoselenazol-2(2H)-ones and evaluation against Mycobacterium tuberculosis, J. Org. Chem, DOI: 10.1021/acs.joc.7b00440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Copeland AR (2005) Irreversible Enzyme Inactivators, in Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, pp 214–219, John Wiley & Sons, Hoboken, NJ. [PubMed] [Google Scholar]
- (14).Klebe G (2015) Applying thermodynamic profiling in lead finding and optimization. Nat. Rev. Drug Discovery 14, 95–110. [DOI] [PubMed] [Google Scholar]
- (15).Marenich AV, Cramer CJ, and Truhlar DG (2009) Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 113 (18), 6378–6396. [DOI] [PubMed] [Google Scholar]
- (16).Takatani T, Hohenstein EG, Malagoli M, Marshall MS, and Sherrill CD (2010) Basis set consistent revision of the S22 test set of noncovalent interaction energies. J. Chem. Phys. 132, 144104. [DOI] [PubMed] [Google Scholar]
- (17).Rezac J, Riley KE, and Hobza P (2014) Erratum to “S66: A Well-balanced Database of Benchmark Interaction Energies Relevant to Biomolecular Structures”. J. Chem. Theory Comput. 10, 1359–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sinnokrot MO, and Sherrill CD (2003) Unexpected Substituent Effects in Face-to-Face π-Stacking Interactions. J. Phys. Chem. A 107 (41), 8377–8379. [Google Scholar]
- (19).Sinnokrot MO, and Sherrill CD (2004) Substituent Effects in π—π Interactions: Sandwich and T-Shaped Configurations. J. Am. Chem. Soc. 126, 7690–7697. [DOI] [PubMed] [Google Scholar]
- (20).Smith T, Slipchenko LV, and Gordon MS (2008) Modeling π—π Interactions with the Effective Fragment Potential Method: The BenzeneDimer and Substituents. J. Phys. Chem. A 112, 5286–5294. [DOI] [PubMed] [Google Scholar]
- (21).Davis MR, and Dougherty DA (2015) Cation-pi interactions: computational analyses of the aromatic box motif and the fluorination strategy for experimental evaluation. Phys. Chem. Chem. Phys. 17, 29262–29270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Khan HM, Grauffel C, Broer R, MacKerell AD, Havenith RW, and Reuter N (2016) Improving the Force Field Description of Tyrosine-Choline Cation-pi Interactions: QM Investigation of Phenol-N(Me)4+ Interactions. J. Chem. Theory Comput. 12, 5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Duffy EM, Kowalczyk PJ, and Jorgensen WL (1993) Do Denaturants Interact with Aromatic Hydrocarbons in Water? J. Am. Chem. Soc. 115, 9271–9275. [Google Scholar]
- (24).Anderson DH, Harth G, Horwitz MA, and Eisenberg D (2001) An interfadal mechanism and a class of inhibitors inferred from two crystal structures of the Mycobacterium tuberculosis 30 kDa major secretory protein (Antigen 85B), a mycolyl transferase. J. Mol. Biol. 307, 671–681. [DOI] [PubMed] [Google Scholar]
- (25).Ronning DR, Vissa V, Besra GS, Belisle JT, and Sacchettini JC (2004) Mycobacterium tuberculosis antigen 85A and 85C structures confirm binding orientation and conserved substrate specificity. J. Biol. Chem. 279, 36771–36777. [DOI] [PubMed] [Google Scholar]
- (26).Ronning DR, Klabunde T, Besra GS, Vissa VD, Belisle JT, and Sacchettini JC (2000) Crystal structure of the secreted form of antigen 85C reveals potential targets for mycobacterial drugs and vaccines. Nat. Struct. Biol. 7 (2), 141–146. [DOI] [PubMed] [Google Scholar]
- (27).Backus KM, Dolan MA, Barry CS, Joe M, McPhie P, Boshoff HI, Lowary TL, Davis BG, and Barry CE 3rd (2014) The three Mycobacterium tuberculosis antigen 85 isoforms have unique substrates and activities determined by non-active site regions. J. Biol. Chem. 289, 25041–25053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Pantoliano MW, Petrella EC, Kwasnoski JD, Lobanov VS, Myslik J, Graf E, Carver T, Asel E, Springer BA, Lane P, and Salemme FR (2001) High-Density Miniaturized Thermal Shift Assays as a General Strategy for Drug Discovery. J. Biomol Screening 6 (6), 429–440. [DOI] [PubMed] [Google Scholar]
- (29).Vedadi M, Arrowsmith CH, Allali-Hassani A, Senisterra G, and Wasney GA (2010) Biophysical characterization of recombinant proteins: a key to higher structural genomics success. J. Struct. Biol. 172 (1), 107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Geders TW, Gustafson K, and Finzel BC (2012) Use of differential scanning fluorimetry to optimize the purification and crystallization of PLP-dependent enzymes. Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 68 (5), 596–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lo MC, Aulabaugh A, Jin G, Cowling R, Bard J, Malamas M, and Ellestad G (2004) Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal. Biochem. 332, 153–159. [DOI] [PubMed] [Google Scholar]
- (32).Scandurra R, Consalvi V, Chiaraluce R, Politi L, and Engel PC (1998) Protein thermostability in extremophiles. Biochimie 80, 933–941. [DOI] [PubMed] [Google Scholar]
- (33).Szilayi A, and Zavodszky P (2000) Structural differences between mesophilic, moderately thermophilic and extremely thermophilic protein subunits: results of a comprehensive survey. Structure 8 (5), 493–504. [DOI] [PubMed] [Google Scholar]
- (34).Sadeghi M, Naderi-Manesh H, Zarrabi M, and Ranjbar B (2006) Effective factors in thermostability of thermophilic proteins. Biophys. Chem. 119 (3), 256–270. [DOI] [PubMed] [Google Scholar]
- (35).Sasindran SJ, and Torrelles JB (2011) Mycobacterium Tuberculosis Infection and Inflammation: what is Beneficial for the Host and for the Bacterium? Front. Microbiol. 2, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Pai M, Behr MA, Dowdy D, Dheda K, Divangahi M, Boehme CC, Ginsberg A, Swaminathan S, Spigelman M, Getahun H, Menzies D, and Raviglione M (2016) Tuberculosis. Nat. Rev. Dis. Primers 2, 16076. [DOI] [PubMed] [Google Scholar]
- (37).Bradshaw JM, McFarland JM, Paavilainen VO, Bisconte A, Tam D, Phan VT, Romanov S, Finkle D, Shu J, Patel V, Ton T, Li X, Loughhead DG, Nunn PA, Karr DE, Gerritsen ME, Funk JO, Owens TD, Verner E, Brameld KA, Hill RJ, Goldstein DM, and Taunton J (2015) Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat. Chem.Biol. 11 (7), 525–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Yun C, Mengwasser KE, Toms AV, Woo MS, Grulich H, Wong K, Meyerson M, and Eck MJ (2008) The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity of ATP. Proc. Natl. Acad. Sci. U. S. A. 105 (6), 2070–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Ibrahim DA, Boucau J, Lajiness DH, Veleti SK, Trabbic KR, Adams SS, Ronning DR, and Sucheck SJ (2012) Design, synthesis, and X-ray analysis of a glycoconjugate bound to Mycobacterium tuberculosis antigen 85C. Bioconjugate Chem. 23 (12), 2403–2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Otwinowski Z, and Minor W (1997) Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- (41).McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40 (4), 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66 (2), 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010) Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66 (4), 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Moriarty NW, Grosse-Kunstleve RW, and Adams PD (2009) electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr., Sect. D: Biol. Crystallogr. 65 (10), 1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr., Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, and Fox DJ (2009) Gaussian 09, revision E.01, Gaussian, Inc., Wallingford, CT. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.