

Abstract
Tuberculosis (TB) continues to be an important public health threat worldwide, due in part to drug resistant Mycobacterium tuberculosis (Mtb) strains. The United States recently reported a shortage of isoniazid (INH), which could drive higher INH resistance rates. Changes in the Mtb proteome before and after acquisition of INH resistance in a clean genetic background remain understudied and may elucidate alternate drug targets. Here, we focused on Mtb clonal strains to characterize the consequences of INH resistance on mycobacterial metabolism. Proteomic analysis was conducted by liquid-chromatography tandem mass spectrometry (LC-MS/MS) of cellular and secreted fractions, followed by a normalized spectral counting (NSAF) analysis (data are available via ProteomeXchange with identifier PXD009549). Two different Mtb clonal pairs representing a specific genetic lineage (one clinical and one generated in the laboratory) but sharing a katG mutation associated with INH resistance, were used in our analysis. Overall, we found 26 Mtb proteins with altered abundances after acquisition of INH resistance across both Mtb genetic lineages studied. These proteins were involved in ATP synthesis, lipid metabolism, regulatory events, and virulence, detoxification, and adaptation processes. Proteomic findings were validated by Western blotting analyses whenever possible. Mycolic acid (MA) analysis through LC/MS in the clonal Mtb pairs did not reveal a common trend in the alteration of these fatty acids across both INHr strains but revealed a significant reduction in levels of the two more abundant α-MA features in the clinical INHr strain. Interestingly, the clinical clonal pair demonstrated more variation in the abundance of the proteins involved in the FAS II pathway. Together, the proteomic and lipidomic data highlight the identification of potential drug targets such as alternative lipid biosynthetic pathways that may be exploited to combat clinically relevant Mtb INHr strains.
Keywords: Tandem Mass Spectrometry, Tuberculosis, Lipidomics, Thin Layer Chromatography, Mass Spectrometry, Drug Resistance
Despite global efforts to control tuberculosis (TB),1 the emergence of drug-resistant cases has been detrimental for the successful control of this disease. Today, TB, a bacterial infection caused by Mycobacterium tuberculosis (Mtb), remains a significant contributor to morbidity in many countries and is the leading cause of death due to an infectious agent worldwide (1). Isoniazid (INH), one of the first-line drugs used to treat TB patients, was thought to be the solution for the “white plague” in the last century. However, INH resistance emerged rapidly, and INH-resistant (INHr) Mtb strains continue to expand globally. The understanding of INH bactericidal activity and bacterial resistance strategies lagged behind the emergence of INH resistance and the spread of INHr Mtb, possibly because of the serendipity of its discovery. Once INH is activated by the bacterial enzyme KatG, INH inhibits the synthesis of mycolic acids (MA) (2, 3), which are hallmark molecules of the Mtb cell wall and are essential for bacterial survival (4). Additional mechanisms such as the inhibition of the electron transport, nucleic acid synthesis, and cell division are recognized as other bactericidal strategies of INH (2, 3). Despite the broad range of potential INH targets, katG mutations are the most common cause of INHr clinical isolates.
The study of bacterial drug resistance, particularly in the context of bacterial physiology, helps us to systematically understand the process and consequences of drug resistance and generates new insights into a very complex phenomenon of clinical relevance. The current approach consisted of comprehensive biochemical exploration of the Mtb phenotype after acquisition of INH resistance, specifically the global analysis of changes in protein and lipid abundances in INHr strains due to clinically relevant mutations. A previous descriptive proteomic comparison of INHr and INHs clonal strains of the Mtb Beijing lineage exhibited altered abundance of proteins involved in central carbon and lipid metabolism in the INHr strains (5). Here, we focused our efforts on describing the proteome and MA-related metabolome of two clonal pairs (INHs and INHr) representing two different Mtb genetic lineages (T and Euro-American) as well as different degrees of virulence in the mouse model (6). Using two distinct clonal pairs allowed us to study the INH resistance phenomenon without confounding factors given by the inherent variability of the different mycobacterial genetic lineages.
EXPERIMENTAL PROCEDURES
Bacterial Strains
One clinical and one laboratory clonal pair of Mtb with different genetic backgrounds were analyzed in this study. The clinical pair was previously identified as members of the T lineage by restriction fragment length polymorphism RFLP—IS6110 (7) and spoligotyping (8) and was obtained from the same patient pre- and post-drug exposure (6). The institutional review board for the clinically isolated Mtb pair has been described previously (6, 9, 10). The laboratory pair consisted of the reference strain H37Rv and its katG mutant generated in the laboratory after exposure to INH in an infected mouse (11). The INH-susceptible and -resistant Mtb strains were denoted as INHs and INHr, respectively. INHr strains from both clinical and laboratory pairs have the katG mutation V1A, while the clinical INHr strain also bears a second katG mutation, E3V (6, 11, 12). A previous whole-genome sequencing study on the T lineage corroborated that only katG mutation was associated with drug resistance and the clonality of these pairs.
Proteomic Analysis
Culture Filtrate Proteins and Subcellular Fractions
For the proteomic analysis, all Mtb strains were cultured in triplicate in a final volume of 1 liter of glycerol alanine salts medium and incubated at 37 °C in constant agitation. Glycerol alanine salts medium was prepared in-house with the following composition per liter: 2 g of ammonium chloride (Fisher Scientific, Waltham, MA, MK338412, U.S.), 1 g of l-alanine (Sigma-Aldrich, A7627, U.S.), 0.3 g of Bacto Casitone (Fisher Scientific, 225930, U.S.), 4 g of dibasic potassium phosphate (Sigma-Aldrich, St. Louis, MO, 60353, U.S.), 2 g of citric acid (ALFA AESAR, Haverhill, MA, AA36664-36, U.S.), 50 mg of ammonium iron (III) citrate (Sigma-Aldrich, F5879, U.S.), 1.2 g of magnesium chloride hexahydrate (J.T. Baker, Center Valley, PA, JT40031, U.S.), 0.6 g of potassium sulfate (J.T. Baker, JT3278–1, U.S.), 1.8 ml of 10 m sodium hydroxide (Fisher Scientific, S318–500, U.S.), 10 ml of glycerol (Fisher Scientific, AC33203–0025, U.S.), and 0.05% Tween 80 (G-Biosciences, St. Louis, MO, 786–519, U.S.). This minimal medium was used for the proteomic analysis purposely guaranteeing the growth of the bacterium with minimal interference for the downstream analytical process (such as the excess of contaminant proteins from a more enriched medium or detergents such as polyethylene glycol). Moreover, we wanted to provide findings regarding the specific adaptive responses of the bacterium after the acquisition of INH resistance to reduce potential confounding factors. Cells and culture filtrate were harvested after three weeks of culture as described elsewhere (14). Culture filtrate proteins (CFP) and subcellular fractions: membrane (MEM), cytosol (CYT) and cell wall (CW) were prepared as previously described by Lucas et al. (15). Each fraction was qualified for protein concentration, total protein content, and marker proteins (where applicable) per standard qualification criteria established by our laboratory and used to qualify and provide similar biological reagents to the mycobacteria research community through BEI resources (https://www.beiresources.org/About/QualityControl.aspx). Before the digestion of proteins, the CW fraction was delipidated as described elsewhere (14). All the resulting proteins were resuspended in 10 mm ammonium bicarbonate (Sigma-Aldrich, A6141, U.S.).
In-solution Protein Digestion
Total protein concentration of all subcellular fractions and CFPs was measured using the bicinchoninic acid method (Thermo Scientific™ Pierce™ BCA Protein Assay, U.S.). In-solution trypsin digestion of 30 μg of each protein sample was performed as described previously, using ProteaseMax surfactant (Fisher Scientific, PRV2071, U.S.) followed by a final desalting step using Pierce® C-18 spin columns (Thermo Fisher Scientific, 89870, U.S.) (14). Peptides were finally resuspended in solvent A (0.1% formic acid (Thermo Scientific™ Pierce™, PI28905, U.S.), 3% acetonitrile (Burdick & Jackson, Mexico City, Mexico, AH015–4PC, U.S.) in HPLC water (Fisher Scientific, P51140, U.S.)).
Liquid Chromatography Coupled with Tandem Mass Spectrometry Assay (LC-MS/MS)
One microliter (0.5 μg) of digested peptides from subcellular fractions and CFPs were randomly injected in duplicate using the Orbitrap Velos MS (Thermo Scientific, U.S.) coupled with the instrument EASY-nLC II nanoflow HPLC (Thermo Scientific, U.S.). Each sample was injected using the nano-HPLC instrument, and peptides were purified and concentrated using an on-line enrichment column EASY-Column, 100 μm inner diameter × 2 cm ReproSil-Pur C18 (Thermo Scientific, U.S.). Subsequent chromatographic separation was performed on a reverse phase nanospray column (EASY-Column, 3 μm, 75 μm inner diameter × 100 mm ReproSil-Pur C18) using a 90-min linear gradient from 5–45% solvent B (100% acetonitrile, 0.1% formic acid) at a flow rate of 400 nl/min. Peptides were eluted directly into the mass spectrometer (Thermo Scientific Orbitrap Velos). The instrument was operated in Orbitrap-LTQ mode where precursor measurements were acquired in the Orbitrap (60,000 resolution) with a m/z range of 400–2,000 and the automatic gain control target for MS1 full scan was 1 × 106. Tandem MS/MS spectra (top 20) were acquired in the LTQ ion trap with a normalized collision energy of 35%. Other mass spectrometer parameters were: signal threshold for triggering MS/MS event of 5,000, charge state screening states accepted for MS2 were 2+ and 3+, rejecting all other charge states as well as the unknown charge state of precursors. Parameters for dynamic exclusion: exclusion after a repeat count of two within 30 s; exclusion size list: 500; exclusion duration: 90 s.
Database Searching
Tandem mass spectra raw data were converted to mzXML files using ProteoWizard (MSConvert version 3.0) (16). All MS/MS samples were analyzed using Sorcerer2TM integrated data analysis platform (Sage-N Research, Milpitas, CA, version 5.0.1) and Sequest (Thermo Fisher Scientific, San Jose, CA, U.S.; version v. 3.5). Sequest was set up to search the Mtb strain H37Rv Tuberculist database, version R25 completed in April 2012 (17) including all reverse entries as decoys (7,992 entries) with trypsin as the digestion enzyme and up to two missed cleavage sites. Sequest was searched with a fragment ion mass tolerance of 1.00 Da and a parent ion tolerance of 20 ppm. Oxidation of methionine and carbamidomethylation of cysteine were specified in Sequest as variable modifications. Fixed modifications were not included in the analysis.
Criteria for Protein Identification and Statistical Analysis
Scaffold (version Scaffold_4.3.2, Proteome Software, Inc., Portland, OR) was used to validate MS/MS-based peptide and protein identifications. Peptide identifications were accepted if they could be established at greater than 95.0% probability by the Scaffold Local FDR algorithm. Protein identifications were accepted if they could be established at greater than 99.0% probability to achieve an FDR less than 1.0% and contained at least two identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm (18). Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony.
Experimental Design and Statistical Rationale
Each experiment included three independent biological replicates (individual 1-liter cultures) for each evaluated condition (INHs and INHr, either clinically or laboratory isolated). Two independent technical replicates (injections) were acquired for each biological replicate. Biological replicates were accepted as part of the experiment data set if the cultures grew to uniform turbidity and the spectra for each technical sample consisted of the same distribution of qualified spectra. A qualified spectrum was determined by evaluation of the normalized spectra abundance factor (NSAF) using inferno (R-based statistical analysis platform), evaluating boxplots (for normalized distribution of spectra), and principal component analysis graphs (for sample set clustering). Three biological replicate sets were accepted for each condition under these quality control criteria, with the exception of the clinical INHr sample, from which two biological replicate sets met the qualification criteria following upscale to 1-liter cultures. Differences between protein abundances, expressed as NSAF values among the two different phenotypes (INHs versus INHr) in each pair (clinical or lab-derived strains) were tested by two-tailed Student's t test. At each pair comparison, the INHs strain was used as the control for the analysis of the protein levels. p values less than or equal to 0.05 were considered significant.
The MS/MS proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the data set identifier PXD009549 and 10.6019/PXD009549 (19, 20).
Western Blotting (WB) Validation
To validate the levels of the proteins Ag85, HspX, and AcpM, 5 μg of total cytosolic protein from two biological replicates for each strain were separated in a SDS-PAGE and then transferred onto a nitrocellulose membrane. For FabG1/MabA and InhA, 5 μg of cell wall protein, and for UmaA, 5 μg of membrane protein fraction, prepared as described previously, were used (15). Mouse monoclonal primary antibodies for Ag85 complex (Clone CS-90) and HspX (Clone CS-49) were obtained through BEI Resources, NIAID, and NIH (https://www.beiresources.org). Rabbit polyclonal anti-AcpM was produced as follows: rAcpM was produced in and purified from Escherichia coli. The gene fragment encoding AcpM was amplified by PCR using the primers 5′-CTTAAGG CATATG CCT GTC ACT CAG GAA GAA AT-3′ (forward) and 5′-GCC GAATTC TCA CTT GGA CTC GGC CTC AAG-3′ (reverse). The amplified gene product was ligated into the multiple cloning site for pET28a (EMD Biosciences, Gibbstown, NJ, U.S.) following digestion with NdeI and EcoRI and the recombinant plasmid was transformed, plated, induced, and purified using methods described previously (21). Purified recombinant protein was used to produce polyclonal antisera through contracted work to Lampire Biological Laboratories (Pipersville, PA) using their standard EXPRESS-LINE Basic 50 day protocol (http://www.lampire.com/products/poly_express.php?section=custom_poly&cat=poly_express), with the modification that only incomplete Freund's adjuvant was used for all injections. Mouse polyclonal antisera for InhA, MabA, and rabbit polyclonal antisera for UmaA were kindly provided by Dr. John Spencer and Dr. Angela Marques, respectively, from Colorado State University. Secondary antibodies Goat anti-rabbit F(ab)2 fragment (Thermo Scientific) and goat anti-mouse IgG (H+L) (Thermo Scientific)-HRP conjugated were used depending on the nature of the primary antibody. The proteins were detected with the chemiluminescent SuperSignal West Pico Stable peroxide solution detection system (Thermo Scientific Pierce).
Lipidomics
Total Lipid Extraction
Clonal pairs were cultured in 100 ml of glycerol alanine salts media and incubated under the same conditions as that for the proteomic analyses. After removing the media by centrifugation (at 3,500 x g for 20 min at 4 °C), the bacterial cell pellets were washed with 1X PBS and incubated with 10 ml of chloroform/methanol/water 10:10:3 overnight at room temperature with constant agitation to inactivate the bacteria and extract the total lipids, as previously described (22). After drying the lipid solutions under the nitrogen bath, samples were diluted in chloroform/methanol 2:1.
One-dimensional Thin-layer Chromatography (TLC)
In order to compare lipids profiles from the different Mtb clonal pairs, 400 μg of total lipid from each strain were added on aluminum-backed silica TLC plates (HPTLC Silica gel 60 F254, EMD Millipore, Burlington, MA). For phthiocerol dimycocerosates (PDIMs) comparison, TLC plates were run in petroleum ether: Ethyl acetate (98:2) and then the lipids were detected with 10% cupric sulfate (in 8% phosphoric acid solution). For the glycolipids comparison, TLC plates were run in 40:8:1 chloroform:methanol:water, and then the lipids were detected with α-naphthol (0.5% α-naphthol in 5% sulfuric acid in ethanol) followed by heat. Lipid standards: trehalose monomycolates (TMM), trehalose dimycolates (TDM), sulfolipids, phosphatidylinositol mannosides (PIM), and PDIMs were obtained from BEI Resources, NIAID, NIH (https://www.beiresources.org/). Intensity of the lipid signal on the TLC plates was measured using the software ImageJ and the statistical comparison was performed in GraphPad Prism 6.
Mycolic Acids Extraction
Mtb cell pellets obtained from the previously described delipidation process were transferred to 13 × 100 mm glass tubes, mixed with 2 ml of 1 m KOH (Sigma-Aldrich, 484016, U.S.) in methanol (Honeywell Research Chemicals, Center Valley, PA, LC230–2.5, U.S.) and incubated for 2 h at 80 °C. After setting the tubes at room temperature for 20 min, pH was adjusted to 5.0 with HCl, and 2 ml of diethyl ether (Sigma-Aldrich, 309966–1L, U.S.) were added to extract the saponified material. The diethyl ether extraction step was repeated, collecting the upper layer in both cases in the same tube. After this, 2 ml of HPLC water (Fisher Scientific, P51140, U.S.) were added, and the upper (organic) layer was carefully transferred to a preweighted glass tube. This resulting solution of MA was dried under nitrogen gas. The MA samples were diluted to 1 μg/μl in solvent B (5 mm ammonium acetate in n-propanol-hexane-water (79:20:1; v/v/v)).
LC-MS Analysis
5 μl of MA solution (1 μg/μl, in solvent B) were used for the LC-Q-TOF-MS analysis, following the method described by Sartain et al. (22) with minor modifications. The autosampler was kept at 20 °C. A Water's XBridge (Ethylene-Bridged Hybrid, BEH, Waters Corporation, Millford, MA) C18 column (2.1 × 150 mm, 3.5 μm) was used for chromatographic separation of lipids at 45 °C, over 35 min with a flow rate of 0.32 ml/min. The system was equilibrated with 100% solvent A (5 mm ammonium acetate in methanol-water 99:1; v/v) and 20% solvent B (described above). Solvent A was maintained at 100% for 2 min, followed by a 30-min linear gradient to 100% solvent B. The conditions for the TOF analyzer were described previously (22); briefly, negative ion data were generated by operation of the mass spectrometer in a dual ESI mode with a capillary voltage of 4,000 V, nebulizer of 45 psig, drying gas of 8.0 liters/min, gas temperature of 300 °C, fragmentor of 125 V, skimmer of 65 V, and octapole radio frequency voltage of 750 V. Mass spectra were acquired at 2.0 spectra/s. Data were collected over a m/z of 250 to 2,500 Da with the Agilent MassHunter WorkStation Data Acquisition software, version B.05. MS/MS spectra of MAs were acquired in targeted mode with isolation of precursor ion width medium (∼4 amu). Fragmentation of targeted molecules was achieved using purified nitrogen as collision gas, applying collision energy of 65 voltage.
Lipidomic Data Analysis and Statistical Comparison
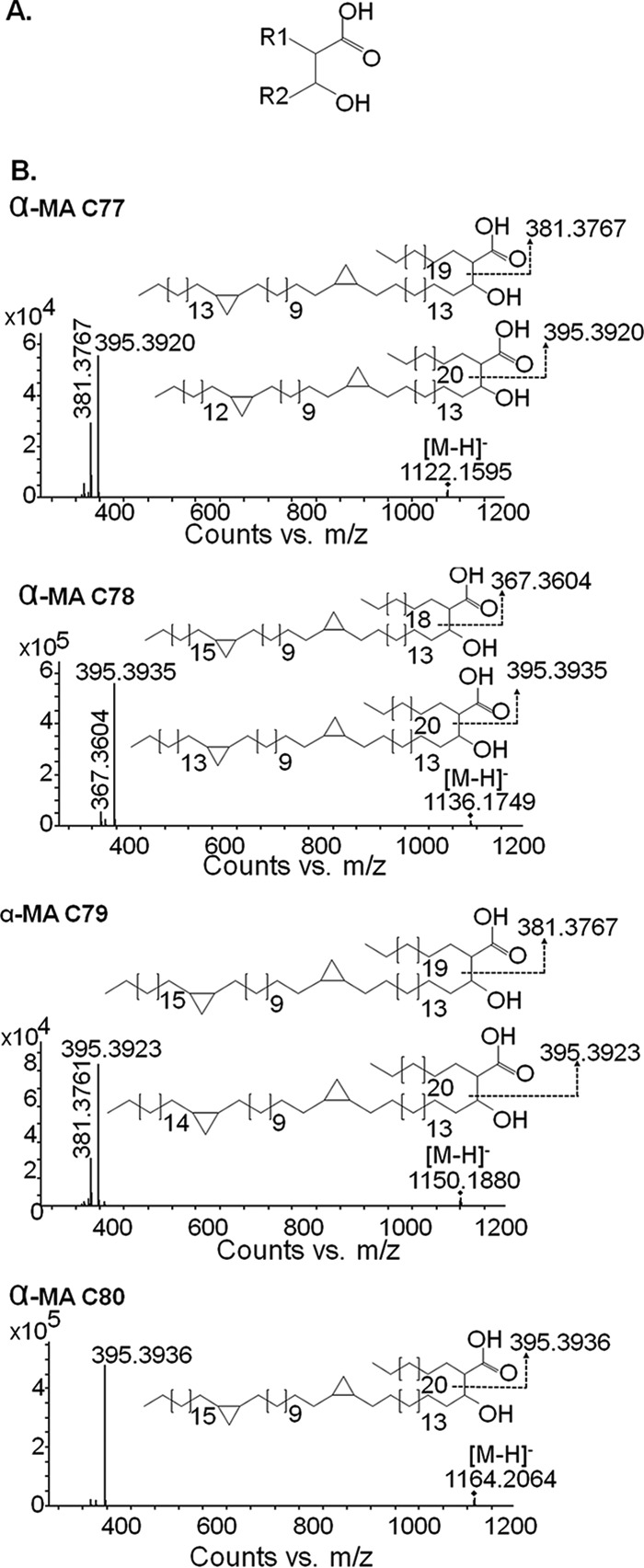
Since α-MA represent more than 70% of the total MA in Mtb (23), the levels of the most abundant α-MA: α-MA C78 and α-MA C80 (24, 25), as well as two other shorter chain and less abundant α-MA (C77 and C79) were compared between the clonal Mtb pairs. The m/z values corresponding to these features were obtained looking at the Mtb lipid database described by Sartain et al. (20). The comparison of the peak area for each extracted ion chromatogram was conducted by two-tailed t test using GraphPad Prism 6. The features were analyzed using Agilent MassHunter Qualitative Analysis B.06.00 and the chemical structures derived from this analysis were represented using Chemdraw Professional 15.1.
RESULTS
The analysis of proteins that presented altered abundances among INHr versus INHs in both clinical and laboratory clonal pairs with the same trend as well as individual results observed only in the clinical clonal pair are described in detail in the following sections. All the proteins identified at each cellular fraction for each strain are described in supplemental Data 1. A total of 41 proteins with different abundances were shared between both clonal pairs; 26 of them had the same trend (Table I). Proteins such as KatG and PpK had statistically significant variation in at least two cellular fractions, with the same tendency for each pair comparison (Table I). Levels of the proteins SecA2, AhpC, AhpD, MetK, LldD2, FadD32, FadD6, and DapB varied significantly not only in both pair comparisons (clinical and laboratory pair comparison), but this variation was statistically significant also in at least another cellular fraction (Table I). The commonly affected proteins belong to different functional categories, where metabolism and respiration as well as lipid metabolism were the most represented categories (Fig. 1A). In addition, a higher number of proteins with significant differences in their abundances (t test, p < 0.05) between INHr and INHs were observed in the clinical pair (Fig. 1).
Table I. Proteins with significant differential abundance (p > 0.05) in both clinical and lab strains after acquisition of INHr.
| Accession number | Gene | Log2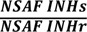 |
Variation in other fractions* | ||
|---|---|---|---|---|---|
| Clinical pair | Lab pair | ||||
| Identified proteins CFP (4) | |||||
| Rv0363c.1 | fba | Fructose-bisphosphate aldolase | 1.20 | −0.15 | |
| Rv1908c.1 | katG | Catalase-peroxidase-peroxynitritase T | 4.65 | 1.43 | All# |
| Rv2495 | pdhC | Dihydrolipoamide S-acetyltransferase E2 component | a | −1.32 | |
| Rv1910c.1 | Rv1910c.1 | Hypothetical exported protein | −2.32 | −0.32 | |
| Identified proteins CW (7) | |||||
| Rv1731 | gabD2 | Succinate-semialdehyde dehydrogenase NADPH-dependent | −0.32 | −0.51 | MEM |
| Rv1908c.1 | katG | Catalase-peroxidase-peroxynitritase T | 6.62 | 2.32 | All# |
| Rv2213 | pepB | Aminopeptidase | −0.51 | −0.51 | MEM |
| Rv2214 | ephD | Short-chain type dehydrogenase | −1.32 | −1.74 | |
| Rv2346 | esxO | Esat-6 like protein | 0.38 | 0.77 | |
| Rv2567 | Rv2567 | Conserved alanine and leucine rich protein | −3.32 | 0.49 | |
| Rv2984 | ppk | Polyphosphate kinase | −2.32 | 4.10 | MEM# |
| Identified proteins CYT (7) | |||||
| Rv0757.1 | phoP | Two component system transcriptional regulator | −1.00 | −2.32 | CW and MEM |
| Rv1454 | qor | Quinone reductase | −0.32 | 0.85 | CW and CFP |
| Rv1821.1 | secA2 | Preprotein translocase ATPase | 2.89 | −3.32 | MEM (lab pair only)# |
| Rv1908c.1 | katG | Catalase-peroxidase-peroxynitritase T | 2.17 | 1.63 | All# |
| Rv2428.1 | ahpC | Alkyl hydroperoxide reductase C | 1.32 | −0.32 | CW and MEM (clinical pair only)# |
| Rv2703.1 | sigA | RNA polymerase sigma factor | −1.00 | −1.00 | CW and MEM |
| Rv3801c.1 | fadD32 | Fatty-acid-Coa ligase | 0.38 | −2.32 | MEM (lab pair only)# |
| Identified proteins MEM (27) | |||||
| Rv0134 | ephF | Epoxide hydrolase | −3.32 | −0.74 | CW |
| Rv0243.1 | fadA2 | Acetyl-coA acyltransferase | −1.00 | 0.38 | CW and CYT |
| Rv0636.1 | hadB | (3R)-hydroxyacyl-ACP dehydratase catalytic subunit | 1.00 | −1.74 | CYT |
| Rv0710.1 | rpsQ | 30S ribosomal protein S17 | 0.68 | 0.58 | CYT and CFP |
| Rv0896.1 | gltA2 | Citrate synthase I | −0.32 | 0.26 | CW |
| Rv1077 | cbs | Cystathionine beta-synthase | a | 0.68 | |
| Rv1206 | fadD6 | fatty-acid-CoA ligase | −3.32 | −1.32 | CW (clinical pair#) |
| Rv1326c.1 | glgB | 1,4-α–glucan branching enzyme | 2.17 | 1.14 | All |
| Rv1392.1 | metK | S-adenosylmethionine synthetase | 0.49 | 0.49 | CYT (clinical pair only)# |
| Rv1647 | Rv1647 | Adenylate cyclase (ATP pyrophosphate-lyase) | −2.32 | −3.32 | CW |
| Rv1872c.1 | lldD2 | l-lactate dehydrogenase | −1.00 | −0.51 | CW (clinical pair only)# |
| Rv1908c.1 | katG | Catalase-peroxidase-peroxynitritase T | 2.41 | 2.30 | All* |
| Rv1978.1 | Rv1978.1 | Conserved hypothetical protein | −3.32 | −1.00 | CW and CFP |
| Rv2037c.1 | Rv2037c.1 | Conserved membrane protein | −3.32 | −1.74 | CW |
| Rv2198 | mmpS3 | Membrane protein mmpS3 | a | b | |
| Rv2225 | panB | 3-methyl-2-oxobutanoate hydroxymethyltransferase | 3.20 | 0.77 | |
| Rv2338c | moeW | Molybdopterin biosynthesis protein | −3.32 | −4.06 | |
| Rv2357 | glyS | glycyl-tRNA synthetase | −1.32 | −0.74 | CW |
| Rv2429.1 | ahpD | Alkyl hydroperoxide reductase D protein | 2.98 | −0.74 | All but CYT and CFP of clinical pair# |
| Rv2564.1 | glnQ | Glutamine-transport ATP-binding protein ABC transporter | −1.00 | −0.74 | |
| Rv2574.1 | Rv2574.1 | Conserved hypothetical protein | −3.32 | −0.74 | |
| Rv2773 | dapB | Dihydrodipicolinate reductase | −5.06 | −0.15 | CFP and CW |
| Rv2951c.1 | Rv2951c.1 | Oxidoreductase | −1.32 | −0.74 | CW |
| Rv2971 | Rv2971 | Oxidoreductase | 0.93 | −3.32 | CW |
| Rv2984 | ppk | Polyphosphate kinase | −1.74 | 0.68 | CW# |
| Rv3050c.1 | Rv3050c.1 | Transcriptional regulator, asnc-family | −3.32 | −1.32 | CW |
| Rv3137.1 | Rv3137.1 | Monophosphatase | 3.17 | 0.68 | CW |
*Variation with the same tendency reported in the previous columns.
#Statistically significant variation.
aNSAF (normalized spectral abundance factor) values for the INHr strain were zero.
bNSAF values for the INHs strain were zero. Negative values indicate proteins with significantly decreased levels while positive values indicate proteins with significantly increased levels in the INHs strain.
Fig. 1.
(A) Number of proteins with significant differences in their abundance in the LC-MS/MS analysis between INHr and INHs pairs, grouped by functional category according to Tuberculist (http://tuberculist.epfl.ch/) (p < 0.05, t test). (B) Number of proteins with significant abundance differences among INH-susceptible and -resistant pairs for each cellular fraction. CFP: secreted proteins, CW: cell wall, CYT: cytosol, MEM: membrane.
Altered Proteins with the Same Trend Among the Mtb Clonal Pairs
KatG Levels Were Decreased in Both INHr Strains
As it was expected based on previous genomic and Western blotting analyses (6, 12), there was a statistically significant reduction of KatG levels in both clinical and laboratory INHr strains compared with their respective INHs progenitor strains (Table I, Fig. 2). KatG deficiency had been noted in a previous virulence study of these strains in the water-soluble fractions (CYT and CFP) by WB (6). Interestingly, the LC-MS/MS analysis revealed that this protein was truly decreased in all the cellular fractions tested for both INHr strains (Fig. 2). It is known that katG mutations distinctively alter the levels of the enzyme besides its catalytic domains (26, 27). Here, we show that the less frequent 5′ end katG mutations also result in a significant reduction of the KatG enzyme levels (Fig. 2). In the same functional category of virulence detoxification and adaptation, we also found increased levels of two other oxidoreductases, Rv1751 and Rv2951c, in both INHr strains (Fig. 2).
Fig. 2.

Proteins in the virulence, detoxification, and adaptation category with significant abundance differences between clonal pairs of Mtb after INHr acquisition. *p < 0.05 (t test) in the laboratory strains comparison only, **p < 0.05 (t test) in the clinical strains comparison only, §p < 0.05 (t test) in both (clinical and lab) comparisons. The black box indicates the proteins that were not only shared but had the same trend in both comparisons. The cellular fraction is only indicated for KatG to emphasize the strong decreased levels of this protein. CFP: culture filtrate; CW: cell wall; MEM: membrane; CYT: cytosol proteins.
Mtb katG Mutant Strains Had Altered Proteins Involved in Energy Metabolism
In the category of energy metabolism and respiration, there was a common significant increase in the abundance of AtpG, an ATP synthase protein, and QcrC, a protein of the cytochrome bc1 complex, in the membrane and cell wall fractions (Figs. 3A–3B). Although AtpH was also increased in both INHr strains, this difference was statistically significant only in the membrane and cell wall fractions of the laboratory pair (Fig. 3A). The dihydrolipoamide dehydrogenase LpdC, also involved in energy metabolism in Mtb, was decreased in both INHr strains (Fig. 3A). Additional proteins in this category were increased in either the laboratory or the clinical INHr strain. These include other members of the ATP synthase (AtpF and D), the cytochrome bc1 complex (QcrA and B), as well as the cytochrome aa3-type oxidase (CtaC and D) (Figs. 3A–3B). All of these increased proteins in the INHr strains participate in the electron-transport chain (Fig. 3B) and suggest a highly reductive environment in the cell with the enzymatic capacity to produce high levels of ATP (28). Additionally, a common increased abundance of three enzymes of the central carbon metabolism, specifically of the tricarboxylic acid cycle, were observed for both INHr strains. These include the enzymes isocitrate dehydrogenase Icd2, succinate-semialdehyde dehydrogenase GabD2, and l-lactate dehydrogenase LldD2 (Fig. 3A).
Fig. 3.

(A) Proteins in the energetic metabolism and respiration category significantly different between clonal pairs of Mtb after INHr acquisition. p < 0.05 (t test) *in the laboratory strains comparison only, **in the clinical strains comparison only, §in both (clinical and lab) comparisons. The black box indicates the proteins that were not only shared but had the same trend in both comparisons. (B) Components of the electron-transport chain and ATP synthase proteins altered in the INHr strains. MK: menaquinone; MKH2: menaquinol.
Increased Abundance of Proteins in the Fatty Acid β-Oxidation in katG Mutants
Overall, there was a net increase in the abundance of enzymes involved in the β oxidation pathway in both INHr strains (Fig. 4). Particularly, FadD6, FadE24, and FadB were all more abundant in the INHr strains. FadD6 and FadE24 catalyze the first and second β-oxidation reaction, respectively. FadB forms a canonical β-oxidation complex with FadA (29), which is increased in the clinical INHr strain only (Fig. 4). In the same way, lipolytic enzymes such as lipases/esterases, phospholipases, and enzymes involved in cholesterol oxidation were mostly increased in the INHr strains (supplemental Fig. S1). Among the lipases, higher levels of LipD and LipW were observed in the clinical INHr strain while only higher levels of LipN were detected in the laboratory INHr strain (supplemental Fig. S1).
Fig. 4.
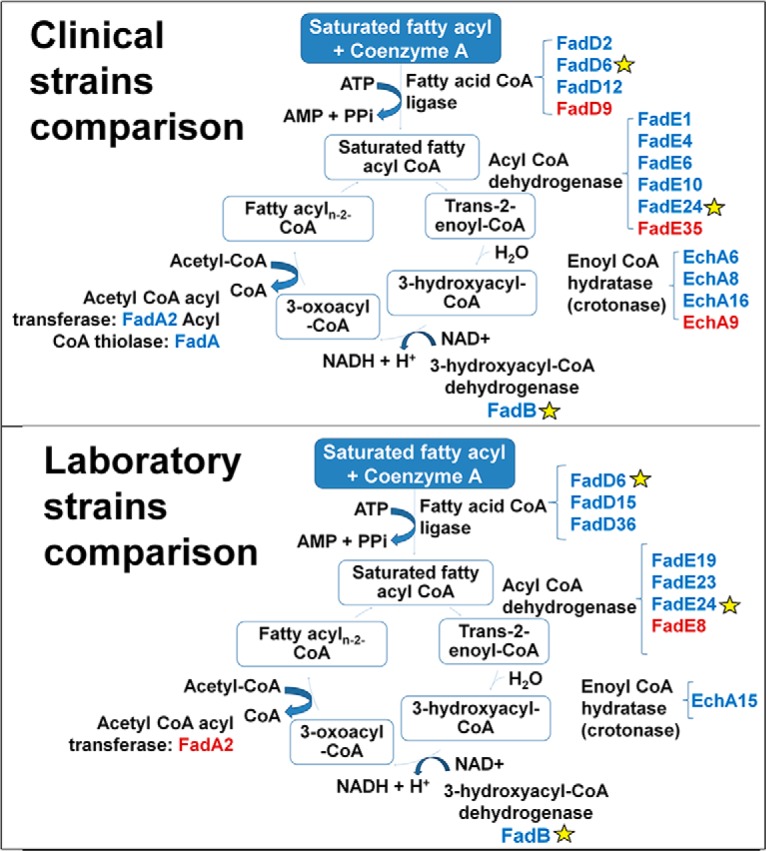
Enzymes involved in the β-oxidation of fatty acids with altered levels in the Mtb INHr strains. For each reaction, the name of the enzyme is described in black; close to them are the different isozymes that were found in the LC-MS/MS analysis significantly higher (blue) or lower (red) in the INHr strain for each comparison. Yellow stars indicate the common proteins in both clinical and laboratory strains comparison.
Short-chain Dehydrogenase EphD and the Epoxide Hydrolase EphF
Increased levels of these two enzymes were observed in both INHr strains. EphF and EphD are grouped in the virulence and detoxification category and are involved in detoxification reactions following the degradation of lipids, which is in relation with the increased levels of enzymes participating in the β-oxidation pathway.
Decreased Levels of Proteins Involved in the MA Synthesis
Three enzymes of the fatty acid synthase pathway II (FAS II), UmaA, FbpA, and InhA had significantly lower levels in both INHr strains (Fig. 5). UmaA is a probable methyl transferase whose function in Mtb remains unknown. As a methyl transferase, UmaA can participate in the modification of mycolates since it has a cyclopropane synthase domain (30). The next enzyme, FbpA is part of the Antigen 85 complex and participates in the transfer of MA to TMMs and arabinogalactan to form cell-wall mycolyl-arabinogalactan-peptidoglycan complex (31–33). Finally, InhA is an NADH-dependent meromycolic acid reductase that participates in the mycolate biosynthesis and is the most accepted target of INH. Inhibition of InhA by INH potentially affects the last elongation steps for the formation of the meromycolate chain, with the possible accumulation of the cis-unsaturated short chain precursors (23). In addition to these three enzymes, other proteins related to MA metabolism specific to each of the clonal pairs were also observed and are presented in supplemental Figs. S2 and S3.
Fig. 5.

Altered enzymes in the fatty acid synthesis pathway of M. tuberculosis (p < 0.05) that were shared between laboratory and clinical pair comparisons. Red squares represent lower levels of enzymes in the INHr strains. Alternative enzymes (Rv3720 and FabG4) depicted with quotation marks (?) are added; see main text in the clinical pair comparison only to find more details. Reprinted with some adaptations from Marrakchi et al. (4) with permission from Elsevier.
Decreased Abundance of Proteins Involved in the S-Adenosyl-l-methionine (SAM) Metabolism
In line with altered levels of methyl transferases (Fig. 5 and supplemental Fig. S2), significantly reduced levels of MetK and SahH were observed in both INHr strains (supplemental Fig. S4). These two proteins are involved in the SAM metabolism. MetK participates in the final conversion of methionine to SAM while SahH is a thioester hydrolase expected to convert S-adenosyl-l-homocysteine into adenosine and l-homocysteine. These differences were observed at different cellular fractions for each pair comparison (supplemental Fig. S4).
Higher Levels of Transcriptional Regulatory Proteins in Both INHr Strains
The regulatory proteins PhoP and Rv3050c.1 were significantly increased in both INHr strains (Table II). PhoP is a transcriptional regulatory protein that works together with PhoQ to positively regulate some genes implied in the growth of fully virulent Mtb strains in the mouse model (34). Increased expression of PhoP has also been shown in transcriptomic studies in response to hypoxia and nutrient starvation (35, 36). Little is known about Rv3050c.1 that is part of the AsnC-family which groups regulators specifically triggered by asparagine binding. Among the possible events regulated by this protein are amino acid metabolism, DNA repair, central carbon metabolism, and persistence (37). Lastly, the regulatory protein Rv3676 was found with increased abundance in our study for our clinical isolate and was also shown to be an important regulatory protein via transcriptomic studies in response to hypoxia and/or nutrient starvation that include discordant results similar to ours, which may be explained by differences between strains (36, 38, 39).
Table II. Regulatory proteins of Mtb that were found altered in both clinical pair comparisons after acquisition of INH resistance.
| Accession number | Gene | Identified proteins (19) | Log2 |
|
|---|---|---|---|---|
| Clinical pair | Lab pair | |||
| Rv0757.1 | phoP | Two component system transcriptional regulator phoP | −1.00 | −2.32 |
| Rv3050c.1 | Rv3050c.1 | Transcriptional regulator, asnC-family | −3.32 | −1.32 |
| Rv0042c.1 | Rv0042c.1 | Transcriptional regulator, marR-family | −2.32 | NS |
| Rv0043c.1 | Rv0043c.1 | Transcriptional regulator, gntR-family | a | NS |
| Rv0474.1 | Rv0474.0 | Transcriptional regulator | b | NS |
| Rv0844c.1 | narL | Nitrate/nitrite response transcriptional regulator narL | b | NS |
| Rv1423.1 | whiA | Transcriptional regulator whiA | −1.00 | NS |
| Rv1479.1 | moxR1 | Transcriptional regulator moxR1 | −1.74 | NS |
| Rv1626.1 | Rv1626.1 | Two-component system transcriptional regulator | −0.32 | NS |
| Rv2919c.1 | glnB | Nitrogen regulatory protein P-II glnB | −1.74 | NS |
| Rv3143.1 | Rv3143.1 | Response regulator | −2.32 | NS |
| Rv3291c.1 | Rv3291c.1 | Transcriptional regulator, asnC-family | −0.51 | NS |
| Rv3295.1 | Rv3295.1 | Transcriptional regulator, tetR-family | −0.51 | NS |
| Rv3676.1 | Rv3676.1 | Transcriptional regulator, crp/fnr-family | −1.00 | NS |
| Rv3692.1 | moxR2 | Methanol dehydrogenase transcriptional regulator moxR2 | b | NS |
| Rv0576.1 | Rv0576 | Transcriptional regulator, arsR-family | NS | −1.32 |
| Rv1267c.1 | embR | Transcriptional regulator embR | NS | −2.32 |
| Rv1379.1 | pyrR | Pyrimidine operon regulatory protein pyrR | NS | −2.32 |
| Rv1719.1 | Rv1719.1 | Transcriptional regulator | NS | −3.32 |
*Log2 fold change values provided only for significant differences, t-test, p≤ 0.05).
aNSAF (normalized spectral abundance factor) values in the INHr strain were zero,
bNSAF values in the INHs strain were zero. NS: not statistically significant. Negative values indicate proteins significantly decreased levels while positive values indicate proteins with significantly increased levels in the INHs strain.
Increased Levels of the Lipoproteins in the INHr Strains
There were no commonly altered lipoproteins in either the clinical and laboratory comparisons; however, there was a common trend of increased lipoprotein abundance. Six lipoproteins were significantly increased in the laboratory INHr strain, while three lipoproteins were significantly increased in the clinical INHr strain. The lipoprotein LppX was the exception in this group, as it was significantly decreased in the clinical INHr strain only (Fig. 6). LppX is a lipoprotein involved in the translocation of PDIMs across the plasma membrane of Mtb (40).
Fig. 6.

Lipoproteins with significant differences between clonal pairs of Mtb after INHr acquisition. p < 0.05 (t test) *in the laboratory strains comparison only, **in the clinical strains comparison only.
Fig. 7.
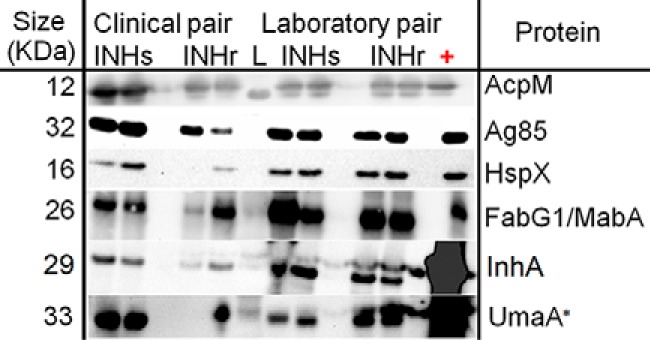
Western blotting (WB) validation of selected proteins with significantly different levels in both clinical and laboratory INHr strains versus their respective INHs pairs. Blots were developed by chemiluminescent reaction. Two biological replicates were used for almost all WB, except for the WB performed for UmaA (*), where only one replicate was used for the clinical INHr sample due to scarcity of the sample. Five μg of the cytosolic fraction of H3Rv were used as positive control for the analysis of Ag85 and HspX and 5 μg of culture filtrate (CFP) of H3Rv for AcpM. For the proteins FabG1/MabA, InhA, and UmaA, 5 μg of recombinant protein for each case were used as positive control. LC-MS/MS levels of HspX were described in Fig. 2.
Protein Differences Showing Opposite Trends Between Clinical and Laboratory Strains
Levels of the Antioxidant Complex AhpD/AhpC Are Not Common Events in All katG Mutants
In the analysis of proteins that may compensate the reduced levels of KatG, the alkyl peroxidases AhpC and AhpD were evaluated in both INHr strains. These proteins form a complex that is part of the antioxidant response in Mtb, with peroxidase and peroxynitrite-reductase functions (41, 42). Both AhpC and D were lower in the clinical INHr but higher in the laboratory INHr strain compared with their susceptible pairs, respectively (Table I, Fig. 2). The distinct AhpC trend was previously observed in these INHr strains by Western blotting in a virulence study, where lower levels of AhpC protein were simultaneously observed in the INHr strain along with reduced fitness and virulent profile in the mouse model. On the contrary, higher levels of AhpC were concomitant with a nonsignificantly altered virulent bacterial phenotype (6). Previous studies have also evidenced the importance of AhpD in the in vivo stress response in INHr Mtb strains (42, 43).
NADH Dehydrogenase Complex I
Although showing a different trend in the INHr strains compared with their INHs parental strains, proteins of the proton pumping I NADH dehydrogenase complex (NDH-I) were altered in both INHr strains. The proteins NuoF and NuoG were significantly decreased in the laboratory INHr while the proteins NuoC, D, and G were significantly increased in the clinical INHr strain (Fig. 3A). These four proteins are encoded by the nuo operon and are involved in respiration with the oxido-reduction of menaquinone (Fig. 3B). This suggests that NDH-1 complex alteration could be a common result of the INHr phenotype, but the trend to which it could be affected may depend on additional factors such as its genetic background or type of drug exposure, for instance, one single drug (as in the case of the laboratory INHr strain) or a combined therapy (as in the case of the clinical INHr strain).
Protein Differences Specific to the Clinical INHr Strain
Decreased Levels of Additional FAS II Enzymes
Members of the core reactions of FAS II route: AcpM, KasB and the HadABC complex, together with FadD32 and AccD5 showed decreased levels in the clinical INHr strain. Parallel to the diminished levels of enzymes in this vital metabolic pathway, increased levels of the enzymes FabG4, HtdX, FadD13, and other members of proteins grouped in the mymA operon (Rv3087 and Rv3088) were observed in the INHr stain.
Increased Level of Lipases
A significant increase of the enzyme cutinase 2 (Cut2 or Cfp25) in the clinical INHr strain was observed (supplemental Fig. S1). Mtb has seven putative cutinases that are esterases with the ability to hydrolyze phospholipids and Tween 80 as was discussed by Parker et al. (45). Cutinases have been experimentally associated with phospholipases (45), which were also increased in the clinical INHr strain (PlcC and PlcB, supplemental Fig. S1). In this category, enzymes involved in lipid degradation and cholesterol catabolism in Mtb, such as FadA5, Rv1106, and ChoD, were higher in the clinical INHr strain in the cell wall and membrane fractions (supplemental Fig. S1).
Lipidomics
Total Lipid Analysis
Given the numerous differences at the protein level of lipid metabolic enzymes in both the clinical and laboratory INHr versus INHs strains, we proceeded to analyze the lipid content of these strains. The clinical INHr strain showed significantly lower levels of PDIM and TMM when compared with its INHs pair (p value < 0.05) as evidenced by TLC analysis (Fig. 8A). Regarding phosphatidylinositol mannosides, both the clinical and laboratory INHr strains had significantly lower levels of this glycolipid compared with their INHs progenitors (Fig. 8B).
Fig. 8.

TLC analysis. Levels of (A) PDIM and (B) glycolipids: TMM (Trehalose monomycolates), TDM (Trehalose dimycolates), and SL (sulfolipids) among two biological replicates of each clinical and laboratory Mtb strains. INHr: isoniazid resistant; INHs: isoniazid susceptible. Ten μg of each H37Rv purified standard was included in the analysis for each evaluated lipid.
MA Comparison
Here, we present the extracted ion chromatogram and spectra of the features evaluated for the clinical pair comparison only because there were no significant differences in α-MA levels in the laboratory clonal pair comparison. There were significantly higher levels of α-MA C78 and C80 in the clinical INHs compared with its INHr pair (Fig. 9). After the LC/MS analysis, four precursor ions belonging to α-MA C77, C78, C79, and C80 with the m/z values: 1,122.1571, 1,136.1746, 1,150.1886, and 1,164.2062, were selected for LC-MS/MS analysis. This analysis revealed diagnostic fatty-acid-derived ions, such as the alpha (alkyl or R1) and meromycolate (beta-hydroxy or R2) chains in the MAs. According to the masses, two cyclopropane rings in the meromycolic acid chain were predicted for all α-MA variants studied (Fig. 10). The alpha chain (R1) lengths varied among the different α-MA species detected, for instance, as the α-MA increases in carbon number; they became a purer class of molecules containing C24H49 in their R1 group (Fig. 10B). This observation was previously described in the analysis of MA of M. bovis Bacillus Calmette–Guérin (BCG) (46). Additionally, we detected other R1 groups in the α-MA with m/z values of 381.376 and 367.36 that correspond to the [M-H]- adduct of fatty acids C23H47 and C22H45 respectively (Fig. 10). The R1 = C23H47 was only observed for the odd number α-MA C77 and C79 among the analyzed species (Fig. 10).
Fig. 9.

MA analysis of the clinical Mtb pair through LC-MS in the negative mode. Extracted ion chromatogram and spectra showing the different features compatible with α-MA according to their m/z value. Only two specific α-MA showed peak areas with statistically significant difference (p < 0.05, paired t test): α-MA C78 and C80. Three replicates were used for these study, black lines represent the results the clinical INHs strain while the red represents the clinical INHr strain. All the features were detected at least with the hydrogen adduct.
Fig. 10.
LC-MS/MS analysis of different α-mycolic acids (MA) among INHr and INHs Mtb clinical pair. (A) R1 represents the alpha, alkyl, or fatty acid, while R2 represents the meromycolic acid (or beta-hydroxy) chains, which compose the MA. (B) MS/MS spectral interpretation, including the most significant different MA (α-MA C78 and C80) in clinical Mtb strains.
DISCUSSION
This study presents a snapshot of genetically related Mtb strains under the same in vitro conditions to detect specific changes related to the INHr phenotype. It is important to emphasize two points that should be considered in the analysis of the findings presented here; first, with the exception of KatG levels, none of the other protein changes described in this study have associated mutations in their respective genes, as previously revealed by whole genomics sequencing of these strains (12). Second, this shotgun proteomics approach presents several limitations in that changes in protein abundance may be attributed to decreased expression, differences in protein half-lives, and losses in protein abundance due to aggregation and post-translational modifications with concomitant changes in protein function. Rather, this study is a first description of brute protein variation in Mtb strains (using two different genotypes as reference: one clinical and one laboratory derived) after acquisition of INH resistance. While this study was also limited in scope in resolving functional protein analyses, the analysis of lipid variation provided additional evidence of the effect of protein level variation in enzymes associated with the lipid biosynthetic pathway similar to that which occurred for protein variation in KatG.
At least two initial known common events occurred in the INHr Mtb strains analyzed here; first both acquired INH resistance after exposure to the drug in vivo, and both contained mutations in the 5′ end of the katG gene (11, 12). These mutations generated an important reduction in KatG levels, which in addition to halting INH activation, also impaired the bacteria's ability to handle oxidative stress, as KatG is an important catalase, peroxidase, and peroxynitritase (47, 48). The reduction of the anti-oxidative stress capacity was particularly notorious for the clinical INHr strain that lacked compensatory production of AhpC and had slower growth in vitro as well as an impaired growth in the mouse model of infection when compared with its INHs pair (6). The findings presented here provide a substantial biochemical description of clinically relevant clonal Mtb strains with the cleanest genetic background possible, using minimal media so as not to artificially bolster those strains with reduced fitness (in particular the clinical INHr stain) in order to help generate data-driven hypothesis to enhance the understanding of INH resistance phenotype in Mtb.
The observed alterations in enzymes involved in NAD+/NADH metabolism are very relevant in this analysis since this is the cofactor required for the synthesis of the active form of INH. NADH is an important nucleotide used in many relevant and essential metabolic reactions, including central carbon, energy, and lipid metabolism, most of which had proteins with a net increased level in the INHr strains. It is possible that the increased or decreased protein levels also depends on the affinity of the different enzymes for the two different cofactors, NADH and NADPH. Many of the altered enzymes found in this study, such as SahH, Rv0753c, Rv2723, FadB2, InhA, Rv2971, and Rv2766c, have a high affinity for the INH-NAD(P) adduct as previously demonstrated by one computational and one experimental study (49, 50).
It is important to highlight that besides SahH, this study found other altered proteins associated with SAM metabolism. SAM is a common substrate in the methylation of different biomolecules (51). For instance, SAM is the substrate for the introduction of trans methyl branches and cyclopropane rings in the meromycolic acid chain of MAs (33). Methionine and SAM metabolism are expected to be a promising drug target in Mtb, as metA mutants are unable to grow in vitro and are rapidly cleared in vivo either in immunocompetent or immunocompromised mice (52). Although we did not find significant differences in the levels of MetA, we did find significantly reduced abundances of MetK, in addition to SahH, both of which are involved in these metabolic pathways (supplemental Fig. S4). In particular, the clinical INHr strain also had increased levels of MetB, which is involved in methionine synthesis, using cystathionine as an intermediate molecule (supplemental Fig. S4). According to these findings, we can hypothesize that INH resistance leads to proteomic changes that can be associated to altered levels of methionine as well as cysteine in Mtb. Previous work by Vilcheze et al. demonstrated that cysteine prevents the formation of INH-resistant and INH-tolerant Mtb strains in vitro, inducing higher oxygen uptake and rendering the cells more sensitive to oxidative stress (53, 54). Therefore, the cysteine/methionine metabolism may be important in the survival of INHr strains in vivo and should be explored as a potential drug target in these strains.
Among the other significantly increased proteins in the INHr strains were those associated with energy metabolism, ATP synthesis (Figs. 3A–3B) and some detoxifying enzymes that are particularly important after the β-oxidation of fatty acids (Table I, Figs. 1–6). The net increase of the fatty acid oxidation enzymes suggests that lipids are an important source of energy for the INHr strains, with the generation of substantial quantities of NADPH (55). The higher β-oxidation activity in Mtb is also related to an altered energetic metabolism, with a possible higher production of ATP molecules. For instance, the β-oxidation of palmitate generates 106 ATP molecules, while the oxidation of glucose yields only 38 ATP molecules (55). The increased fatty acid oxidation pathway is concomitant with the increase of some proteins in the ATP synthase machinery and the adenylate cyclase enzyme (Rv1647) in both INHr strains (Table I, Fig. 3A), which also suggests a higher ATP production and hydrolysis. The increase of β-oxidation is also associated with the reduced levels of the pyruvate kinase (PykA) in the INHr strains (although only statistically significant for the clinical INHr, Fig. 3A) that is related with the use of nonglycolytic carbon sources such as lipids (56). It is important to emphasize that β oxidation of fatty acids in Mtb is a metabolic route with great redundancy (29). Multiple enzymes have been identified with the ability to participate in one single reaction of the β oxidation of fatty acids pathway (Fig. 4). Due to this high redundancy, the β oxidation of fatty acids is not considered an attractive drug target against Mtb. This is also confirmed by our proteomics findings, where the decrease of one isozyme was highly compensated by the increase of others (Fig. 4).
On the other hand, the most common drug targets known for Mtb are proteins in the FAS-II pathway. Regarding lipid biosynthesis, low levels of FbpA (Ag85-a), InhA, and UmaA were observed in both INHr strains (Fig. 5). Here, concurrently with the reduced FbpA levels, we found reduced abundance of both TMM and TDM in the clinical INHr strain only, albeit only statistically significant for TMM evaluated through TLC (Fig. 8B). Those findings are supported by a previous study that demonstrated that low levels of FbpA have been associated with a lower production of TDM (59). In the same line, both INHr strains had reduced levels of InhA, which shares up to 26% of identity with other short chain dehydrogenases (23). Additionally, the epoxide hydrolase EphF shares 18.7% identity with InhA according to the Multiple Sequence Alignment platform MUSCLE. In our study, the short chain dehydrogenase EphD, as well as EphF were increased in both INHr strains. It is possible that EphD and EphF could compensate for the reduced abundance of InhA in these INHr strains. Further functional analysis would confirm if the increased levels of these proteins play a role in the synthesis of fatty acids in the INHr phenotype as well as if they can be used as potential drug targets for INHr Mtb strains. Additionally, the reduced levels of UmaA, which is believed to use SAM as substrate, were concomitant with the alteration of proteins in the SAM metabolism pathway (supplemental Fig. S4).
The reduced levels of several FAS II enzymes in the clinical INHr strain only (i.e. KasB, AcpM, HadABC, and FadD32) seem to be partially compensated by the increase of alternative enzymes that catalyze similar reactions (supplemental Fig. S3B). An alternative fatty acid biosynthetic pathway, called the “fatty acid elongation system II,” was first observed in M. smegmatis in 1983 (60). The proposed alternative enzymes that participate in this alternative pathway, FabG4, HtdX, FadD13, and other members of the mymA operon, were increased in the INHr strain only (supplemental Figs. S2 and S3B). These enzymes can perform the same reactions but have a different affinity for the NADH and NADPH cofactors and substrates. The significant lower levels of AcpM in the clinical INHr strain suggests a slow entrance of the acyl-carrier protein (ACP) derivatives into the FAS II pathway. The decreased production of AcpM derivatives may induce the increased levels of FabG4. FabG4 has affinity for both NADH and NADPH cofactors and also for both coenzyme A and ACP fatty acid derivatives. FabG1 in turn, has a higher affinity for NADPH and almost exclusively ACP derivatives (61, 62). In addition to this, reduced levels of ACP derivatives (due to the reduced levels of AcpM) as well as the reduced levels of the HadABC complex enables the catalytic role of HtdX in this alternative FAS II route. Specifically the role of the HtdX has been proposed in the dehydration step of the hydroxyacyl-ester to an enoyl intermediate regularly catalyzed by the HadABC complex (Fig. 5) (63). HtdX is strictly specific for coenzyme A substrates rather than ACP derivatives, which gives more support to our hypothesis (64). Finally, the fatty-acid-AMP ligase FadD13 was significantly increased in the clinical INHr strain (supplemental Figs. S2 and S3B). FadD13 is proposed to participate in the biosynthetic pathway of MAs through activation of fatty acids (similarly to the role of FadD32, which levels are decreased in the INHr clinical strain). FadD13 is part of the mymA operon, which is up-regulated under acidic growth conditions simultaneous to a down-regulation of the FAS II pathway (65). The acyl transferases Rv3087 and Rv3088 that are significantly increased in the clinical INHr strain (supplemental Fig. S3B) are also part of the mymA operon. These last two enzymes are proposed to transfer FadD13 activated fatty acids to an acceptor in the mycobacterial cell wall (66). Again, this suggests that an alternative biosynthetic route for MAs is up-regulated when the levels of canonical FAS II enzymes are diminished in the INHr strains due to an unbalanced redox environment.
According to the LC-MS/MS analysis of the α-MA, only two features compatible with α-MA C78 and C80 were significantly decreased in the clinical INHr strain only (Figs. 8 and 9). This trend was not observed for the α-MA C77 and C79 nor for any of the MA features studied in the laboratory Mtb pair. This finding could be partially explained by the level of alteration of the FAS II pathway observed in the clinical INHr strain only (supplemental Fig. S3B). The proteomics and lipidomics data together suggest that although there is an increase in enzymes of the alternative MA biosynthetic pathway, this alternative route is not as efficient as the canonical FAS II pathway in producing α-MA. Proteins of this alternative pathway were also found to be increased in two previous proteomics studies, one involving clinical isogenic strains of Beijing genotype (one of them INHr) and other evaluating clinic clonal pairs of the Central-Asian 1-Delhi lineage that transition from drug susceptible to multidrug-resistant (INHr plus rifampicin resistant) Mtb strains. The clinical Beijing INHr strain also exhibited increased levels of FabG4 and HtdX (5), while the multidrug-resistant strain of Central-Asian 1 lineage had increased levels of FabG4 (67). Taken together, the alternative lipid biosynthetic pathway should be further explored in a clinical context to evaluate if the previous exposure to the combined therapy (that only occurred in the clinical INHr strain of this study) is able to induce this alternative lipid biosynthetic route in INHr strains as well as its implications in the survival of those drug resistant strains.
Additionally, the lower levels of LppX in the clinical INHr strain is in line with decreased levels of phenolpthiocerol synthesis type-I polyketide synthase, as well as the significantly lower levels of PDIM observed only for this strain in the TLC analysis (Figs. 6 and 8A and supplemental Fig. S1). These -omics results, together with the role of PDIM in Mtb virulence, suggest that the overall reduction of PDIM levels may be another reason for the reduced virulence previously reported for the clinical INHr strain of this study (6). From the biochemical standpoint, this reduced virulence can be explained as a cumulative effect of reduced levels of KatG (the responsible for the INHr phenotype), FbpA, and LppX, among other proteins included in the virulence, detoxification, and adaptation category (such as AhpC, Fig. 2) (40, 42, 68).
In summary, after acquisition of INHr due to katG N terminus mutations, Mtb strains have a proteomic rearrangement that includes common changes in the β-oxidation of fatty acids and energy metabolism, as well as individual changes that appear to be dependent on the genetic background of the bacterium and/or the previous exposure to a combined anti-TB therapy. Taken together, the proteomic and lipidomic changes may be exploited in the search for potential drug targets to combat INH resistant Mtb strains.
DATA AVAILABILITY
Raw mass spectrometry data deposited at data at http://proteomecentral.proteomexchange.org/cgi/GetDataset, using the project identifier PXD009549.
Supplementary Material
Acknowledgments
We have no conflict of interest to declare. This work was supported by the scholarship Francisco Jose de Caldas-convocatoria 512 from the Colombian Administrative Department of Science, Technology, and Innovation Colciencias (recipient: Luisa Maria Nieto) and by the American Type Culture Collection fund #2010-0516-0005 (recipient: Karen Dobos). We thank Dr. Michael McNeil, Reem Almubarak, Prithwiraj De, Gustavo Diaz, Corey Broeckling, Megan Lucas, Anne Simpson, and Kala Early for their technical support and advice. We thank Dr. John Spencer and Dr. Angela Marques for their kind provision of the polyclonal antibodies for the proteins of the FAS II pathway. Finally, we thank also Dr. Ian Orme's and Marcos Burgos for facilitating the clinical strains and Dr. Gyanu Lamichhane, Dr. Eric Nuermerberg, and Dr. Rokeya Tasneen from the Center of Tuberculosis Research at Johns Hopkins University for providing the laboratory INHr strain.
Footnotes
* This work was supported by Departamento Administrativo de Ciencia, Tecnología e Innovación (COLCIENCIAS) and American Type Culture Collection.
 This article contains supplemental material Figs. S1–S5.
This article contains supplemental material Figs. S1–S5.
1 The abbreviations used are:
- TB
- tuberculosis
- Mtb
- Mycobacterium tuberculosis
- INH
- isoniazid
- INHr
- isoniazid resistant
- INHs
- isoniazid susceptible
- CFP
- culture filtrate proteins
- CYT
- cytosol
- MEM
- membrane
- CW
- cell wall
- MS/MS
- tandem mass spectrometry
- TLC
- thin-layer chromatography
- MA
- mycolic acids
- PDIM
- phthiocerol dimycocerosates
- TMM
- trehalose monomycolate
- TDM
- trehalose dimycolate
- SAM
- S-adenosyl-methionine
- ACP
- acyl carrier protein
- RFLP
- restriction fragment length polymorphism
- LTQ
- linear triple quadrapole
- ESI
- electro-spray ionization
- NIAID
- National Institutes of Allergy and Infectious Diseases.
REFERENCES
- 1. World Health Organization. (2015) Global tuberculosis report. Available: http://apps.who.int/iris/bitstream/10665/191102/1/9789241565059_eng.pdf?ua=1
- 2. Timmins G. S., and Deretic V. (2006) Mechanisms of action of isoniazid. Mol. Microbiol. 62, 1220–1227 [DOI] [PubMed] [Google Scholar]
- 3. Vilchèze C., and Jacobs W. R. Jr. (2007) The mechanism of isoniazid killing: Clarity through the scope of genetics. Annu. Rev. Microbiol. 61, 35–50 [DOI] [PubMed] [Google Scholar]
- 4. Marrakchi H., Lanéelle M. A., and Daffé M. (2014) Mycolic acids: Structures, biosynthesis, and beyond. Chem. Biol. 21, 67–85 [DOI] [PubMed] [Google Scholar]
- 5. Nieto R. L. M., Mehaffy C., and Dobos K. M. (2016) Comparing isogenic strains of Beijing genotype Mycobacterium tuberculosis after acquisition of isoniazid resistance: A proteomics approach. Proteomics 16, 1376–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nieto R. L. M., Mehaffy C., Creissen E., Troudt J., Troy A., Bielefeldt-Ohmann H., Burgos M., Izzo A., and Dobos K. M. (2016) Virulence of Mycobacterium tuberculosis after acquisition of isoniazid resistance: Individual nature of katG mutants and the possible role of AhpC PLoS ONE 11, e0166807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. van Soolingen D., de Haas P. E., and Kremer K. (2001) Restriction fragment length polymorphism typing of mycobacteria. Methods Mol. Med. 54, 165–203 [DOI] [PubMed] [Google Scholar]
- 8. Kamerbeek J. Schouls L., Kolk A., van Agterveld M., van Soolingen D., Kuijper S., Bunschoten A., Molhuizen H., Shaw R., Goyal M., and van Embden J. (1997) Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J. Clin. Microbiol. 35, 907–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Burgos M., DeRiemer K., Small P. M., Hopewell P. C., and Daley C. L. (2003) Effect of drug resistance on the generation of secondary cases of tuberculosis. J. Infect. Dis. 188, 1878–1884 [DOI] [PubMed] [Google Scholar]
- 10. Gagneux S., Burgos M. V., DeRiemer K., Encisco A., Muñoz S., Hopewell P. C., Small P. M., and Pym A. S. (2006) Impact of bacterial genetics on the transmission of isoniazid-resistant Mycobacterium tuberculosis. PLoS Pathog. 2, e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Almeida D., Nuermberger E., Tasneen R., Rosenthal I., Tyagi S., Williams K., Peloquin C., and Grosset J. (2009) Paradoxical effect of isoniazid on the activity of rifampin-pyrazinamide combination in a mouse model of tuberculosis. Antimicrob. Agents Chemother. 53, 4178–4184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Datta G., Nieto L. M., Davidson R. M., Mehaffy C., Pederson C., Dobos K. M., and Strong M. (2016) Longitudinal whole genome analysis of pre and post drug treatment Mycobacterium tuberculosis isolates reveals progressive steps to drug resistance. Tuberculosis 98, 50–55 [DOI] [PubMed] [Google Scholar]
- 13. Deleted in proof.
- 14. Bisson G. P., Mehaffy C., Broeckling C., Prenni J., Rifat D., Lun D. S., Burgos M., Weissman D., Karakousis P. C., and Dobos K. (2012) Upregulation of the phthiocerol dimycocerosate biosynthetic pathway by rifampin-resistant, rpoB mutant Mycobacterium tuberculosis. J. Bacteriol. 194, 6441–6452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lucas M. C., Wolfe L. M., Hazenfield R. M., Kurihara J., Kruh-Garcia N. A., Belisle J., and Dobos K. M. (2015) Fractionation and analysis of mycobacterial proteins. Methods Mol. Biol. 1285, 47–75 [DOI] [PubMed] [Google Scholar]
- 16. Chambers M. C., Maclean B., Burke R., Amodei D., Ruderman D. L., Neumann S., Gatto L., Fischer B., Pratt B., Egertson J., Hoff K., Kessner D., Tasman N., Shulman N., Frewen B., Baker T. A., Brusniak M. Y., Paulse C., Creasy D., Flashner L., Kani K., Moulding C., Seymour S. L., Nuwaysir L. M., Lefebvre B., Kuhlmann F., Roark J., Rainer P., Detlev S., Hemenway T., Huhmer A., Langridge J., Connolly B., Chadick T., Holly K., Eckels J., Deutsch E. W., Moritz R. L., Katz J. E., Agus D. B., MacCoss M., Tabb D. L., and Mallick P. (2012) A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lew J. M., Mao C., Shukla M., Warren A., Will R., Kuznetsov D., Xenarios I., Robertson B. D., Gordon S. V., Schnappinger D., Cole S. T., and Sobral B. (2013) Database resources for the tuberculosis community. Tuberculosis 93, 12–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nesvizhskii A. I., Keller A., Kolker E., and Aebersold R. (2003). A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 [DOI] [PubMed] [Google Scholar]
- 19. Vizcaíno J. A., Csordas A., del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q. W., Wang R., and Hermjakob H. (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vizcaino J. A., Deutsch E. W., Wang R., Csordas A., Reisinger F., Ríos D., Dianes J. A., Sun Z., Farrah T., Bandeira N., Binz P. A., Xenarios I., Eisenacher M., Mayer G., Gatto L., Campos A., Chalkley R. J., Kraus H. J., Albar J. P., Martinez-Bartolomé S., Apweiler R., Omenn G. S., Martens L., Jones A. R., Hermjakob H. (2014) ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nature Biotechnol. 32, 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang H., Troudt J., Grover A., Arnett K., Lucas M., Cho Y. S., Bielefeldt-Ohmann H., Taylor J., Izzo A., and Dobos K. M. (2011) Three protein cocktails mediate delayed-type hypersensitivity responses indistinguishable from that elicited by purified protein derivative in the guinea pig model of Mycobacterium tuberculosis infection. Infect. Immun. 79, 716–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sartain M. J., Dick D. L., Rithner C. D., Crick D. C., and Belisle J. T. (2011) Lipidomic analyses of Mycobacterium tuberculosis based on accurate mass measurements and the novel “Mtb LipidDB.” J. Lipid Res. 52, 861–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Takayama K., Wang C., and Besra G. S. (2005) Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin. Microbiol. Rev. 18, 81–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Song S. H., Park K. U., Lee J. H., Kim E. C., Kim J. Q., and Song J. (2009) Electrospray ionization-tandem mass spectrometry analysis of the mycolic acid profiles for the identification of common clinical isolates of mycobacterial species. J. Microbiol. Methods 77, 165–177 [DOI] [PubMed] [Google Scholar]
- 25. Teramoto K., Suga M., Sato T., Wada T., Yamamoto A., and Fujiwara N. (2015) Characterization of mycolic acids in total fatty acid methyl ester fractions from Mycobacterium species by high resolution MALDI-TOFMS. Mass Spectrom. 4, A0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pym A. S., Saint-Joanis B., and Cole S. T. (2002) Effect of katG mutations on the virulence of Mycobacterium tuberculosis and the implication for transmission in humans. Infect. Immun. 70, 4955–4960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ando H., Kitao T., Miyoshi-Akiyama T., Kato S., Mori T., and Kirikae T. (2011) Downregulation of katG expression is associated with isoniazid resistance in Mycobacterium tuberculosis. Mol. Microbiol. 79, 1615–1628 [DOI] [PubMed] [Google Scholar]
- 28. Cook G. M., Hards K., Vilchèze C., Hartman T., and Berney M. (2014) Energetics of respiration and oxidative phosphorylation in mycobacteria. Microbiol. Spectr. 2, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cole S. T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S. V., Eiglmeier K., Gas S., Barry C. E. 3rd, Tekaia F., Badcock K., Basham D., Brown D., Chillingworth T., Connor R., Davies R., Devlin K., Feltwell T., Gentles S., Hamlin N., Holroyd S., Hornsby T., Jagels K., Krogh A., McLean J., Moule S., Murphy L., Oliver K., Osborne J., Quail M. A., Rajandream M. A., Rogers J., Rutter S., Seeger K., Skelton J., Squares R., Squares S., Sulston J. E., Taylor K., Whitehead S., and Barrell B. G. (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544 [DOI] [PubMed] [Google Scholar]
- 30. Finn R. D., Bateman A., Clements J., Coggill P., Eberhardt R. Y., Eddy S. R., Heger A., Hetherington K., Holm L., Mistry J., Sonnhammer E. L., Tate J., and Punta M. (2014) Pfam: The protein families database. Nucleic Acids Res. 42, D222–D230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Belisle J. T., Vissa V. D., Sievert T., Takayama K., Brennan P. J., and Besra G. S. (1997) Role of the major antigen of Mycobacterium tuberculosis in cell wall biogenesis. Science 276, 1420–1422 [DOI] [PubMed] [Google Scholar]
- 32. Puech V., Guilhot C., Perez E., Tropis M., Armitige L. Y., Gicquel B., and Daffé M. (2002) Evidence for a partial redundancy of the fibronectin-binding proteins for the transfer of mycoloyl residues onto the cell wall arabinogalactan termini of Mycobacterium tuberculosis. Mol. Microbiol. 44, 1109–1122 [DOI] [PubMed] [Google Scholar]
- 33. Takayama K., Wang C., and Besra G. S. (2005) Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin. Microbiol. Rev. 18, 81–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pérez E., Samper S., Bordas Y., Guilhot C., Gicquel B., and Martín C. (2001) An essential role for phoP in Mycobacterium tuberculosis virulence. Mol. Microbiol. 41, 179–187 [DOI] [PubMed] [Google Scholar]
- 35. Galagan J. E., Minch K., Peterson M., Lyubetskaya A., Azizi E., Sweet L., Gomes A., Rustad T., Dolganov G., Glotova I., Abeel T., Mahwinney C., Kennedy A. D., Allard R., Brabant W., Krueger A., Jaini S., Honda B., Yu W. H., Hickey M. J., Zucker J., Garay C., Weiner B., Sisk P., Stolte C., Winkler J. K., Van de Peer Y., Iazzetti P., Camacho D., Dreyfuss J., Liu Y., Dorhoi A., Mollenkopf H. J., Drogaris P., Lamontagne J., Zhou Y., Piquenot J., Park S. T., Raman S., Kaufmann S. H., Mohney R. P., Chelsky D., Moody D. B., Sherman D. R., Schoolnik G. K. (2013) The Mycobacterium tuberculosis regulatory network and hypoxia. Nature 499, 178–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Matern W. M., Rifat D., Bader J. S., and Karakousis P. C. (2018) Gene enrichment analysis reveals major regulators of Mycobacterium tuberculosis gene expression in two models of antibiotic tolerance. Front. Microbiol. 9, 610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Deng W., Wang H., and Xie J. (2011) Regulatory and pathogenesis roles of Mycobacterium Lrp/AsnC family transcriptional factors. J. Cell. Biochem. 112, 2655–2662 [DOI] [PubMed] [Google Scholar]
- 38. Voskuil M. I., Visconti K. C., and Schoolnik G. K. (2004) Mycobacterium tuberculosis gene expression during adaptation to stationary phase and low-oxygen dormancy. Tuberculosis 84, 218–227 [DOI] [PubMed] [Google Scholar]
- 39. Betts J. C., Lukey P. T., Robb L. C., McAdam R. A., and Duncan K. (2002) Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 43, 717–731 [DOI] [PubMed] [Google Scholar]
- 40. Sulzenbacher G., Canaan S., Bordat Y., Neyrolles O., Stadthagen G., Roig-Zamboni V., Rauzier J., Maurin D., Laval F., Daffé M., Cambillau C., Gicquel B., Bourne Y., and Jackson M. (2006) LppX is a lipoprotein required for the translocation of phthiocerol dimycocerosates to the surface of Mycobacterium tuberculosis. EMBO J. 25, 1436–1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Maksymiuk C., Balakrishnan A., Bryk R., Rhee K. Y., and Nathan C. F. (2015) E1 of alpha-ketoglutarate dehydrogenase defends Mycobacterium tuberculosis against glutamate anaplerosis and nitroxidative stress. Proc. Natl. Acad. Sci. U.S.A. 112, E5834–E5843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hillas P. J., del Alba F. S., Oyarzabal J., Wilks A., and Ortiz de Montellano P. R. (2000) The AhpC and AhpD antioxidant defense system of Mycobacterium tuberculosis. J. Biol. Chem. 275, 18801–18809 [DOI] [PubMed] [Google Scholar]
- 43. Koshkin A., Zhou X. T., Kraus C. N., Brenner J. M., Bandyopadhyay P., Kuntz I. D., Barry C. E. 3rd, and Ortiz de Montellano P. R. (2004) Inhibition of Mycobacterium tuberculosis AhpD, an element of the peroxiredoxin defense against oxidative stress. Antimicrob. Agents Chemother. 48, 2424–2430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Deleted in proof.
- 45. Parker S. K., Curtin K. M., and Vasil M. L. (2007) Purification and characterization of mycobacterial phospholipase A: An activity associated with mycobacterial cutinase. J. Bacteriol. 189, 4153–4160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Steck P. A., Schwartz B. A., Rosendahl M. S., and Gray G. R. (1978) Mycolic acids. A reinvestigation. J. Biol. Chem. 253, 5625–5629 [PubMed] [Google Scholar]
- 47. Cade C. E., Dlouhy A. C., Medzihradszky K. F., Salas-Castillo S. P., and Ghiladi R. A. (2010) Isoniazid-resistance conferring mutations in Mycobacterium tuberculosis KatG: Catalase, peroxidase, and INH-NADH adduct formation activities. Protein Sci. 19, 458–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vilchèze C., and Jacobs W. R. Jr. (2014) Resistance to isoniazid and ethionamide in Mycobacterium tuberculosis: Genes, mutations, and causalities. Microbiol. Spectr. 2, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jena L., Deshmukh S., Waghmare P., Kumar S., and Harinath B. C. (2015) Study of mechanism of interaction of truncated isoniazid–nicotinamide adenine dinucleotide adduct against multiple enzymes of Mycobacterium tuberculosis by a computational approach. Int. J. Mycobacteriol. 4, 276–283 [DOI] [PubMed] [Google Scholar]
- 50. Argyrou A., Jin L., Siconilfi-Baez L., Angeletti R. H., and Blanchard J. S. (2006) Proteome-wide profiling of isoniazid targets in Mycobacterium tuberculosis. Biochemistry 45, 13947–13953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chiang P. K., Gordon R. K., Tal J., Zeng G. C., Doctor B. P., Pardhasaradhi K., and McCann P. P. (1996) S-Adenosylmethionine and methylation. FASEB J. 10, 471–480 [PubMed] [Google Scholar]
- 52. Berney M., Berney-Meyer L., Wong K. W., Chen B., Chen M., Kim J., Wang J., Harris D., Parkhill J., Chan J., Wang F., and Jacobs W. R. Jr. (2015) Essential roles of methionine and S-adenosylmethionine in the autarkic lifestyle of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 112, 10008–10013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vilchèze C., Hartman T., Weinrick B., Jain P., Weisbrod T. R., Leung L. W., Freundlich J. S., and Jacobs W. R. Jr. (2017) Enhanced respiration prevents drug tolerance and drug resistance in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 114, 4495–4500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vilchèze C., Hartman T., Weinrick B., and Jacobs W. R. (2013) Mycobacterium tuberculosis is extraordinarily sensitive to killing by a vitamin C-induced Fenton reaction. Nature Commun. 4, 1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kumar A., Farhana A., Guidry L., Saini V., Hondalus M., and Steyn A. J. (2011) Redox homeostasis in mycobacteria: The key to tuberculosis control? Expert Rev. Mol. Med. 13, e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chavadi S., Wooff E., Coldham N. G., Sritharan M., Hewinson R. G., Gordon S. V., and Wheeler P. R. (2009) Global effects of inactivation of the pyruvate kinase gene in the Mycobacterium tuberculosis complex. J. Bacteriol. 191, 7545–7553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Deleted in proof.
- 58. Deleted in proof.
- 59. Nguyen L., Chinnapapagari S., and Thompson C. J. (2005) FbpA-dependent biosynthesis of trehalose dimycolate is required for the intrinsic multidrug resistance, cell wall structure, and colonial morphology of Mycobacterium smegmatis. J. Bacteriol. 187, 6603–6611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kikuchi S., and Kusaka T. (1983) Purification and characterization of 3-oxoacyl-CoA synthase of Mycobacterium smegmatis. J. Biochem. 94, 1045–1051 [DOI] [PubMed] [Google Scholar]
- 61. Gurvitz A. (2009) The essential mycobacterial genes, fabG1 and fabG4, encode 3-oxoacyl-thioester reductases that are functional in yeast mitochondrial fatty acid synthase type 2. Mol. Genetics Genomics 282, 407–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Dutta D., Bhattacharyya S., Roychowdhury A., Biswas R., and Das A. K. (2013) Crystal structure of hexanoyl-CoA bound to β-ketoacyl reductase FabG4 of Mycobacterium tuberculosis. Biochem. J. 450, 127–139 [DOI] [PubMed] [Google Scholar]
- 63. Gurvitz A., Hiltunen J. K., and Kastaniotis A. J. (2009) Heterologous expression of mycobacterial proteins in Saccharomyces cerevisiae reveals two physiologically functional 3-hydroxyacyl-thioester dehydratases, HtdX and HtdY, in addition to HadABC and HtdZ. J. Bacteriol. 191, 2683–2690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sacco E., Slama N., Bäckbro K., Parish T., Laval F., Daffé M., Eynard N., and Quémard A. (2010) Revisiting the assignment of Rv0241c to fatty acid synthase type II of Mycobacterium tuberculosis. J. Bacteriol. 192, 4037–4044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fisher M. A., Plikaytis B. B., and Shinnick T. M. (2002) Microarray analysis of the Mycobacterium tuberculosis transcriptional response to the acidic conditions found in phagosomes. J. Bacteriol. 184, 4025–4032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Singh A., Jain S., Gupta S., Das T., and Tyagi A. K. (2003) mymA operon of Mycobacterium tuberculosis: Its regulation and importance in the cell envelope. FEMS Microbiol. Lett. 227, 53–63 [DOI] [PubMed] [Google Scholar]
- 67. Singh A., Gopinath K., Sharma P., Bisht D., Sharma P., Singh N., and Singh S. (2015) Comparative proteomic analysis of sequential isolates of Mycobacterium tuberculosis from a patient with pulmonary tuberculosis turning from drug sensitive to multidrug resistant. Indian J. Med. Res. 141, 27–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Armitige L. Y., Jagannath C., Wanger A. R., and Norris S. J. (2000) Disruption of the genes encoding antigen 85A and antigen 85B of Mycobacterium tuberculosis H37Rv: Effect on growth in culture and in macrophages. Infect. Immun. 68, 767–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Deleted in proof.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw mass spectrometry data deposited at data at http://proteomecentral.proteomexchange.org/cgi/GetDataset, using the project identifier PXD009549.