

Abstract
Sun exposure is the main risk factor for cutaneous malignant melanoma (CMM). However, the UV-related pathogenetic mechanisms leading to CMM are far to be fully elucidated. In this paper we will focus on what we still don't fully know about the relationship between UVR and CMM. In particular, we will discuss: the action spectrum of human CMM, how different modalities of exposure (continuous/ intermittent; erythemal/ suberythemal) relate to different CMM variants, the preferential UVR induced DNA mutations observed in different CMM variants, the role of UV-related and UV-unrelated genetic damages in the same melanoma cells. Moreover, we will debate the importance of UVA induced oxidative and anaerobic damages to DNA and other cell structures and the role of melanins, of modulation of innate and acquired immunity, of vitamin D and of chronic exposure to phototoxic drugs and other xenobiotics. A better understanding of these issues will help developing more effective preventative strategies and new therapeutic approaches.
Keywords: melanoma, sun exposure, vitamin D, UVA, UVB
Introduction
It is widely accepted that ultraviolet light radiation (UVR) is the major—but not the only—risk factor for the development of cutaneous malignant melanoma (CMM) (1). It is thought that genotoxic, inflammatory, and immunosuppressive properties of UVR contribute together to initiation, progression, and metastasis of CMM. However, several important mechanistic details regarding how sunlight causes CMM remain to be fully elucidated. As a consequence, we still cannot provide fully effective preventative behavioral strategies. In the present paper, we will focus on the main weaknesses of the present understanding of UVR-CMM relationships.
Modality of UVR exposure and CMM variants
Cutaneous malignant melanoma (CMM) is not a single tumor entity with a homogeneous profile of risk factors and prognosis. Consequently we recognize a few variants. It is completely unclear why different modalities of UVR exposure (erythemal/suberythemal doses; chronic/intermittent exposures) induce different molecular damages in the same cell population (2) and why these different molecular damages lead to different clinical CMM variants. For example, we do not know why chronic lifetime sun damage, seen in elderly people and outdoor workers, is related to the specific pattern of DNA mutations characteristic of Lentigo Maligna Melanoma (LMM) (3), while sunburns do not seem to be a significant risk factor for this CMM variant (4). In contrast, Superficial Spreading Melanoma (SSM) and Nodular Melanoma (NM), that have a different spectrum of UV related DNA mutations, usually develop on intermittently exposed healthy skin of younger subjects (3) and a history of sunburns (particularly during childhood) was found to double the risk (5).
Waveband dependency of genotoxic damages and CMM
The most relevant cromophore for skin carcinogenesis is DNA. Its absorption peak is in the UVB region. The different types of UV-DNA photoproducts and their waveband- dependency are summarized in Figure 1. Cyclo-butane pyrimidine dimers (CPD) (T < >T, C < >C, C < >T, and T < >C)-and 6–4 photoproducts (6–4PP) are the most frequent. Until few years ago, it was assumed that UVB biological effects were mainly caused by oxygen-independent reactions, whereas UVA reactions were considered exclusively oxygen dependent. As a consequence, the terms “UVB effects” and “UVA effects” were used as synonyms for anaerobic (synonyms: direct, anoxic or type 1) and aerobic (synonyms: indirect, oxidative or type 2) effects, respectively. However, it was demonstrated that UVA induces DNA type 1 damages as well (6). Indeed, UVA seems able to produce oxidative DNA damage directly or after the oxidative sensitization of not yet identified endogenous photosensitizers (7). Concerning DNA damages, it is therefore clear that the sharp distinction between UVB and UVA, should be reconsidered, because C -> T transitions and CC -> TT tandem mutations (8) are no more to be considered as only UVB damages. Therefore, the term “UVB signatures” should be avoided because potentially misleading.
Figure 1.

UV- induced DNA damages and their waveband dependency.
UVA genotoxic activity is about 1,000 times weaker than UVB's one if considered on a per photon basis (6–8). However, its importance is partially compensated by the UVA environmental irradiance that is about 20–40 times higher, depending on some factors, including time of day, season, latitude and altitude (9). In addition, UVA irradiation is even higher when UV exposure happens through a window glass or in sunbeds. Also, the application of non-broad-spectrum sunscreens, not able to filter UVA as much as UVB, can cause UVA over- exposure. In the same way, aerobic damages to DNA, leading to the formation of 8-oxo-7,8-dihydro-2′deoxyguanosine (8oxo-dG) and consequent reparation with G → T transversions and G → A transitions, cannot be considered “UVA signatures” because UVB is able to produce aerobic damages as well (6, 8).
Other mutations, induced by both UVA and UVB, are DNA-protein cross-links and single and double strand breaks, but their role in CMM development remains to be clarified (6, 8). Anaerobic UV DNA damages are repaired by the nucleotide excision repair (NER) system, while oxidative DNA damages are repaired by the base excision repair (BER) system. It is interesting to note that the mutation rate per DNA photoproduct is higher with UVA: in fact UVA damages are not followed by as many protective, anti-mutagenic and reparative responses as UVB damages are (10).
Anaerobic and aerobic photoproducts of DNA, together with other biomolecules, induce a cascade of pro-inflammatory signals and suppress pro apoptotic pathways (Figure 2). However, the respective relevance and interplay of UVA- and UVB- activities are still largely unknown. Finally, we emphasize how important it is to extrapolate in a very careful and critical manner the results of experimental studies about photo-genotoxicity made with cell cultures: during “in-vivo” UV exposure, while UVA has a deeper penetration, only a small fraction of the incident UVB radiation reaches the level of the dermal-epidermal junction of human skin, where melanocytes are located. We should also be careful with findings of studies on animals, because penetration into human skin may be different.
Figure 2.

Selected inflammatory molecular pathways that are triggered by UVR in melanocytes.
UVR and gene mutations
Gene mutations found in CMM cells are more frequent at selected loci. Aiming to understand their possible diagnostic and prognostic meaning, they have been divided in two main groups: mutations providing no selective advantage to the tumor growth (that occur stochastically during cancer development), and genomic alterations that have a role in cancer development or in the determination of cancer phenotype. In order to assess a correlation between sun exposure and mutations at hot-spots within promoters, the detection of canonical UV signatures (C to T and CC to TT mutations) is mandatory.
UVR-induced mutations are frequently found in the CDKN2A gene in all CMM variants. In humans, this gene encodes for the tumor suppressor proteins p16 and p14ARF (11).
N-RAS is the most frequently affected RAS family member in CMM. Both anaerobic and oxidative, as well as non-UV related, damages have been detected on this gene (12). These mutations can lead to the production of a permanently activated RAS protein, causing the consequent activation of phosphatidylinositol 3′ kinase and mitogen-activated protein kinase (MAPK) pathways. The constant activation of these two pathways leads to unintended and overactive signaling for cell growth, differentiation and survival even in the absence of incoming signals (13). If the frequency of NRAS mutations in LMM, in comparison to SSM and NM, is higher (14, 15) or not (16, 17) is still debated.
The BRAF gene encodes for a serine/threonine kinase that plays a key role in the MAPK signaling pathway. BRAF mutation is more often associated with SSM and NM and it is particularly common in younger patients (18–22). KIT mutations are often found in acral lentiginous melanoma (ALM) and mucosal melanoma (MuM), less frequently in LMM, and rarely in SSM and NM (23). The gene encoding for the pro-apoptotic p53 is another frequent target of UVB damage. Mutated p53 is often observed in melanoma metastases (24), while it is less frequent in primary CMM. This clearly indicates a role for UVR in CMM progression (25, 26). The Nucleotide Excision Repair (NER), and in particular the global genome repair (GGR) damage recognition sub-group, may also be damaged by UVR in melanoma cells, leading to a defective DNA repair response (27, 28).
Therefore, we can conclude that clinical variants of CMM are differently associated to different driver mutations (Figure 3). However, the biological mechanism for which selected DNA mutations drive the mutated melanocyte to a specific clinical variant of CMM is unknown. Furthermore, a significant correlation between clinical outcome and genomic damages is yet to be found (29).
Figure 3.

Associations of the clinical variants of CMM with modalities of exposures and driver mutations. Legend: SSM, superficial spreading melanoma; LMM, Lentigo Maligna Melanoma; ALM, acral lentiginous melanoma; MuM, mucosal melanoma; UvM, uveal melanoma (references in the text).
UV-related and non UV-related DNA damages in melanoma cells
Even if the great majority of BRAF, RAS, and NF1 mutations harbored UV signatures (26, 30) they also showed a high burden of non-UV related mutations (31). This suggests that UV has a role in melanoma pathogenesis, but UV-unrelated mutations can play a role as well. The pathogenic contribution of these UV-independent mutations is still to be clarified. In addition, analysis of whole-genome sequences reveals different carcinogenic processes across the CMM subtypes, some unrelated to sun exposure, and extends potential involvement of the non-coding genome in its pathogenesis (31).
UVR, melanoma and melanins
The protective role of eumelanin is suggested by the evidence that people with dark skin are less prone to develop CMM. Indeed, eumelanin has UV-filtering properties. However, experimental findings have shown that the relationship between CMM and melanins is more complex. The action spectrum of human CMM is unknown but, in the 90 s, Setlow et al. demonstrated that UVA and UVB wavebands have similar pathogenetic activity for CMM in the xiphophorus fish model. Furthermore, they demonstrated that melanin-photosensitized radical production is the major causative step of CMM of this fish (32). Recent experimental work in transgenic mice, confirmed that UVA dependent eumelanin's pro-oxidative activity has a significant pathogenetic role for CMM (33). In addition, unlike UVB, that initiates CMM in a “pigment-independent” manner through direct DNA damage, UVA was found to require the presence of melanin (33). Furthermore, it is worrying to know that, in a preliminary study, it was found that the majority of UVA- induced CPDs in melanocytes are generated after more than 3 h from exposure (34). These “dark CPDs” arise when UVA-induced reactive oxygen and nitrogen species combine to excite melanin that induces CPDs by energy transfer to DNA, in a radiation-independent manner (34). Studies on albino African people provide more evidence that melanin is important for CMM development. The incidence of CMM in this population is low while they still early produce several non melanoma skin cancers (NMSCs).
The highest risk of CMM belongs to people with a ‘red hair/fair skin' phenotype, who synthesize a great amount of phaeomelanin. People with homozygote and heterozygote red hair MC1R variants have eumelanin/ pheomelanin ratio of 1.46 and 4.44 respectively, while wild types have 5.81. Unlike eumelanin, phaeomelanin has poorer protective activity against UVR and greater oxidative potential, both in the dark and after UV exposure (35). Phaeomelanin synthesis is regulated by the MC1R gene. Its variants could play a role in CMM development, also via non-pigmentary pathways (36), including a defective control of α- melanocortin (α-MSH)-mediated DNA repair (37, 38), repair of oxidative DNA damage (39), Nucleotide excision repair system and PTEN- dependent pro-apoptotic pathway (40).
Beside red hair subjects, specific MC1R variants may be also found in people with dark hair. These people have an increased risk to develop CMM. Finally, MC1R regulates the expression of the transcription factor MITF that, in addition to pigment biosynthesis enzymes, regulates genes that control DNA repair (APEX nuclease1) (41), cell cycle (CDKN2A, CDK2) (42, 43), apoptosis (BCL2) (44), and invasion (DIA1) (45).
UVR, phototoxic drugs and melanoma
Experimental and clinical findings suggest that drugs (e.g., azathioprine, vemurafenib, fluoroquinolone antibiotics, propionic acid derivative NSAIDs and voriconazole) can favor melanomagenesis following activation by repeated sub-erythemal UVA exposure (46–48). Also, high levels of folic acid are claimed to have a genotoxic potential because of their pro-oxidative activity that can be further enhanced by pre-treatment with methotrexate (49). The relevance of these findings in the general population is however still to be elucidated.
UVR induced metabolic changes in melanoma
UVA radiation can play a very important role at an early stage of metastasis through mechanisms that are not directly depending on DNA damage. After repetitive exposures to low doses of UVA, glycolysis and lactate production are increased (Warburg effect) (50). This effect persists for at least 5 days after the last UV exposure and is associated with an up-regulation of several matrix metalloproteinases (MMP2, MMP3, MMP9, MMP13, MMP15), with a consequent increment of melanoma invasiveness (51, 52). Warburg effect increases the speed of tumoral cell mitosis too, because part of the glycolysis-derived pyruvate can be used for anabolic pathways (amino acid or fatty acid synthesis) (53).
Photoimmunology
CMM is a potentially highly immunogenic tumor due to its multiple auto-antigens (54). However, UV exposure produces a partial loss of immuno-surveillance by decreasing the number and functionality of antigen presenting cells (both Langerhans and dendritic cells) (55, 56). This leads to a shift of the immune response from Th1 to Th2 (57) and the impairment of the activation of effector T cells and NK-T cells (58). The activation of antigen specific regulatory T cells leads to an antigen-specific suppressive effect on the anti-tumor immune response and it creates an environment where skin tumors can grow (59, 60). However, some degree of immune-surveillance is still preserved as shown by the evidence that the risk for CMM is much higher for patients who are immune-suppressed by drugs. Therefore, the main difference between UVR-induced immunosuppression and drug-induced immunosuppression is probably to be identified in antigen specificity (54, 60).
A key problem is the identification of the UV dose that can be significantly dangerous in humans (61). In early studies, immunosuppression in mice was reached with chronic exposures at erythemal doses (62). Later on, it was proved that a single high irradiation (above the erythemal dose) was also capable of producing the same effect (61).
Afterwards, it was demonstrated that low UVB doses (lower than the MED) - as well as UVA - could promote immunosuppression both in mice (61) and human beings (23, 63–65). Consequently, it seems that immunosuppression can be obtained even while normally walking outdoors, in daylight, during summer. However, everyone is frequently exposed to a very low dose of UV. Therefore, the most important question becomes: how much these exposures are dangerous for CMM development (61)? Chronic low-dose exposures were not found to represent a risk factor for CMM in melano-competent subjects (2). Even more surprisingly, it was recently found that a history of sunny holidays, before CMM diagnosis, was associated with lower mean Breslow thickness (66) and intermittent or regular sun exposures, after CMM diagnosis, were associated with lower mean relapse rates (66, 67). A possible explanation is that low/ physiologic doses of UVR inhibit the adaptive immune system but induce parts of the innate immune system (68). However, UVR effects on the presence and activity of innate immune system cells, e.g., macrophages, tumor associated macrophages (TAMs), dendritic cells (DCs), mast cells and NK cells remain to be explored (69).
UVR, vitamin D and melanoma
Vitamin D and its receptor polymorphisms might play an important role as risk factors for CMM (70). It is very well known that UV radiation is essential for Vitamin D synthesis, in particular for the photoconversion of 7-dehydrocholesterol to cholecalciferol in the epidermis. The production of this vitamin represents one of the most important beneficial effects of sunlight exposure.
Vitamin D bond to its receptor (VDR) results in the transcription of different genes that play a role in the inhibition of MAPK signaling, the induction of apoptosis and cell-cycle inhibition. Therefore, vitamin D has anti-proliferative and pro-apoptotic effects in many kinds of cells, including melanoma cells (71). Vitamin D has also several other positive effects against melanoma, e.g., increase of tumor suppressor PTEN, increase of metastasis suppressor NDRTG1, anti-inflammatory and anti-angiogenic effects and inhibition of “in vivo” melanoma cell proliferation, migration and metastasis (71). However, studies about Vitamin D immunological activity apparently show contrasting findings (61). It was found that Vitamin D might have both a suppressive (58) and a protective activity (72). In other studies it was reported that it is not necessary to immune-suppress UV irradiated animals with Vitamin D to induce CMM (58, 73). A possible explanation of these contrasting effects could be found in different vitamin D concentrations and/or particular pre-activated pathways (58). For example: topically applied 0.1 μg of 1,25(OH)2VitD3 (diluted in acetone/olive oil, 4:1), which represents 240 pmoles, seems able to induce immunosuppression. On the other hand, Dixon et al. (72) used 159.6 and 44.8 pmoles (diluted in ethanol, propylene glycol, and water to a final solvent ratio of 2: 1: 1, resp.) of the vitamin, in order to obtain significant protection against UV-induced immunosuppression. Even though the concentration of Vitamin D used in these experiences is different, an important question arises: which one best represents the concentration of vitamin D in the skin after UV exposure? A conclusive answer has not been found so far (73–75) but it seems likely that biological Vitamin D increments after UV exposure are not sufficient to justify the suppression of specific immune responses. Findings of recent clinical studies have suggested (but by no means proved) that vitamin D might also have a role in melanomagenesis and tendency to metastatic dissemination (71). Godar et al. have suggested that low cutaneous vitamin D3 levels with high environmental and low ratio of UVB/UVA doses are the two main drivers for CMM development. In fact, both Europeans and Americans, in some age groups, have a significant increase of CMM incidence if this ratio decreases (76). If this is true, we could explain the curious relationship between melanoma risk and sun exposure, where sunburn is a factor but occupational sun exposure is not (at least in temperate climes). In MM patients, decreased 25(OH)D serum levels are associated with increased tumor thickness and advanced tumor stage (77).
Conclusions
The life of human beings depends on the sun. This relationship may be beneficial but, at the same time, dangerous. CMM is one of the most deadly tumor and sunlight is, for sure, the main risk factor, although genetic, UV-unrelated and stochastic factors could also play a pathogenetic role (78) (Figure 4). Worldwide, CMM incidence is progressively increasing and two over simplified hypotheses are often put forward as possible explanations: (1) increasing sun exposures and (2) increasing aging of the population. However, we have no data to support the first hypothesis (when exposing to the sun, people tend to be more careful now than before), and the increase of life expectancy in western countries in the last 2 decades seems rather stable1. In addition, a growing number of data point out that the relationship between sun exposure and CMM is not simple and straightforward. In particular, even if there is not a light dose that can always be considered either dangerous or beneficial, we know that some UVR doses and some UVA/UVB combinations have a better ratio of beneficial rather than dangerous effects. It is reasonable to conclude that the assessment of the optimal UVR exposure level for each individual will be one of the major future challenges.
Figure 4.
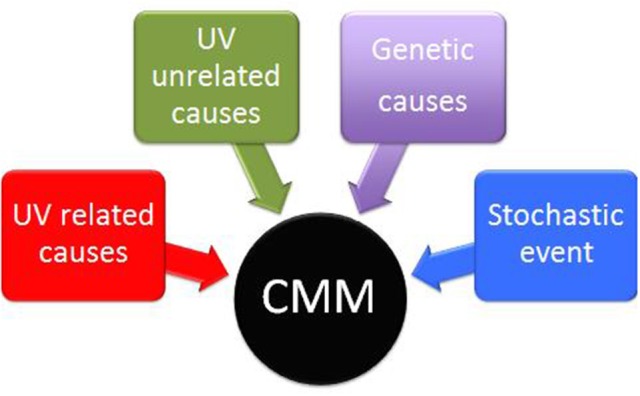
Interplay of UV related, UV unrelated, genetic and stochastic factors in the pathogenesis of cutaneous Malignant Melanoma (CMM).
Author contributions
PC-P, MV, MA, AZ, SC, and CZ researched literature to gather necessary information and contributed in the writing of the paper. PC-P, MV, MA, AZ, SC, CR, and CZ reviewed the article several times to reorganize the concepts and provide a more solid structure and reviewed the bibliography.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
- 1.Arnold M, de Vries E, Whiteman DC, Jemal A, Bray F, Parkin DM, et al. Global burden of cutaneous melanoma attributable to ultraviolet radiation in 2012. Int J Cancer (2018) 143:1305–14. 10.1002/ijc.31527 [DOI] [PubMed] [Google Scholar]
- 2.Whiteman DC, Pavan WJ, Bastian BC. The melanomas: a synthesis of epidemiological, clinical, histopathological, genetic, and biological aspects, supporting distinct subtypes, causal pathways, and cells of origin. Pigment Cell Melanoma Res. (2011) 24:879–97. 10.1111/j.1755-148X.2011.00880.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Franceschi S, Levi F, Randimbison L, La Vecchia C. Site distribution of different types of skin cancer: new aetiological clues. Int J Cancer (1996) 67:24–8. [DOI] [PubMed] [Google Scholar]
- 4.Kvaskoff M, Siskind V, Green AC. Risk factors for lentigo maligna melanoma compared with superficial spreading melanoma: a case-control study in Australia. Arch Dermatol. (2012) 148:164–70. 10.1001/archdermatol.2011.291 [DOI] [PubMed] [Google Scholar]
- 5.Gandini S, Autier P, Boniol M. Reviews on sun exposure and artificial light and melanoma. Prog Biophys Mol Biol. (2011) 107:362–6. 10.1016/j.pbiomolbio.2011.09.011 [DOI] [PubMed] [Google Scholar]
- 6.Kappes UP, Luo D, Potter M, Schulmeister K, Rünger TM. Short and long-wave ultraviolet light (UVB and UVA) induce similar mutations in human skin cells. J Invest Dermatol. (2006) 126:667–75. 10.1038/sj.jid.5700093 [DOI] [PubMed] [Google Scholar]
- 7.Drobetsky EA, Turcotte J, Chateauneuf A. A role for ultraviolet A in solar mutagenesis. Proc Natl Acad Sci USA. (1995) 92:2350–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harris RS. Cancer mutation signatures, DNA damage mechanisms, and potential clinical implications. Genome Med. (2013) 5:87. 10.1186/gm490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marionnet C, Tricaud C, Bernerd F. Exposure to non-extreme solar UV daylight: spectral characterization, effects on skin and photoprotection. Int J Mol Sci. (2014) 16:68–90. 10.3390/ijms16010068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larsson P1, Andersson E, Johansson U, Ollinger K, Rosdahl I. Ultraviolet A and B affect human melanocytes and keratinocytes differently. A study of oxidative alterations and apoptosis. Exp Dermatol. (2005) 14:117–23. 10.1111/j.0906-6705.2005.00238.x [DOI] [PubMed] [Google Scholar]
- 11.Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, et al. 5' CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med. (1995) 1:686–92. [DOI] [PubMed] [Google Scholar]
- 12.Albino AP, Nanus DM, Mentle IR, Cordon-Cardo C, McNutt NS, Bressler J, et al. Analysis of ras oncogenes in malignant melanoma and precursor lesions: correlation of point mutations with differentiation phenotype. Oncogene (1989) 4:1363–74. [PubMed] [Google Scholar]
- 13.Kubo Y, Matsudate Y, Fukui N, Nakasuka A, Sogawa M, Oshima M, et al. Molecular tumorigenesis of the skin. J Med Invest. (2014) 61:7–14. 10.2152/jmi.61.7 [DOI] [PubMed] [Google Scholar]
- 14.van 't Veer LJ, Burgering BM, Versteeg R, Boot AJ, Ruiter DJ, Osanto S, et al. N-ras mutations in human cutaneous melanoma from sun-exposed body sites. Mol Cell Biol. (1989) 9:3114–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ball NJ, Yohn JJ, Morelli JG, Norris DA, Golitz LE, Hoeffler JP. Ras mutations in human melanoma: a marker of malignant progression. J Invest Dermatol. (1994) 102:285–90. [DOI] [PubMed] [Google Scholar]
- 16.Broekaert SM, Roy R, Okamoto I, van den Oord J, Bauer J, Garbe C, et al. Genetic and morphologic features for melanoma classification. Pigment Cell Melanoma Res. (2010) 23:763–70. 10.1111/j.1755-148X.2010.00778.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wagner SN, Ockenfels HM, Wagner C, Höfler H, Goos M. Ras gene mutations: a rare event in non metastatic primary malignant melanoma. J Invest Dermatol. (1995) 104:868–71. [DOI] [PubMed] [Google Scholar]
- 18.Edlundh-Rose E, Egyházi S, Omholt K, Månsson-Brahme E, Platz A, Hansson J, et al. NRAS and BRAF mutations in melanoma tumours in relation to clinical characteristics: a study based on mutation screening by pyrosequencing. Melanoma Res. (2006) 16:471–8. 10.1097/01.cmr.0000232300.22032.86 [DOI] [PubMed] [Google Scholar]
- 19.Goel VK, Lazar AJ, Warneke CL, Redston MS, Haluska FG. Examination of mutations in BRAF, NRAS, and PTEN in primary cutaneous melanoma. J Invest Dermatol. (2006) 126:154–60. 10.1038/sj.jid.5700026 [DOI] [PubMed] [Google Scholar]
- 20.Liu W, Kelly JW, Trivett M, Murray WK, Dowling JP, Wolfe R, et al. Distinct clinical and pathological features are associated with the BRAF(T1799A(V600E)) mutation in primary melanoma. J Invest Dermatol. (2007) 127:900–5. 10.1038/sj.jid.5700632 [DOI] [PubMed] [Google Scholar]
- 21.Maldonado JL, Fridlyand J, Patel H, Jain AN, Busam K, Kageshita T, et al. Determinants of BRAF mutations in primary melanomas. J Natl Cancer Inst. (2003) 95:1878–90. [DOI] [PubMed] [Google Scholar]
- 22.Thomas NE, Edmiston SN, Alexander A, Millikan RC, Groben PA, Hao H, et al. Number of nevi and early-life ambient UV exposure are associated with BRAF-mutant melanoma. Cancer Epidemiol Biomarkers Prev. (2007) 16:991–7. 10.1158/1055-9965.EPI-06-1038 [DOI] [PubMed] [Google Scholar]
- 23.Beadling C, Jacobson-Dunlop E, Hodi FS, Le C, Warrick A, Patterson J, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res. (2008) 14:6821–8. 10.1158/1078-0432.CCR-08-0575 [DOI] [PubMed] [Google Scholar]
- 24.Shain AH, Yeh I, Kovalyshyn I, Sriharan A, Talevich E, Gagnon A, et al. The genetic evolution of melanoma from precursor lesions. N Engl J Med. (2015) 373:1926–36. 10.1056/NEJMoa1502583 [DOI] [PubMed] [Google Scholar]
- 25.Matsumura Y, Ananthaswamy HN. Short-term and long-term cellular and molecular events following UV irradiation of skin: implications for molecular medicine. Expert Rev Mol Med. (2002) 4:1–22. 10.1017/S146239940200532X [DOI] [PubMed] [Google Scholar]
- 26.Matsumura Y, Sato M, Nishigori C, Zghal M, Yagi T, Imamura S, et al. High prevalence of mutations in the p53 gene in poorly differentiated squamous cell carcinomas in xeroderma pigmentosum patients. J Invest Dermatol. (1995) 105:399–401. [DOI] [PubMed] [Google Scholar]
- 27.Bowden NA, Ashton KA, Avery-Kiejda KA, Zhang XD, Hersey P, Scott RJ. Nucleotide excision repair gene expression after Cisplatin treatment in melanoma. Cancer Res. (2010) 70:7918–26. 10.1158/0008-5472.CAN-10-0161 [DOI] [PubMed] [Google Scholar]
- 28.Belanger F, Rajotte V, Drobetsky EA. A majority of human melanoma cell lines exhibits an S phase-specific defect in excision of UV-induced DNA photoproducts. PLoS ONE (2014) 9:e85294. 10.1371/journal.pone.0085294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cancer Genome Atlas Network Genomic classification of cutaneous melanoma. Cell (2015) 161:1681–96. 10.1016/j.cell.2015.05.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, et al. A landscape of driver mutations in melanoma. Cell (2012) 150:251–63. 10.1016/j.cell.2012.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, Nones K, et al. Whole-genome landscapes of major melanoma subtypes. Nature (2017) 545:175–80. 10.1038/nature22071 [DOI] [PubMed] [Google Scholar]
- 32.Wood SR, Berwick M, Ley RD, Walter RB, Setlow RB, Timmins GS. UV causation of melanoma in Xiphophorus is dominated by melanin photosensitized oxidant production. Proc Natl Acad Sci USA. (2006) 103:4111–5. 10.1073/pnas.0511248103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Noonan FP, Zaidi MR, Wolnicka-Glubisz A, Anver MR, Bahn J, Wielgus A, et al. Melanoma induction by ultraviolet A but not ultraviolet B radiation requires melanin pigment. Nat Commun. (2012) 3:884 10.1038/ncomms1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Premi S, Wallisch S, Mano CM, Weiner AB, Bacchiocchi A, Wakamatsu K, et al. Photochemistry. Chemiexcitation of melanin derivatives induces DNA photoproducts long after UV exposure. Science (2015) 347:842–7. 10.1126/science.1256022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mitra D, Luo X, Morgan A, Wang J, Hoang MP, Lo J, et al. An ultraviolet-radiation independent pathway to melanoma carcinogenesis in the red hair/fair skin background. Nature (2012) 491:449–53. 10.1038/nature11624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fargnoli MC, Gandini S, Peris K, Maisonneuve P, Raimondi S. MC1R variants increase melanoma risk in families with CDKN2A mutations: a meta-analysis. Eur J Cancer (2010) 46:1413–20. 10.1016/j.ejca.2010.01.027 [DOI] [PubMed] [Google Scholar]
- 37.Robinson SJ, Healy E. Human melanocortin 1 receptor (MC1R) gene variants alter melanoma cell growth and adhesion to extracellular matrix. Oncogene (2002) 21:8037–46. [DOI] [PubMed] [Google Scholar]
- 38.Kadekaro AL, Leachman S, Kavanagh RJ, Swope V, Cassidy P, Supp D, et al. Melanocortin 1 receptor genotype: an important determinant of the damage response of melanocytes to ultraviolet radiation. FASEB J. (2010) 24:3850–60. 10.1096/fj.10-158485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song X, Mosby N, Yang J, Xu A, Abdel-Malek Z, Kadekaro AL. alpha-MSH activates immediate defense responses to UV-induced oxidative stress in human melanocytes. Pigment Cell Melanoma Res. (2009) 22:809–18. 10.1111/j.1755-148X.2009.00615.x [DOI] [PubMed] [Google Scholar]
- 40.Davies JR, Randerson-Moor J, Kukalizch K, Harland M, Kumar R, Madhusudan S, et al. Inherited variants in the MC1R gene and survival from cutaneous melanoma: a BioGenoMEL study. Pigment Cell Melanoma Res. (2012) 25:384–94. 10.1111/j.1755-148X.2012.00982.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu F, Fu Y, Meyskens FL, Jr. MiTF regulates cellular response to reactive oxygen species through transcriptional regulation of APE-1/Ref-1. J Invest Dermatol. (2009) 129:422–31. 10.1038/jid.2008.255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Du J, Widlund HR, Horstmann MA, Ramaswamy S, Ross K, Huber WE, et al. Critical role of CDK2 for melanoma growth linked to its melanocyte-specific transcriptional regulation by MITF. Cancer Cell. (2004) 6:565–76. 10.1016/j.ccr.2004.10.014 [DOI] [PubMed] [Google Scholar]
- 43.Loercher AE, Tank EM, Delston RB, Harbour JW. MITF links differentiation with cell cycle arrest in melanocytes by transcriptional activation of INK4A. J Cell Biol. (2005) 168:35–40. 10.1083/jcb.200410115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McGill GG, Horstmann M, Widlund HR, Du J, Motyckova G, Nishimura EK, et al. Bcl2 regulation by the melanocyte master regulator Mitf modulates lineage survival and melanoma cell viability. Cell (2002) 109:707–18. 10.1016/S0092-8674(02)00762-6 [DOI] [PubMed] [Google Scholar]
- 45.Carreira S, Goodall J, Denat L, Rodriguez M, Nuciforo P, Hoek KS, et al. Mitf regulation of Dia1 controls melanoma proliferation and invasiveness. Genes Dev. (2006) 20:3426–39. 10.1101/gad.406406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peacock M, Brem R, Macpherson P, Karran P. DNA repair inhibition by UVA photoactivated fluoroquinolones and vemurafenib. Nucleic Acids Res. (2014) 42:13714–22. 10.1093/nar/gku1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robinson KS, Traynor NJ, Moseley H, Ferguson J, Woods JA. Cyclobutane pyrimidine dimers are photosensitised by carprofen plus UVA in human HaCaT cells. Toxicol In Vitro. (2010) 24:1126–32. 10.1016/j.tiv.2010.03.007 [DOI] [PubMed] [Google Scholar]
- 48.Miller DD, Cowen EW, Nguyen JC, McCalmont TH, Fox LP. Melanoma associated with long-term voriconazole therapy: a new manifestation of chronic photosensitivity. Arch Dermatol. (2010) 146:300–4. 10.1001/archdermatol.2009.362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Butzbach K, Epe B. Photogenotoxicity of folic acid. Free Radic Biol Med. (2013) 65:821–7. 10.1016/j.freeradbiomed.2013.08.168 [DOI] [PubMed] [Google Scholar]
- 50.Kamenisch Y, Baban TS, Schuller W, von Thaler AK, Sinnberg T, Metzler G, et al. UVA-irradiation induces melanoma invasion via the enhanced Warburg effect. J Invest Dermatol. (2016) 136:1866–75. 10.1016/j.jid.2016.02.815 [DOI] [PubMed] [Google Scholar]
- 51.Quintanilla-Dieck MJ, Codriansky K, Keady M, Bhawan J, Rünger TM. Cathepsin K in melanoma invasion. J Invest Dermatol. (2008) 128:2281–8. 10.1038/jid.2008.63 [DOI] [PubMed] [Google Scholar]
- 52.Codriansky KA, Quintanilla-Dieck MJ, Gan S, Keady M, Bhawan J, Rünger TM. Intracellular degradation of elastin by cathepsin K in skin fibroblasts–a possible role in photoaging. Photochem Photobiol. (2009) 85:1356–63. 10.1111/j.1751-1097.2009.00592.x [DOI] [PubMed] [Google Scholar]
- 53.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324:1029–33. 10.1126/science.1160809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pawelec G. Immune correlates of clinical outcome in melanoma. Immunology (2018) 153:415–22. 10.1111/imm.12870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Garmyn M, Yarosh DB. The Molecular and Genetic Effects of Ultraviolet Radiation Exposure on Skin Cells. New York, NY: Photodermatology, Informa Health Care; (2007). p. 41–54. [Google Scholar]
- 56.Schwarz T. 25 years of UV-induced immunosuppression mediated by T cells-from disgregated T suppressor cells to highly respected regulatory T cells. Photochem Photobiol (2008) 84:10–8. 10.1111/j.1751-1097.2007.00223.x [DOI] [PubMed] [Google Scholar]
- 57.Garibyan L, Fisher DE. How sunlight causes melanoma. Curr Oncol Rep. (2010) 12:319–26. 10.1007/s11912-010-0119-y [DOI] [PubMed] [Google Scholar]
- 58.Moodycliffe AM, Nghiem D, Clydesdale G, Ullrich SE. Immune suppression and skin cancer development: regulation by NKT cells. Nat Immunol. (2000) 1:521–5. 10.1038/82782 [DOI] [PubMed] [Google Scholar]
- 59.Schwarz A, Maeda A, Wild MK, Kernebeck K, Gross N, Aragane Y, et al. Ultraviolet radiation-induced regulatory T cells not only inhibit the induction but can suppress the effector phase of contact hypersensitivity. J Immunol. (2004) 172:1036–43. 10.4049/jimmunol.172.2.1036 [DOI] [PubMed] [Google Scholar]
- 60.Schwarz A, Navid F, Sparwasser T, Clausen BE, Schwarz T. In vivo reprogramming of UV radiation-induced regulatory T-cell migration to inhibit the elicitation of contact hypersensitivity. J Allergy Clin Immunol. (2011) 128:826–33. 10.1016/j.jaci.2011.06.005 [DOI] [PubMed] [Google Scholar]
- 61.González Maglio DH, Paz ML, Leoni J. Sunlight effects on immune system: is there something else in addition to UV-induced immunosuppression? BioMed Research International. (2016) 2016:1934518. 10.1155/2016/1934518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kripke ML. Antigenecity of murine skin tumors induced by ultraviolet light. J Natl Cancer Inst. (1974) 53:1333–6. [DOI] [PubMed] [Google Scholar]
- 63.Damian DL, Matthews YJ, Phan TA, Halliday GM. An action spectrum for ultraviolet radiation-induced immunosuppression in humans. Br J Dermatol. (2011) 164:657–9. 10.1111/j.1365-2133.2010.10161.x [DOI] [PubMed] [Google Scholar]
- 64.Halliday GM, Byrne SN, Damian DL. Ultraviolet A radiation: its role in immunosuppression and carcinogenesis. Semin Cutan Med Surg. (2011) 30:214–21. 10.1016/j.sder.2011.08.002 [DOI] [PubMed] [Google Scholar]
- 65.Halliday GM, Lyons JG. Inflammatory doses of UV may not be necessary for skin carcinogenesis. Photochem Photobiol. (2008) 84:272–83. 10.1111/j.1751-1097.2007.00247.x [DOI] [PubMed] [Google Scholar]
- 66.Gandini S, De Vries E, Tosti G, Botteri E, Spadola G, Maisonneuve P, et al. Sunny holidays before and after melanoma diagnosis are respectively associated with lower Breslow thickness and lower relapse rates in Italy. PLoS ONE (2013) 8:e78820. 10.1371/journal.pone.0078820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lindqvist PG, Epstein E, Nielsen K, Landin-Olsson M, Ingvar C, Olsson H. Avoidance of sun exposure as a risk factor for major causes of death: a competing risk analysis of the Melanoma in Southern Sweden cohort. J Intern Med. (2016) 280:375–87. 10.1111/joim.12496 [DOI] [PubMed] [Google Scholar]
- 68.Cela EM, Gonzalez CD, Friedrich A, Ledo C, Paz ML, Leoni J, et al. Daily very low UV dose exposure enhances adaptive immunity, compared with a single high-dose exposure. Consequences for the control of a skin infection. Immunology (2018) 154:510–21. 10.1111/imm.12901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mignogna C, Scali E, Camastra C, Presta I, Zeppa P, Barni T, et al. Innate immunity in cutaneous melanoma. Clin Exp Dermatol. (2017) 42:243–50. 10.1111/ced.13023 [DOI] [PubMed] [Google Scholar]
- 70.Cauci S, Maione V, Buligan C, Linussio M, Serraino D, Stinco G. BsmI (rs1544410) and FokI (rs2228570) vitamin D receptor polymorphisms, smoking, and body mass index as risk factors of cutaneous malignant melanoma in northeast Italy. Cancer Biol Med. (2017) 14:302–18. 10.20892/j.issn.2095-3941.2017.0064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Field S, Newton-Bishop JA. Melanoma and vitamin D. Mol Oncol. (2011) 5:197–214. 10.1016/j.molonc.2011.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dixon KM, Norman AW, Sequeira VB, Mohan R, Rybchyn MS, Reeve VE, et al. 1α,25(OH)2-vitamin D and a nongenomic vitamin D analogue inhibit ultraviolet radiation-induced skin carcinogenesis. Cancer Prevent Res. (2011) 4:1485–94. 10.1158/1940-6207.CAPR-11-0165 [DOI] [PubMed] [Google Scholar]
- 73.Gorman S, Scott NM, Tan DHW, Weeden CE, Tuckey RC, Bisley JL, et al. Acute erythemal ultraviolet radiation causes systemic immunosuppression in the absence of increased 25-hydroxyvitamin D3 levels in male mice. PLoS ONE (2012) 7:e46006. 10.1371/journal.pone.0046006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Narbutt J, Lesiak A, Sysa-Jedrzejowska A, Wozniacka A, Cierniewska-Cieslak A, Boncela J, et al. Repeated lowdose ultraviolet (UV) B exposures of humans induce limited photoprotection against the immune effects of erythemal UVB radiation. Br J Dermatol. (2007) 156:539–47. 10.1111/j.1365-2133.2006.07670.x [DOI] [PubMed] [Google Scholar]
- 75.Petersen B, Wulf HC, Triguero-Mas M, Philipsen PA, Thieden E, Olsen P, et al. Sun and ski holidays improve vitamin D status, but are associated with high levels of DNA damage. J Invest Dermatol. (2014) 134:2806–13. 10.1038/jid.2014.223 [DOI] [PubMed] [Google Scholar]
- 76.Godar DE, Subramanian M, Merrill SJ. Cutaneous malignant melanoma incidences analyzed worldwide by sex, age, and skin type over personal Ultraviolet-B dose shows no role for sunburn but implies one for Vitamin D3. Dermato Endocrinol. (2017) 9:e1267077. 10.1080/19381980.2016.1267077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gambichler T, Bindsteiner M, Höxtermann S, Kreuter A. Serum 25-hydroxyvitamin D serum levels in a large German cohort of patients with melanoma. Br J Dermatol. (2013) 168:625–8. 10.1111/j.1365-2133.2012.11212.x [DOI] [PubMed] [Google Scholar]
- 78.Magoni M, Apostoli P, Donato F, Manganoni A, Comba P, Fazzo L, et al. Plasma levels of polychlorinated biphenyls and risk of cutaneous malignant melanoma: a hospital-based case-control study. Environ Int. (2018) 113:20–5. 10.1016/j.envint.2018.01.018 [DOI] [PubMed] [Google Scholar]