

Abstract
This protocol describes the construction and functional studies of a bispecific antibody (bsAb), GPC3-S-Fab. bsAbs can recognize two different epitopes through their two different arms. bsAbs have been actively studied for their ability to directly recruit immune cells to kill tumor cells. Currently, the majority of bsAbs are produced in the form of recombinant proteins, either as Fc-containing bsAbs or as smaller bsAb derivatives without the Fc region. In this study, GPC3-S-Fab, an antibody fragment (Fab) based bispecific antibody, was designed by linking the Fab of anti-GPC3 antibody GC33 with an anti-CD16 single domain antibody. The GPC3-S-Fab can be expressed in Escherichia coli and purified by two affinity chromatographies. The purified GPC3-S-Fab can specifically bind to and kill GPC3 positive liver cancer cells by recruiting natural killer cells, suggesting a potential application of GPC3-S-Fab in liver cancer therapy.
Keywords: Cancer Research, Issue 137, Bispecific antibody, Antibody, Single domain, VHH, GPC3-S-Fab, GPC3, Liver cancer
Introduction
Monoclonal antibodies are now broadly used for cancer treatment1. Due to the flexibility of antibodies, various antibody-based formats have been actively explored. Compared with monoclonal antibodies, bsAbs have two different antigen binding modules, enabling them to recognize two different targets simultaneously and efficiently trigger the recruitment of immune effector cells to target and kill tumor cells2.
Current recombinant bsAb formats can be generally assigned to two classes: Fc-containing bsAbs and bsAbs without an Fc region. Compared with Fc-containing formats that are mostly produced in mammalian cells, bsAbs without an Fc region have the advantages of smaller sizes, are more readily produced in microorganism expression systems, and can penetrate tumor tissues more efficiently3.
bsAbs without an Fc region are commonly formed by linking individual binding moieties, such as single-chain variable fragments (scFvs) or Fabs3. Without the stabilizing domains, bsAbs based on scFv fragments often have compromised thermal stability, low solubility, or an increased potential for aggregation4,5. In contrast, Fab-based bsAbs are more stable due to the heterodimerization of the CH1 and CL in the native Fab moiety4,6.
Variable domain from heavy-chain-only antibodies (VHHs, also referred to as single domain antibodies) are the active antigen-binding fragment of natural heavy chain antibodies7. VHHs have the characteristics of high affinity, specificity of conventional IgGs8, low immunogenicity, and high yields in bacterial expression9. Compared with Fv fragments, VHHs have higher thermal stability10. Compared with Fab moieties, VHHs have smaller sizes due to the lack of CH1 and CL. Thus, S-Fab, the bsAb format obtained by linking the Fab with a single domain antibody, VHH, was designed and studied for its anti-tumor effects11,12.
In this study, the construction of GPC3-S-Fab by linking the Fab of hGC3313 with an anti-CD16a VHH14 was described. The GPC3-S-Fab can be efficiently produced by periplasmic expression in Escherichia coli (E. coli). Functional studies of GPC3-S-Fab suggested that GPC3-S-Fab is a promising strategy for liver cancer therapy. Thus, the advantages of GPC3-S-Fab over alternative techniques with applicable references to previous studies include easy production and purification, and more stable bsAbs.
Mammalian expression systems and prokaryotic expression systems have been used to express various formats of BsAbs. In contrast to mammalian expression systems, E. coli-based protein expression systems have many benefits, including high yields, low cost and labor-saving, the ease of genetic manipulations, and high transformation efficiency15. For bsAbs expression in E. coli, there are two basic strategies: expression in the cytoplasm and expression in the periplasm between the cytoplasm and outer cell membranes15. Compared to the reducing environment of cytoplasm, the periplasm is a more oxidizing environment, which promotes the correct folding and co-expression of proteins16. Correct folding plays a key role in solubility, stability and function generation of bsAbs. Therefore, a signal sequence pelB was added to the N-terminus of the S-Fab to direct secretion to the periplasm of E. coli17. To ensure correct folding, solubility, thermal stability, and conformational stability, reducing the complexity and the size of an antibody is frequently employed16. The S-Fab format consists of one Fab and one VHH, which is expressed very well in bacterial systems likely due to the simple structure and small size.
GPC3 was chosen in this GPC3-S-Fab bispecific antibody format. Glypican-3 (GPC3) is a member of the heparin sulfate (HS) proteoglycan family that is anchored to the cell surface through glycosylphosphatidylinositol (GPI)18. GPC3 is overexpressed in 70% of hepatocellular carcinoma (HCC) cases, which account for the majority of liver cancers19,20,21,22. Because GPC3 is rarely expressed in normal tissues, GPC3 has been proposed as a potential target for HCC. Multiple mouse mAbs have been produced against GPC3. However, only GC33 exhibited limited anti-tumor activity 22, and it failed to exhibit clinical efficacy in patients. In this study, GPC3-S-Fab was shown to be able to recruit NK cells to kill GPC3 tumor cells14.
To recruit NK cells, anti-CD16 VHH was used. CD16a is a low affinity IgG receptor, expressed mainly on natural killer (NK) cells, macrophages, monocytes and some subtypes of T cells. It is involved in antibody-dependent cell cytotoxicity (ADCC) by NK cells23. Human NK cells can be categorized into two types, CD56-CD16+ and CD56+CD16-. In contrast to CD56+CD16− NK cells, CD56-CD16+ NK cells can release higher levels of perforin and granzyme B and thus present a strong cytotoxicity24. Kupffer cells (KCs), expressing CD16a, are the resident macrophages in liver. Kupffer cells play an important role in the suppression of liver cancer25. Thus, bsAbs targeting CD16a may be a more promising strategy than engaging T cells against liver cancer.
Protocol
All of the procedures including human blood collection were approved by the Sun Yat-Sen University Ethics Committee.
1. GPC3-S-Fab Design Strategy
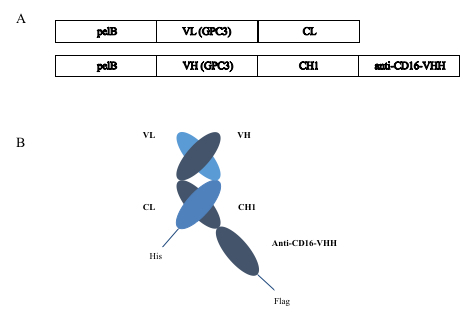
Design GPC3-S-Fab by linking a Fab of anti-GPC3 (humanized GC3313) with an anti-CD16 VHH14 (Figure 1).
Synthesize and clone the VH-CH1-CD16-VHH and VL-CL into the pET26b and pET21a vectors as previously reported12.
2. Transformation and Culture
Add 100 ng of pET26b containing VH-CH1-CD16 (Figure 1A) and 100 ng of pET21a containing VL-CL (Figure 1A) into 100 µL of competent BL21 (DE3) cells. Mix well and incubate on ice for 30 min. Incubate in a 42 °C water bath for 45 - 90 s, transfer, and incubate on ice for 3 - 5 min.
- Add 500 µL of lysogeny broth (LB) medium into a tube containing BL21 (DE3) cells and then incubate at 37 °C, 150 rpm for 45 - 60 min. Spread 100 µL of BL21 (DE3) cells on LB-agar plates containing 50 µg/mL of kanamycin and 100 µg/mL of ampicillin, and then incubate at 37 °C for 12 - 16 h.
- Prepare lysogeny broth (LB) medium: 1% wt/vol tryptone, 0.5% wt/vol yeast extract, 1% wt/vol NaCl.
- Inoculate cells from a single colony into 5 mL of super broth (SB) medium containing 50 µg/mL of kanamycin and 100 µg/mL of ampicillin, and culture at 37 °C, 220 rpm, overnight. Then, inoculate 1 mL of culture into 100 mL of SB medium containing 50 µg/mL of kanamycin and 100 µg/mL of ampicillin and culture at 37 °C, 220 rpm for 4 h. Then, transfer 10 mL of the culture into 1 L of SB medium containing 50 µg/mL of kanamycin and 100 µg/mL of ampicillin and culture at 37 °C, 220 rpm.
- Prepare super broth medium: 3.2% wt/vol tryptone, 2% wt/vol yeast extract, 0.5% wt/vol NaCl.
When the OD600 reaches 0.6 - 0.8, add isopropyl-b-D-thio-galactopyranoside (IPTG) to a final concentration of 0.2 mM. Culture at 16 °C, 180 rpm for 36 - 48 h for periplasmic expression.
3. GPC3-S-Fab Periplasmic Purification
- Periplasmic fraction preparation
- Collect cells by centrifuging at 4,000 x g, 4 °C for 30 min, discard the medium, and weigh the cells.
- Resuspend the cells thoroughly in 4 mL of ice-cold sucrose solution (20 mM of Tris-HCl, pH 7.5; 25% (wt/vol) sucrose and 1 mM of EDTA) per gram of cells, and incubate on ice for 15 min.
- Centrifuge at 8,500 x g (table top centrifuge, fixed angle rotor), 4 °C for 20 min. Remove the supernatant (the sucrose fraction) and save it on ice.
- Resuspend the pellets in a mix of ice-cold 5 mM of MgCl2 and 1 mM of PMSF (4 mL per gram cells). Add 40 µL of the lysozyme stock (15 mg/mL) per gram, and incubate on ice for 30 min.
- Centrifuge at 8,500 x g (table top centrifuge, fixed angle rotor), 4 °C for 20 min. Transfer the supernatant (the periplasmic fraction) and save it on ice.
- Combine the sucrose fraction and periplasmic fraction, and centrifuge at 30,000 x g, 4 °C for 30 min. Remove the supernatant and save it in a 50 mL conical tube on ice.
- Ni-NTA affinity purification
- Resuspend 1 mL of Ni-NTA agarose by mixing thoroughly to achieve a homogeneous suspension, remove 1 mL of Ni-NTA agarose to a fresh 15 mL conical tube, and add 10 column volumes (CV) equilibration buffer (25 mM of Tris-HCl, pH 7.5; 1 M of NaCl) to equilibrate.
- Centrifuge at 400 x g for 5 min, and remove the supernatant carefully. Then, add the Ni-NTA agarose into the 50 mL tube containing the sucrose fraction and periplasmic fraction. Rock at 4 °C for 2 h.
- Centrifuge the mixture at 400 x g, 4 °C for 5 min. Carefully remove the supernatant to another fresh ice-cold tube as the unbound fraction. Transfer the Ni-NTA agarose into a gravity column.
- Add 10 CV of washing buffer (25 mM of Tris-HCl, pH 7.5; 1 M of NaCl; 20 mM, 30 mM or 40 mM of imidazole) to the column, and collect the elute as the washing fraction. NOTE: These solutions should be added in order of increasing concentrations of imidazole.
- Add 3 CV of elution buffer (25 mM of Tris-HCl, pH 7.5; 300 mM of NaCl; 100 mM, 200 mM, 300 mM or 400 mM of imidazole) to the column and collect the elute as Elution fraction 1, 2, 3, and 4. NOTE: These solutions should be added in order of increasing concentrations of imidazole.
- Dialysis the eluted fractions in 2 L of PBS (137 mM of NaCl, 2.7 mM of KCl, 10 mM of Na2HPO4, 2 mM of KH2PO4, pH 7.4) using dialysis tubing (12.4 kDa) at 4 °C for 2 h. Check the presence of GPC3-S-Fab by 12% SDS-PAGE and then by Coomassie blue staining.
- IgG-CH1 affinity purification
- Transfer 1 mL of IgG-CH1 affinity resin into a 50 mL conical tube, and wash the beads with PBS first. Then, add the dialyzed solution containing GPC3-S-Fab after Step 3.2.6. Rock at 4 °C for 2 h.
- Centrifuge at 400 x g for 5 min, 4 °C. Carefully remove the supernatant to another fresh ice-cold tube, and transfer the resin to a gravity column.
- Add 10 CV of washing buffer (PBS) to the column, and collect the elution as the wash fraction.
- Prepare the collection tubes by adding 1 M of Tris pH 9.0 (0.1 mL per mL of each eluted fraction). Elute with 3 CV elution buffer (0.1 M of glycine-HCl, pH 2.7), and collect the eluted fraction; repeat 3 times.
- Dialysis the eluted fractions in 2 L of PBS using dialysis tubing (12.4 kDa) at 4 °C for 2 h. Repeat twice.
- Determine the protein concentration by ultramicrospectrophotometer (Molar extinction coefficient: 91105. 1 A280 = 0.69 mg/L).
4. SDS-PAGE and Western-blotting Analysis
- Take a 10 µL of the sample into 5x SDS loading buffer and mix well. Boil the protein mixture at 100 °C for 5 min.
- Prepare 5x SDS loading buffer: 250 mM of Tris-HCl, pH 6.8, 10% wt/vol SDS, 0.5% wt/vol bromophenol blue, 50% vol/vol glycerol, 5% vol/vol beta-mercaptoethanol.
Prepare an SDS-PAGE gel with a loading gel (5%) and separation gel (12%) as previously described26,27,28.
Load 10 µL of the boiled protein mixture and protein marker, and run the SDS-PAGE.
Stain the gel by shaking for 20 min with Coomassie brilliant blue solution, and then perform destaining.
For western blotting, after transferring to a PVDF membrane, block the membrane with TBST (20 mM of Tris-HCl, 150 mM of NaCl, 0.1% vol/vol Tween 20) + 5% wt/vol skim milk for 1 h at room temperature while shaking.
Incubate the membrane with primary antibodies (anti-His or anti-Flag) diluted 1:2,000 in TBST + 5% skim milk for 1 h.
Wash with TBST for 5 min at room temperature, and repeat 3 times.
Incubate the membrane in secondary antibody (HRP-linked anti-mouse Fc antibody) diluted 1:2,000 in TBST + 5% skim milk for 1 h.
Wash with TBST for 5 min at room temperature, and repeat 3 times.
Add 1 mL of HPR substrate per film, develop and take images.
5. Gel Filtration Analysis
Use the following column: Column volume of 24 mL; Equilibration buffer of PBS pH 7.4; Flow rate of 0.5 mL/min at room temperate; Sample volume of 200 µL.
Use the following protein volume and concentration: 500 µL of 1.2 mg/mL GPC3-S-Fab. Centrifuge at 20,000 x g for 30 min, and take 200 µL to load.
For the protein marker volume and concentration, take 100 µL of cytochrome C (2 mg/mL, 12.4 kDa), 140 µL of anhydrase (3 mg/mL, 29 kDa), 140 µL of albumin (10 mg/mL, 66 kDa) and 200 µL of alcohol dehydrogenase (5 mg/mL, 150 kDa) into one tube. Centrifuge at 20,000 x g for 30 min, and take 200 µL to load.
Before each run, wash with at least 2 CV of distilled water at a flow rate of 0.5 mL/min.
Equilibrate the column with at least 2 CV of equilibration buffer (PBS, pH 7.4) at a flow rate of 0.5 mL/min.
Automatically inject the sample at 0.5 mL/min to load the sample.
Elute with equilibration buffer at 0.5 mL/min and detect at 280 and 215 nm.
6. Flow Cytometry Analysis
Culture the HepG2 (GPC3 positive), Hep3B (GPC3 positive), Huh7 (GPC3 positive), and MHCC-97H (GPC3 negative) cells in 100 mm x 20 mm plastic dishes containing DMEM medium with 10% fetal bovine serum (FBS), penicillin (100 µg/mL), and streptomycin (50 µg/mL) at 37 °C, 5% CO2.
Culture CHO (GPC3 negative) cells and CHO/GPC3 cells harboring the full-length human GPC3 cDNA (GPC3 positive) in 100 mm x 20 mm plastic dishes containing RPMI 1640 medium with 10% FBS, penicillin (100 µg/mL), and streptomycin (50 µg/mL) at 37 °C, 5% CO2.
Culture the NK92/CD16 harboring the full-length human CD16 cDNA (CD16 positive) in 25cm2 flask containing RPMI 1640 medium with 10% FBS, penicillin (100 µg/mL), and streptomycin (50 µg/mL) at 37 °C, 5% CO2.
When the cells are 70 - 90% confluent, discard the medium and wash the cells with 2 mL of PBS. Add 1 mL of 0.25% trypsin and incubate at 37 °C for 2 - 3 min (or until the cells start to detach from the surface). Add 1 mL of the complete growth medium to neutralize the trypsin and re-suspend the cells by gently pipetting up and down.
Count the cell numbers using a Neubauer hemocytometer and detect the cell viability by Trypan Blue.
Collect 3 x 106 cells for each cell line. Add 1 mL of PBS + 0.2% BSA to re-suspend the cells. Centrifuge at 200 x g, 4 °C for 5 min. Repeat twice.
Add 0.3 mL of PBS + 0.2% BSA to re-suspend the cells, and transfer 100 µL (approximately 1 million) to each 5 mL polystyrene round-bottom tube. NOTE: Please make sure all steps from here to the tubes should be kept cold on ice. When the antibody is binding to the cell surface antigen, the complex will start to internalize. By keeping it cold, the process is slowed significantly.
Add 10 µL of primary antibodies (GPC3-S-Fab or human IgG1, final concentration 5 µg/mL) to every tube, and incubate on ice for 1 h.
Add 1 mL of PBS + 0.2% BSA to wash, and centrifuge at 200 x g, 4 °C for 5 min. Repeat three times.
Re-suspend in 100 µL of PBS + 0.2% BSA, and then add 0.5 µL of secondary antibody (anti-human IgG(H+L)-488, final concentration 10 µg/mL) and incubate on ice for 1 h. NOTE: From this step, please keep the cells from the light.
Add 1 mL of PBS + 0.2% BSA to wash, and centrifuge at 200 x g, 4 °C for 5 min. Repeat three times.
Analyze the cells by flow cytometer according to the standard protocol29. Acquire cells with a 488 nm laser for excitation.
7. Cytotoxic Assays
- Prepare tumor cells.
- Culture HepG2, Hep3B, Huh7, MHCC-97H, and CHO cell lines as described in steps 6.1 - 6.5.
- Dilute cells to 0.5 × 105/mL, and plate 100 µL cells (5,000 cells per well) on 96-well plates. Culture 6 - 8 h to allow the cells to attach.
- Prepare human peripheral blood mononuclear cells (PBMCs).
- Dilute fresh prepared blood with an equal volume of PBS in a 50 mL conical tube. For example, dilute 10 mL of blood with 10 mL of PBS.
- Take 15 mL of lymphocyte separation medium into a fresh 50 mL conical tube. Transfer the diluted blood onto the lymphocyte separation medium along the tube side slowly. NOTE: Keep the tube vertical. Do not break the surface of the lymphocyte separation medium.
- Centrifuge at 400 x g for 35 min at room temperature with the brake off.
- Slowly remove the upper plasma layer, and take the white blood layer containing PBMCs at the plasma-lymphocyte separation medium interface.
- Dilute the PBMCs with a double volume of PBS + 2% FBS. Centrifuge at 400 x g for 5 min.
- Wash the cells twice with PBS + 2% FBS. Centrifuge at 400 x g for 5 min. Count the cell numbers as described in step 6.5.
- Prepare NK cells.
- Prepare the PBMCs suspension at a concentration of 5 x 107 cells/mL in PBS + 2% FBS, and place them in a 5 mL polystyrene round-bottom tube.
- Add Human NK cell Enrichment Cocktail at 50 µL/mL of cells. Mix well, and incubate at room temperature for 10 min.
- Resuspend the Magnetic Particles from the NK cell Enrichment kit, and mix well to ensure they are uniformly suspended by pipetting up and down vigorously more than 5 times. Transfer the resuspended Magnetic Particles at 100 µL/mL to the cell suspension. Mix well, and incubate at room temperature for 5 min.
- Bring the cell suspension up to a total volume of 2.5 mL by adding PBS + 2% FBS. Mix the cells in the tube by gently pipetting up and down 2 - 3 times. Place the tube (without a cap) into the magnet. Let the magnetic beads settle down for 2.5 min at room temperature.
- Remove the magnet. In one continuous motion, invert the tube, pouring off the enriched cell suspension into a new tube.
- Count the cell numbers as described in step 6.5.
- Set up the cytotoxic reaction.
- Dilute the NK cells to 5 × 105 cells/mL using the corresponding culture medium. Add 100 µL of NK cell suspension to the tumor cells plated on a 96-well plate.
- Add 10 µL of GPC3-S-Fab at different concentrations, with the highest dose at a final concentration of 30 µg/mL, 3-fold dilution, 9 point (three replicates).
- Incubate at 37 °C, 5% CO2 for 72 h.
- After 72 h of incubation, remove the medium, and add 100 µL of DMEM medium containing 10 µL of CCK8 reagent to each well in 96-well plates, and then incubate at 37 °C.
- Read the plate at 450 nm at 1, 2, 3, and 4 h after adding the CCK8 reagent.
- Analyze the data. Calculate the survival rate as (OD450 GPC3-S-Fab + Effector + tumor - OD450 medium)/(OD450 tumor - OD450 medium) × 100% and (OD450 GPC3-S-Fab + tumor - OD450 medium)/(OD450 tumor - OD450 medium) × 100%. The effector cells are NK cells.
Representative Results
GPC3-S-Fab purification
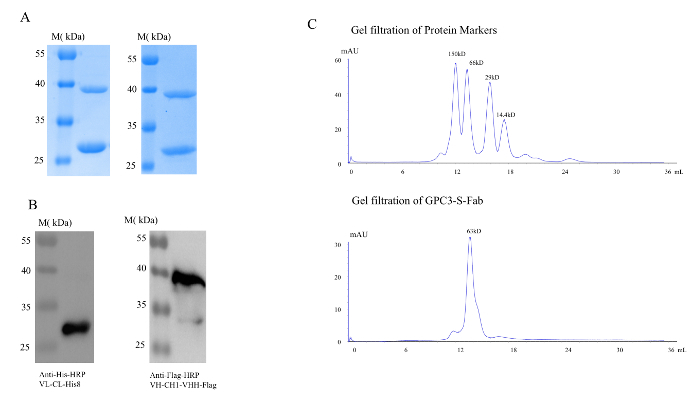
GPC3-S-Fab was purified from E. coli by a two-step affinity purification, first with Ni-NTA-agarose, followed by IgG-CH1 affinity purification. After the two-step affinity purification, GPC3-S-Fab was purified to homogeneity with the two chains close to 1:1 (Figure 2A). The presence of both VH-CH1-CD16 VHH and VL-CL polypeptides can be identified by their distinct C-terminal tags, anti-His for the VL-CL approximately 25 kDa, and anti-Flag for the VH-CH1-VHH approximately 38 kDa (Figure 2B). After the two-step affinity purification, ~1.2 mg protein can be obtained from 1 L of SB culture. To further characterize the purified GPC3-S-Fab, gel filtration was performed. Based on the standard markers (Figure 2C), GPC3-S-Fab was identified as a homogenous monomer in the form of a heterodimer with a molecular size of approximately 63 kDa (Figure 2C).
GPC3-S-Fab binding activities
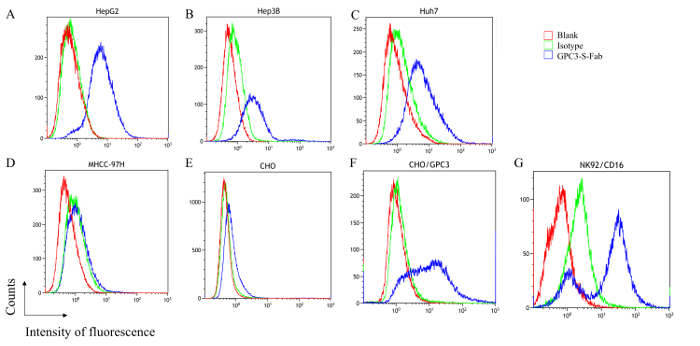
To evaluate whether the GPC3-S-Fab recognizes the GPC3-positive cells and CD16-positive cells, flow cytometry analysis was conducted using the GPC3-positive liver cancer cell lines HepG2, Hep3B, Huh7, CHO/GPC3 and the GPC3-negative liver cancer cell line, MHCC-97H, and CHO cells. The GPC3-S-Fab could bind to all the GPC3-positive cells, although with weaker binding to Hep3B (Figure 3B) and Huh7 cells (Figure 3C) than to HepG2 (Figure 3A) and CHO/GPC3 (Figure 2F). No or minimal binding to GPC3-negative MHCC-97H and CHO cells was observed (Figure 3D, 3E). The GPC3-S-Fab also could bind to the CD16-positive cells, NK92/CD16 cells (Figure 3G). These results are consistent with previous results using other anti-GPC3 antibodies30, suggesting that GPC3-S-Fab can specifically bind GPC3-positive cell lines and CD16-positive cells.
To evaluate the cytotoxicity of GPC3-S-Fab, tumor cells were incubated with fresh isolated NK cells with different concentrations of GPC3-S-Fab. Without NK cells, GPC3-S-Fab had no cytotoxicity against tumor cells regardless of the GPC3 expression status (Figure 4). In the presence of NK cells, GPC3-S-Fab triggered strong cytotoxicity against HepG2, Hep3B, and Huh7 cells in a dose-dependent manner but showed no effect on GPC3-negative MHCC-97H cells (Figure 4), suggesting that the cytotoxicity of GPC3-S-Fab depends on GPC3 expression on the tumor cell surface.
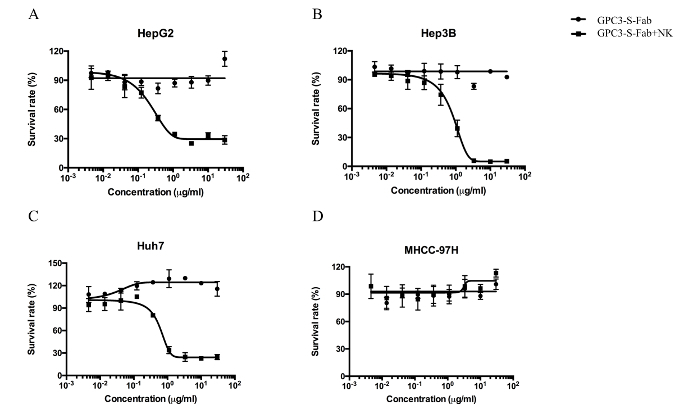
Figure 1. Constructs of GPC3-S-Fab (A) The bacterial expression constructs of GPC3-S-Fab. The constructs contain a pelB signal sequence, a humanized anti-GPC3 (GC33) VH-CH1-anti-CD16 VHH (heavy chain), or a VL-CL (light chain). To facilitate protein detection and purification, a Flag tag or a His8 tag was added to the C-terminus. (B) Diagram of GPC3-S-Fab after co-expression.
Figure 2. GPC3-S-Fab purification from E. coli. (A) Left panel, after Ni-NTA affinity chromatography; Right panel, after anti-IgG-CH1 affinity chromatography; M, molecular weight ladder; Coomassie blue staining. (B) Left panel, after Ni-NTA affinity chromatography; Right panel, after anti-IgG-CH1 affinity chromatography; M, molecular weight ladder; Western-Blotting. (C) Gel filtration analysis of GPC3-S-Fab. Top panel, standard markers; bottom panel, GPC3-S-Fab. Please click here to view a larger version of this figure.
Figure 3. GPC3-S-Fab recognizes GPC3 positive cells and CD16-positive cells. Flow cytometry analysis of HepG2 (A), Hep3B (B), Huh7 (C), MHCC-97H (D), CHO (E), CHO/GPC3 (F) and NK92/CD16 (G) cells was performed as described in the protocol. Red line: tumor cells only; Green line: isotype control, tumor cell + human IgG1 + anti-human IgG(H+L)-488; blue line: tumor cell + GPC3-S-Fab + anti-human IgG (H+L)-488. Please click here to view a larger version of this figure.
Figure 4. GPC3-S-Fab promotes the cell death of GPC3-positive cancer cells. Dose-dependent cytotoxicity assays were performed as described in the protocol for HepG2 (A), Hep3B (B), Huh7 (C), and MHCC-97H (D). The concentrations of GPC3-S-Fab were from 0.0045 µg/mL to 30 µg/mL. The data are the mean of triplicates with error bars representing the standard deviation. Solid circle, only GPC3-S-Fab; solid square, GPC3-S-Fab+ NK, NK cells (50,000 per well) and target cells (5,000 per well). The mixtures were incubated for 72 h before cytotoxicity measurement. Please click here to view a larger version of this figure.
Discussion
In this study, we present a strategy to construct a new format of bsAbs, GPC3-S-Fab, which can recruit NK cells targeting GPC3 positive tumor cells. The S-Fab is based on the natural Fab format by adding an anti-CD16 VHH11,12. Compared with the bsAbs containing Fc region, GPC3-S-Fab can easily be produced in the periplasm of bacteria on a large scale.
Using the expression and purification strategy described in the protocol, we obtained a soluble and functional GPC3-S-Fab in large quantities. To facilitate periplasmic expression, a signal sequence pelB was added to the N-terminus to direct secretion to the periplasm of E. coli17. In contrast to the cytoplasm, the more oxidizing environment in the periplasmic space between the cytoplasmic and outer membranes is equipped with a number of proteins important for protein folding and assembly and thus promotes the correcting folding and solubility of recombinant proteins16. However, the machinery for protein folding and export to the periplasm in E. coli has limited capacity. High expression of recombinant proteins often results in the accumulation of insoluble product in the periplasm16. To avoid the high expression of protein over a short period of time, low IPTG (0.2 mM) and low temperature (16 °C) in nutritious medium were applied in this protocol to avoid the accumulation of insoluble protein. In this study, ~1.2 mg protein was obtained from 1 L of medium, improving the yields at least 2-fold compared with previous works12.
To avoid protein degradation, all fractions containing GPC3-S-Fab in various steps should be kept on ice. With the His tag at the C-terminal of VL-CL, Ni-NTA-agarose was used for purification. However, single Ni-NTA-agarose purification is not sufficient to remove unwanted proteins, such as unpaired VL-CL protein. To purify the homogenous heterodimeric GPC3-S-Fab, the second step, anti-IgG-CH1 affinity purification, was applied. The purified protein had high purity and two polypeptides close to 1:1 (Figure 2A-2B).
Gel filtration chromatography can determine the molecular weights of proteins based on their molecular sizes and shapes; analyze the degree of purity; and determine whether the purified protein is a monomer, dimer, or composed of aggregates15. By gel filtration chromatography, GPC3-S-Fab was identified with the expected molecular size of approximately 63 kDa, which is in the form of a monomer with heterodimerization of GPC3-S-Fab (Figure 2C).
Flow cytometry can determine whether an antibody can bind its antigen on the cell membrane, compared with traditional western-blotting and ELISA, which only detect antigen-antibody binding reactions. By flow cytometry analysis, GPC3-S-Fab recognized the GPC3 on the cell membrane, suggesting the GPC3-Fab moiety was functional. When performing flow cytometry analysis, it is critical to keep all of the steps on ice or at 4 °C after the antibody is added. When the antibody binds to the cell surface antigen, the antibody-antigen complex will initiate internalization. By keeping it cold, the internalization process is slowed significantly.
PBMCs and NK cells were efficiently isolated from human peripheral blood by centrifugation using lymphocyte separation medium, followed by magnetic-activated cell sorting strategies. GPC3-S-Fab showed potent cytotoxicity against the GPC3 positive cells when the ratio of NK cells to target cells (tumor cells) was 10:1, and the ratio of NK cells to target cells could vary from 50:1 to 5:1.
In summary, GPC3-S-Fab, which can be produced in a microorganism expression system, provides a new avenue to create functional formats of bsAbs against tumors.
Disclosures
The authors declare no conflicts of interest.
Acknowledgments
This work was financially supported by the R&D Plan of Guangdong Province (PR China) (2016A050503028).
References
- Weiner GJ. Building better monoclonal antibody-based therapeutics. Nature Reviews Cancer. 2015;15(6):361–370. doi: 10.1038/nrc3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathi C, Meibohm B. Clinical pharmacology of bispecific antibody constructs. The Journal of Clinical Pharmacology. 2015;55(Suppl 3):S21–S28. doi: 10.1002/jcph.445. [DOI] [PubMed] [Google Scholar]
- Weidle UH, Kontermann RE, Brinkmann U. Tumor-antigen-binding bispecific antibodies for cancer treatment. Seminars in Oncology. 2014;41(5):653–660. doi: 10.1053/j.seminoncol.2014.08.004. [DOI] [PubMed] [Google Scholar]
- Demarest SJ, Glaser SM. Antibody therapeutics, antibody engineering, and the merits of protein stability. Current Opinion in Drug Discovery and Development. 2008;11(5):675–687. [PubMed] [Google Scholar]
- Mabry R, Snavely M. Therapeutic bispecific antibodies: The selection of stable single-chain fragments to overcome engineering obstacles. IDrugs. 2010;13(8):543–549. [PubMed] [Google Scholar]
- Rothlisberger D, Honegger A, Pluckthun A. Domain interactions in the Fab fragment: a comparative evaluation of the single-chain Fv and Fab format engineered with variable domains of different stability. Journal of Molecular Biology. 2005;347(4):773–789. doi: 10.1016/j.jmb.2005.01.053. [DOI] [PubMed] [Google Scholar]
- Kijanka M, Dorresteijn B, Oliveira S, van Bergen en Henegouwen PM. Nanobody-based cancer therapy of solid tumors. Nanomedicine (London) 2015;10(1):161–174. doi: 10.2217/nnm.14.178. [DOI] [PubMed] [Google Scholar]
- Muyldermans S, Cambillau C, Wyns L. Recognition of antigens by single-domain antibody fragments: the superfluous luxury of paired domains. Trends in Biochemical Sciences. 2001;26(4):230–235. doi: 10.1016/s0968-0004(01)01790-x. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Sapienza G, Rossotti MA, Tabares-da Rosa S. Single-Domain Antibodies As Versatile Affinity Reagents for Analytical and Diagnostic Applications. Frontiers in Immunology. 2017;8:977. doi: 10.3389/fimmu.2017.00977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes CFC, et al. Camelid Single-Domain Antibodies As an Alternative to Overcome Challenges Related to the Prevention, Detection, and Control of Neglected Tropical Diseases. Frontiers in Immunology. 2017;8:653. doi: 10.3389/fimmu.2017.00653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, et al. A novel bispecific antibody, S-Fab, induces potent cancer cell killing. Journal of Immunotherapy. 2015;38(9):350–356. doi: 10.1097/CJI.0000000000000099. [DOI] [PubMed] [Google Scholar]
- Li A, et al. A single-domain antibody-linked Fab bispecific antibody Her2-S-Fab has potent cytotoxicity against Her2-expressing tumor cells. AMB Express. 2016;6(1):32. doi: 10.1186/s13568-016-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano K, et al. Anti-glypican 3 antibodies cause ADCC against human hepatocellular carcinoma cells. Biochemical and Biophysical Research Communications. 2009;378(2):279–284. doi: 10.1016/j.bbrc.2008.11.033. [DOI] [PubMed] [Google Scholar]
- Behar G, et al. Isolation and characterization of anti-FcgammaRIII (CD16) llama single-domain antibodies that activate natural killer cells. Protein Engineering, Design, and Selection. 2008;21(1):1–10. doi: 10.1093/protein/gzm064. [DOI] [PubMed] [Google Scholar]
- Baral TN, Arbabi-Ghahroudi M. Expression of single-domain antibodies in bacterial systems. Methods in Molecular Biology. 2012;911:257–275. doi: 10.1007/978-1-61779-968-6_16. [DOI] [PubMed] [Google Scholar]
- Arbabi-Ghahroudi M, Tanha J, MacKenzie R. Prokaryotic expression of antibodies. Cancer and Metastasis Reviews. 2005;24(4):501–519. doi: 10.1007/s10555-005-6193-1. [DOI] [PubMed] [Google Scholar]
- Spiess C, et al. Bispecific antibodies with natural architecture produced by co-culture of bacteria expressing two distinct half-antibodies. Nature Biotechnology. 2013;31(8):753–758. doi: 10.1038/nbt.2621. [DOI] [PubMed] [Google Scholar]
- Filmus J, Selleck SB. Glypicans: proteoglycans with a surprise. Journal of Clinical Investigation. 2001;108(4):497–501. doi: 10.1172/JCI13712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumhoer D, et al. Glypican 3 expression in human nonneoplastic, preneoplastic, and neoplastic tissues: a tissue microarray analysis of 4,387 tissue samples. American Journal of Clinical Pathology. 2008;129(6):899–906. doi: 10.1309/HCQWPWD50XHD2DW6. [DOI] [PubMed] [Google Scholar]
- Llovet JM, et al. A molecular signature to discriminate dysplastic nodules from early hepatocellular carcinoma in HCV cirrhosis. Gastroenterology. 2006;131(6):1758–1767. doi: 10.1053/j.gastro.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Zhu ZW, et al. Enhanced glypican-3 expression differentiates the majority of hepatocellular carcinomas from benign hepatic disorders. Gut. 2001;48(4):558–564. doi: 10.1136/gut.48.4.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu HC, Cheng W, Lai PL. Cloning and expression of a developmentally regulated transcript MXR7 in hepatocellular carcinoma: biological significance and temporospatial distribution. Cancer Research. 1997;57(22):5179–5184. [PubMed] [Google Scholar]
- Flaherty MM, et al. Nonclinical evaluation of GMA161--an antihuman CD16 (FcgammaRIII) monoclonal antibody for treatment of autoimmune disorders in CD16 transgenic mice. Toxicological Sciences. 2012;125(1):299–309. doi: 10.1093/toxsci/kfr278. [DOI] [PubMed] [Google Scholar]
- Sharma R, Das A. Organ-specific phenotypic and functional features of NK cells in humans. Immunological Research. 2014;58(1):125–131. doi: 10.1007/s12026-013-8477-9. [DOI] [PubMed] [Google Scholar]
- Li XY, et al. Effect of CD16a, the surface receptor of Kupffer cells, on the growth of hepatocellular carcinoma cells. International Journal of Molecular Medicine. 2016;37(6):1465–1474. doi: 10.3892/ijmm.2016.2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala GJ, Schamel WW, Blumenthal B. Blue native polyacrylamide gel electrophoresis (BN-PAGE) for analysis of multiprotein complexes from cellular lysates. Journal of Visualized Experiments. 2011. [DOI] [PMC free article] [PubMed]
- Hwang AC, Grey PH, Cuddy K, Oppenheimer DG. Pouring and running a protein gel by reusing commercial cassettes. Journal of Visualized Experiments. 2012. [DOI] [PMC free article] [PubMed]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227(5259):680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Menon V, Thomas R, Ghale AR, Reinhard C, Pruszak J. Flow cytometry protocols for surface and intracellular antigen analyses of neural cell types. Journal of Visualized Experiments. 2014. [DOI] [PMC free article] [PubMed]
- Feng M, et al. Therapeutically targeting glypican-3 via a conformation-specific single-domain antibody in hepatocellular carcinoma. Proceedings of the National Academy of Science U S A. 2013;110(12):E1083–E1091. doi: 10.1073/pnas.1217868110. [DOI] [PMC free article] [PubMed] [Google Scholar]