

Abstract
The presence of ribonucleotides in nuclear DNA has been shown to be a source of genomic instability. The extent of ribonucleotide incorporation can be assessed by alkaline hydrolysis and gel electrophoresis as RNA is highly susceptible to hydrolysis in alkaline conditions. This, in combination with Southern blot analysis can be used to determine the location and strand into which the ribonucleotides have been incorporated. However, this procedure is only semi-quantitative and may not be sensitive enough to detect small changes in ribonucleotide content, although strand-specific Southern blot probing improves the sensitivity. As a measure of one of the most striking biological consequences of ribonucleotides in DNA, spontaneous mutagenesis can be analyzed using a forward mutation assay. Using appropriate reporter genes, rare mutations that results in the loss of function can be selected and overall and specific mutation rates can be measured by combining data from fluctuation experiments with DNA sequencing of the reporter gene. The fluctuation assay is applicable to examine a wide variety of mutagenic processes in specific genetic background or growth conditions.
Keywords: Biology, Issue 137, Ribonucleotide incorporation, DNA replication, RNase H2, alkaline gel electrophoresis, fluctuation test, mutagenesis
Introduction
During eukaryotic nuclear DNA replication, ribonucleotides are incorporated into the genome by all three major DNA replicases, DNA polymerases (Pols) α, ε, and δ1,2. RNase H2-dependent ribonucleotide excision repair (RER3) removes the majority of these embedded ribonucleotides.
A ribonucleotide in DNA is susceptible to hydrolysis, as the 2' hydroxyl group of the sugar moiety can attack the adjacent phosphodiester bond, releasing one end with a 2'-3' cyclic phosphate and the other with a 5'-OH4. Alkaline conditions can greatly accelerate this reaction. Thus, the hydrolysis of embedded ribonucleotides during incubation in a basic solution causes fragmentation of genomic DNA, which can be visualized by alkaline-agarose electrophoresis5. This DNA can be transferred to a membrane and probed by Southern blot analysis using strand-specific probes that allow the identification of alkali-sensitive sites caused by ribonucleotides incorporated into the nascent leading- or lagging-strand DNA, respectively.
In yeast cells lacking RNase H2 activity, removal of ribonucleotides can be initiated when topoisomerase I (Top1) nicks the DNA at the embedded ribonucleotide6,7. However, when Top1 cleaves on the 3' side of the ribonucleotide, this generates 5'-OH and 2'-3' cyclic phosphate DNA ends that are refractory to religation. Failure to repair, or aberrant processing of these 'dirty ends' can lead to genomic instability. In addition, if the incision occurs within a repeat DNA sequence, the repair process can lead to deletion mutations. This is particularly problematic for tandem repeats, where short deletions (of between two and five base pairs) are commonly observed in RNase H2-deficient cells. The Top1-dependent deleterious effects in the absence of yeast RNase H2 activity are exacerbated in a DNA polymerase ε mutant (pol2-M644G) promiscuous for ribonucleotide incorporation during nascent leading strand synthesis.
Processing of ribonucleotides in DNA leads to spontaneous mutations and this mutagenesis can be detected by using appropriate reporter genes and selecting for the accompanying phenotypic change. A fluctuation test or Luria and Delbrück experiment is one of the most commonly used methods to measure spontaneous mutation rates using selectable reporter genes8,9. In yeast, the URA3 and CAN1 genes can be used as reporters in a forward mutation assay, which allows for the detection of all mutation types that result in the loss of gene function. The spontaneous mutation rate is estimated as the median of that observed for multiple parallel cultures started from single colonies without mutations in the target reporter gene. A yeast RNase H2-deficient strain such as rnh201Δ has a moderately elevated overall spontaneous mutation rate that is largely caused by an elevated incidence of 2 - 5 bp deletions in tandem repeat sequences. Thus, to fully characterize the mutagenic effects of ribonucleotides in the genome, specific mutation rates need to be determined. In this case, the URA3 or CAN1 reporter genes can be amplified and sequenced to determine the types and locations of the mutations, and specific mutation rates can be calculated. Compiling mutations identified in multiple independent URA3 or CAN1 mutants can then be used to generate a mutation spectrum.
Protocol
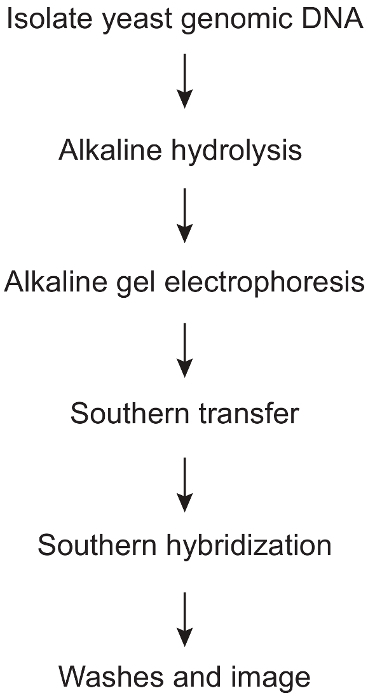
1. Alkaline Hydrolysis and Strand-specific Southern Blot (Figure 1)
- Cell growth and genomic DNA isolation
- Isolate yeast genomic DNA using a yeast DNA purification kit following the manufacturer’s instructions. Use the cultures that are in the exponential phase of growth (between an optical density of 0.5 and 1). Resuspend the final DNA pellet in 35 mL of 1x TE buffer (10 mM Tris-HCl, pH 7.5, 1 mM ethylenediaminetetraacetic acid (EDTA)).
- Add 1 mL of 5 mg/mL RNase A to the DNA and incubate for 30 min at 37 °C.
- Precipitate the DNA using 0.1 volumes of 3 M sodium acetate (pH 5.2) and 2.5 volumes of 100% ethanol for 20 min on ice.
- Centrifuge at 16,000 x g for 20 min at 4 °C. Remove the supernatant using a pipette.
- Wash the pellet twice with 500 mL of 70% ethanol. Remove the supernatant using a pipette.
- Dry the pellet on bench and resuspend in 35 ml of 1x TE buffer.
- Quantitate the DNA using a fluorimeter (Table of Materials) with the dsDNA BR Assay kit.
- Precipitate 5 mg of DNA from each sample using 0.1 volumes of 3 M sodium acetate, pH 5.2 and 2.5 volumes of 100% EtOH for a total volume of 200 mL (add H2O if necessary, additional 5 mg of DNA is needed if the neutral gel control is necessary). Incubate on ice for 20 min.
- Centrifuge at 16,000 x g for 20 min at 4 °C. Remove the supernatant using a pipette.
- Dry the pellet on bench. Resuspend the DNA in 14 mL of H2O.
- Alkaline hydrolysis and gel electrophoresis
- Add 6 mL of 1 M KOH and incubate at 55 °C for 2 h.
- Incubate the samples on ice for 5 min.
- Add 4 mL of 6x alkaline DNA loading buffer: 300 mM KOH, 6 mM EDTA, pH 8.0, 18% polysucrose (Table of Materials), 0.15% bromocresol green, and 0.25% xylene cyanol FF.
- Cast an alkaline gel that is comprised of 1% agarose, 50 mM NaOH, 1 mM EDTA, pH 8.0. Use a comb that allows for 24 mL of DNA sample to be loaded per lane.
- Run the gel at room temperature in 1x alkaline electrophoresis buffer: 50 mM NaOH, 1 mM EDTA, pH 8.0. Include an appropriate DNA size marker.
- Spin down the tubes briefly and load the entire 24 mL sample into the alkaline agarose gel. Electrophorese for 30 min at 30 V, followed by 18 - 20 h at 10 V.
- Neutralize the gel for 2 incubations of 45 min each at room temperature with gentle agitation in Neutralization Buffer I: 1 M Tris-HCl, 1.5 M NaCl.
- Stain the DNA by soaking the neutralized gel in 200 mL of water containing a 1/10,000 dilution of SYBR Gold stain for 2 h at room temperature with gentle shaking.
- Visualize the DNA using a UV transilluminator. Increased alkali-sensitivity causes the appearance of smaller DNA fragments, indicating a higher density of unrepaired ribonucleotides in DNA5.
- Southern transfer
- Soak the alkaline gel for 15 min at room temperature in Alkaline Transfer Buffer: 0.4 M NaOH, 1 M NaCl.
- Wet the nylon membrane (cut to the size of the gel) briefly with water, and then soak for 5 min in Alkaline Transfer Buffer.
- Set up the transfer platform by inverting 3 glass beakers (100 mL) in a glass baking dish. This dish should be slightly larger than the size of the agarose gel. Set a glass plate over top of them.
- Drape 2 large pieces of 3 MM CHR chromatography paper (cut to the size of the width of the gel plus longer sides that can drape over the edges into the buffer reservoir) over the glass plate.
- Pour Alkaline Transfer Buffer over the chromatography paper and half fill the glass dish. Smooth out the bubbles using a 5 mL glass pipette.
- Place the agarose gel on top, again smoothing out the bubbles. Surround all edges of the gel with parafilm. Pour a small amount of Alkaline Transfer Buffer onto the gel.
- Place the pre-soaked nylon membrane on top of the gel. Smooth out the bubbles.
- Wet 2 pieces of paper cut to the size of the agarose gel. Place them on top of the membrane, and smooth out any bubbles.
- Place a large stack of paper towels (cut to a size slightly larger than the agarose gel) on top of the transfer sandwich. Carefully place a glass plate on top of the paper towels. Add a weighted object to the top (a book with a mass of 400 g works well).
- Let the transfer proceed overnight at room temperature.
- Carefully take the transfer stack apart and soak the membrane for 15 min at room temperature in Neutralization Buffer II: 0.5 M Tris-HCl, pH 7.2, 1 M NaCl.
- Crosslink the DNA to the charged nylon membrane using a UV crosslinker. Place the membrane in the cross-linker and use the autocrosslink setting.
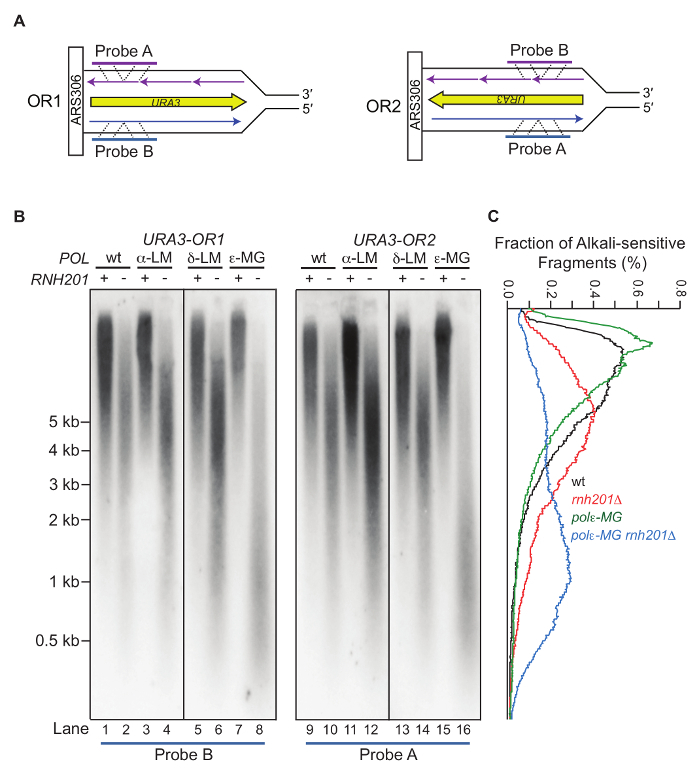
- Southern probe preparation Note: The approach described here to detect alkali-sensitive sites in the nascent leading- versus lagging-strand of DNA strand is based on a protocol published previously11. For our experiment, probing is performed at the URA3 reporter gene which has been inserted in one of two orientations (OR1 or OR2) adjacent to the ARS306 replication origin on chromosome III (Figure 3A). The presence of alkali-sensitive sites in yeast genomic DNA is an indicator of ribonucleotide incorporation, allowing the use of ribonucleotides as the biomarkers of DNA polymerase activity12,13,14. This approach, using a target DNA sequence adjacent to the efficient ARS306 origin of replication, should be amenable to other genomic locations and target genes as well.
- PCR-amplify a 520-base pair segment of the URA3 reporter gene using the following primer pair: URA3-F1 (5´-GCTACATATAAGGAACGTGCTGC) and URA3-R1 (5´-CTTTGTCGCTCTTCGCAATGTC).
- Perform the PCR reaction using 10 - 20 ng of yeast genomic DNA as a template, 4 μL of each primer (10 μM stock), 10 μL of 10x Ex Taq Buffer (Mg2+ plus), 8 μL of dNTP mixture (2.5 mM each), 0.5 μL of Ex Taq DNA Polymerase (5 U/μL), and adjust the final volume to 100 μL with H2O.
- Run the following PCR program: 95 °C for 5 min; 30 cycles of 95 °C for 1 min, 55 °C for 1 min, 72 °C for 2 min; 72 °C for 10 min and hold at 4 °C.
- Purify the PCR product using a PCR purification kit and elute the DNA using 50 mL of H2O. This can now be used as the template in the radiolabeling reaction.
- Use the following primers for strand-specific radiolabeling: Use URA3-A (5´-CTCATCCTAGTCCTGTTGCTGCC) for visualizing DNA that anneals to probe A. This corresponds to the nascent leading strand DNA when URA3 is in OR2 and the tnascent lagging strand DNA when URA3 is in OR1. Use URA3-B (5´-CAGTACCCTTAGTATATTCTCCAG) for visualizing nascent DNA that anneals to probe B. This corresponds to the nascent leading strand DNA when URA3 is in OR1 and nascent lagging strand DNA when URA3 is in OR2 (Figure 3).
- Perform the radiolabeling reaction using 10 - 20 ng amplified URA3 fragment as template DNA, 2 μL of primer (10 μM stock), 5 μL of 10x Ex Taq Buffer (Mg2+ plus), 4 μL of 2.5 mM dNTPs (minus dCTP), 5 μL (50 μCi) of a-32P-dCTP, 0.5 μL of ExTaq DNA Polymerase (5 U/μL) and adjust the final volume to 50 μL with H2O. Run the following program for radiolabeling: 94 °C for 5 min; 25 cycles of 94 °C for 1 min, 55 °C for 30 s, 72 °C for 1 min; 72 °C for 5 min and hold at 4 °C.
- Use a G-25 spin column to remove unincorporated radioactive label. Prior to adding the radiolabeled sample, centrifuge the column for 1 min at 720 x g and removethe buffer by pipetting. Load the radiolabeled reaction product and centrifuge for 2 min at 720 x g to elute the labeled probe.
- Incubate at 95 °C for 5 min to denature the probe immediately prior to hybridization. Cool briefly on ice.
- Southern Hybridization
- Roll the wetted membrane into a hybridization tube and add 25 mL of freshly prepared Hybridization buffer: 0.5 M sodium phosphate buffer, pH 7.2, 7% sodium dodecyl sulfate (SDS), 1% bovine serum albumin (BSA). Incubate at 65 °C for 1 - 2 h with rotation in a hybridization oven.
- Carefully pour off the solution and add 25 mL of Hybridization buffer plus the radiolabelled probe. Incubate at 65 °C for 16 - 18 h with rotation.
- Pour off the solution into a radioactive waste container. Perform 5 washes of 5 min each at room temperature with Phosphate-SDS Washing Solution I: 40 mM sodium phosphate buffer, pH 7.2, 5% SDS, 0.5% BSA, 1 mM EDTA, pH 8.0.
- Perform 2 washes of 15 min each at 65 °C with Phosphate-SDS Washing Solution II: 40 mM sodium phosphate buffer, pH 7.2, 1% SDS, 1 mM EDTA, pH 8.0.
- Remove the membrane from the hybridization tube using tweezers and place in an opened plastic sleeve protector. Cover and expose to an imaging plate. Try a number of different exposure times ranging from 4 h to 4 d.
- If necessary, strip and re-probe the blot using a different probe approximately 3 - 4 times. To strip, incubate the membrane for 2 h at 65 °C in 25 mL of a 50% formamide, 2x SSPE solution. Note: 20x SSPE stock solution: 3 M NaCl, 0.2 M NaH2PO4, 0.02 M EDTA, pH 7.4.
- Pour off the solution into a radioactive waste container. Perform 2 washes of 15 min each at 65 °C in Phosphate-SDS Washing Solution II.
- Check the membrane with a Geiger counter and repeat the stripping protocol if signal remains.
- Perform pre-hybridization and hybridization as described above.
2. Measuring Mutation Rate and Specificity in S. cerevisiae Strains
- Prepare the cell culture
- Inoculate a single colony (healthy yeast cells takes 2 - 3 days at 30 °C to form proper sized colonies) grown on YPD agar supplemented with 100 mg/mL adenine into 5 mL of YPDA broth supplemented with 250 mg/mL adenine (the supplementation of adenine is only necessary if the strain used is an adenine auxotroph). Grow in a shaker incubator or on a rotator at 30 °C until the culture reaches saturation (36 - 48 h for a strain without a severe growth defect).
- Spin down the cells at ~2000 x g for 5 min.
- Discard the supernatant and wash the cells with 5 mL of sterile water.
- Resuspend the cells in 500 mL of sterile water.
- Dilution of the cell culture and plating
- For non-selection controls, dilute the cells in a 96-well plate by a factor of 1,600,000 and plate 100 mL of each culture onto a complete (COM) plate.
- To select for ura3 mutants, plate 100 mL of undiluted cells onto a 5-FOA plate for a wild-type (WT) strain. Dilute the cells accordingly if the mutation rate of the strain of interest is expected to be higher than in a WT strain.
- To select for can1 mutants, dilute the cells 5-fold and plate 100 mL onto a canavanine-containing plate (CAN) for a WT strain. The dilution factor is 0.5.
- Incubate the plates at 30 °C until yeast colonies form. For strains without a growth defect, 2 - 3 days is usually sufficient for COM and CAN plates and 3 - 5 days for 5-FOA plates. To compare the mutation rates between different strains, it is best to choose the same day of growth to count colonies. Store the plates ready to be counted in the cold room (~4 °C).
- Calculation of spontaneous mutation rate
- Count visible colonies on each plate and take notes of colony numbers
- Calculate the mutation rate using Drake’s formula: μ = f ÷ ln (μNt). f is the mutation frequency calculated by number of mutants divided by number of cells in the final culture. Nt is the number of cells in the final culture and μ is the mutation rate. This equation can be solved by an iterative method such as the Newton-Raphson method. There are other estimator methods, including the Lea and Coulson test, to calculate the mutation rate15.
- Calculate the confidence interval assuming log normal distribution of the utation rates of individual isolates. The 95% confidence interval is most often used.
- Analysis of mutation spectrum
- Pick a single colony from each 5-FOA or CAN plate and refresh on a YPDA plate.
- Perform colony PCR to amplify the URA3 or CAN1 gene and sequence in order to detect mutations using the following primers: URA3-F (5’-CGCATATGTGGTGTTGAAGAA-3’) and URA3-R (5’-TGTGAGTTTAGTATACATGCA-3’) for both PCR amplification and sequencing of the 800 bp URA3 gene; CAN1-F (5’-CTTCTACTCCGTCTGCTTTC-3’) and CAN1-R (5’-CAGAGTTCTTCAGACTTC-3’) for amplification and CAN1-AR (5’- TGAAATGTGAAGGCAGCGTT-3’), CAN1-9R (5’- CCTGCAACACCAGTGATA-3’), CAN1-10R (5’- GAGGATGTAACAGGGATGAAT-3’) and CAN1-BR (5’-CGGTGTATGACTTATGAGGG-3’) for sequencing of the 1800 bp CAN1 reporter gene.
- To perform colony PCR, resuspend a single colony in 5 μL of H2O and use 1 μL as template. Add 1 μL of each of the primers (5 μM stock), 2 μL of 5x KAPA2G Buffer (Mg2+ plus), 0.5 μL of dNTP (2.5 mM), 0.5 μL of KAPA2G Fast DNA Polymerase, 12 μL of H2O.
- For the amplification of URA3 gene, run the following PCR program: 95 °C for 5 min; 5 cycles of 95 °C for 10 s, 61 °C for 15 s, 72 °C for 30 s; 35 cycles of 95 °C for 10 s, 61 °C for 15 s, 72 °C for 30 s; 72 °C for 2 min and hold at 4 °C. For the amplification of CAN1 gene, run the following PCR program: 95 °C for 5 min; 5 cycles of 95 °C for 10 s, 61 °C for 15 s, 72 °C for 45 s; 35 cycles of 95 °C for 10 s, 61 °C for 15 s, 72 °C for 45 s; 72 °C for 2 min and hold at 4 °C.
- Align the sequencing results to a reference sequence using a program such as DNASTAR Seqman Pro or Sequencher to identify mutations.
Representative Results
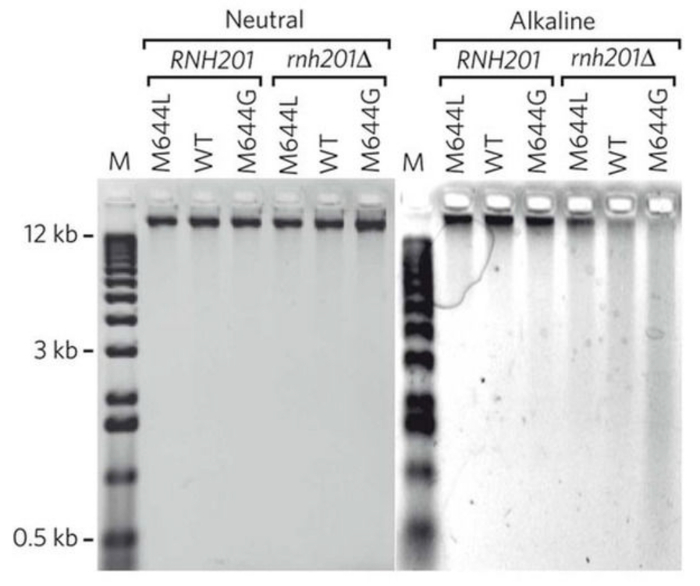
Treatment of genomic DNA with alkali followed by alkaline gel electrophoresis allows for semi-quantitative detection of the DNA fragmentation due to the abundance of stably incorporated ribonucleotides. Figure 2 shows the gel images of yeast genomic DNA treated with or without KOH5. The M644L variant of Pol2, the catalytic subunit of Polε, has reduced the ability to incorporate ribonucleotides while the M644G mutant incorporates more ribonucleotides than the WT polymerase. As detailed in the protocol, the alkaline gel can be further probed by strand-specific Southern blot analysis. With the knowledge of the location of the probed genomic site relative to its adjacent origins, we can selectively probe ribonucleotides incorporated into the nascent leading or lagging strands. Figure 3 shows the Southern blot results probing the URA3 reporter gene inserted in two opposite orientations close to an early-firing replication origin, ARS30616. By using leading strand-specific probes, we observe the DNA fragmentation pattern caused by alkali-cleavage at ribonucleotides in nascent leading strand DNA in a WT strain and in strains expressing the variants of polymerases α, δ, and ε that are promiscuous for ribonucleotide incorporation.
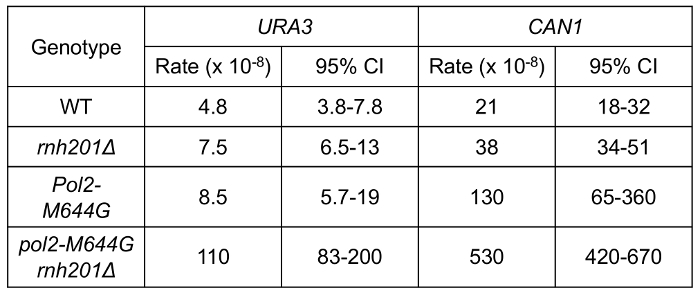
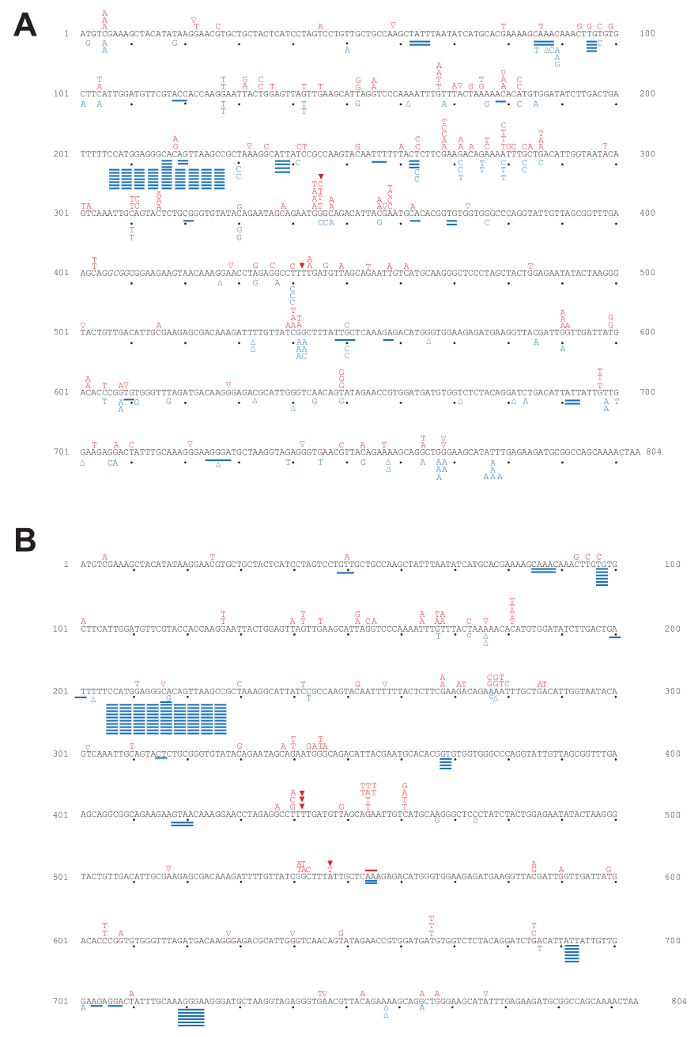
Using a fluctuation experiment, we can measure the mutation rates of yeast strains containing reporter genes. Figure 4 shows the mutation rates at the URA3 and CAN1 loci of strains expressing WT polymerase or the pol2-M644G mutant with or without functional RNase H2. The lack of RNase H2 causes a moderate increase in overall mutation rates in the both backgrounds. However, closer examination of mutation specificity reveals that in strains lacking RNase H2 (Figure 5), there is a strong increase in the mutation rate for short deletions, particularly in tandem repeat sequences. Figure 5A compares the mutation specificity in WT and rnh201Δ strains and Figure 5B displays the mutational effect of further elevated ribonucleotide incorporation into the nascent leading DNA strand by Pol ε.
Figure 1: Protocol steps involved in strand-specific detection of alkali-sensitive sites caused by ribonucleotide incorporation into yeast genomic DNA. An overview of the various major steps involved in this procedure, beginning with yeast genomic DNA isolation and proceeding through the final Southern blot analysis. Please click here to view a larger version of this figure.
Figure 2: A representative image of results obtained by alkaline-agarose gel electrophoresis. This figure has been adapted from Nick McElhinny, S. A. et al.5. Yeast genomic DNA from strains of different genotypes was treated with KCl (neutral) or KOH (alkaline) and then separated on a neutral or alkaline agarose gel. "WT" and "M644G" indicate the status of Pol ε. Please click here to view a larger version of this figure.
Figure 3: Strand-specific probing of an alkaline-agarose gel by Southern blot. This figure has been adapted fromWilliams, J. S. et al.13. (A) The radiolabeled probes anneal to the nascent leading DNA strand within the URA3 reporter gene that has been inserted close to ARS306 in two opposite orientations (OR1 and OR2). (B) Representative results of Southern blotting using the nascent leading strand-specific probes. Displayed are the probing results for the nascent leading strand in a strain with WT DNA polymerases, or the pol1-L868M, pol2-M644G or the pol3-L612M variant strains with or without RNase H2. The POL1, POL2, and POL3 genes encode catalytic subunits of Pols α, ε, and δ, respectively. The variants are more promiscuous for ribonucleotide incorporation5,12,17. (C) Quantification of the radioactive signal by fraction of the total in each lane from (B). Values are expressed as a percentage of the total. Please click here to view a larger version of this figure.
Figure 4: Spontaneous mutation rates in yeast strains. This figure has been adapted from Nick McElhinny, S. A. et al.5. The URA3 and CAN1 reporter genes were used in the forward mutation assay to measure the mutation rates of the indicated strains. URA3 reporter is in "orientation 2" (OR2, Figure 3A). The 95% confidence interval (CI) is included for each measurement. Please click here to view a larger version of this figure.
Figure 5: Mutation spectra for the URA3-OR2 reporter gene. This figure has been adapted from previous publications5,18,19,20. The position and mutation type observed in each independent 5-FOA-resistant colony selected in the forward mutation assay are depicted in the 804 bp URA3 coding sequence. Letters indicate base substitutions, open triangles indicate single base deletions, closed triangles indicate single base insertions, and solid lines indicate short deletions. (A) Mutation spectra in WT and rnh201Δ strains. Red labels above the sequence are mutations observed in WT while blue labels below the sequence are those observed in an rnh201Δ strain. (B) Mutation spectra in pol2-M644G and pol2-M644G rnh201Δ strains. Red labels above the sequence are mutations observed in pol2-M644G while blue below the sequence for pol2-M644G rnh201Δ strains. Please click here to view a larger version of this figure.
Discussion
Here, we describe the protocols for two sets of experiments that are frequently used to semi-quantitatively analyze ribonucleotides incorporated during DNA replication and the mutagenic effects of unrepaired ribonucleotides. Although these approaches involve the model eukaryote S. cerevisiae, these techniques can be easily adapted to other microbes and even higher eukaryotes.
Probing for unrepaired ribonucleotides in DNA using alkaline-agarose electrophoresis coupled with Southern blotting provides important information regarding the number of ribonucleotides present and the strand into which they have been incorporated. However, it is important to note that alkali-sensitivity can also be caused by other DNA lesions, such as an abasic site. Another means of detecting ribonucleotides in DNA is by the treatment with purified RNase H2 enzyme, as has been done in mammalian cells21. Although our approach involves probing for alkali-sensitive sites in nascent leading versus lagging strands at the URA3 reporter gene used in our mutational analysis experiments, it is possible to design the probes that anneal at other locations in order to gain important information regarding ribonucleotide density at other genomic sites. The Southern blot part of the procedure can also be used as reference for other generic DNA probing experiments.
Measurement of mutation rates using a Fluctuation test has been widely used for the analysis of various mutagenic events in microorganisms, and thus is not confined to analysis of the mutagenic consequences of ribonucleotides in DNA. For this experiment, increasing the number of independent cultures can greatly enhance the accuracy of measurement. Independent colonies can be obtained by streaking a strain of interest onto YPDA medium or by the analysis of independent spore colonies obtained by tetrad dissection of a diploid yeast stain. Ideally, the use of multiple independent haploid yeast strains (e.g., from tetrad dissection) is encouraged to minimize the impact of strain-to-strain variation. This is particularly important when the spontaneous mutation rate is high or when the strain has a growth defect. An additional method that can be employed to measure spontaneous the mutation rate is a mutation accumulation experiment22. In this analysis, mutation frequency is determined for the cells after extensive growth and can be used to calculate for the mutation rate. Coupling of a mutation accumulation experiment with deep sequencing technology has been demonstrated to be a powerful tool to analyze the mutation rate and specificity across the genome.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank all current and former Kunkel Lab members for their work and discussions related to protocol and reused data presented here. This work was supported by Project Z01 ES065070 to T.A.K. from the Division of Intramural Research of the National Institutes of Health (NIH), National Institute of Environmental Health Sciences (NIEHS).
References
- Joyce CM. Choosing the right sugar: how polymerases select a nucleotide substrate. Proceedings of the National Acaddemy of Sciences U S A. 1997;94(5):1619–1622. doi: 10.1073/pnas.94.5.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nick McElhinny SA, et al. Abundant ribonucleotide incorporation into DNA by yeast replicative polymerases. Proceedings of the National Acaddemy of Sciences U S A. 2010;107(11):4949–4954. doi: 10.1073/pnas.0914857107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks JL, et al. RNase H2-initiated ribonucleotide excision repair. Molecular Cell. 2012;47(6):980–986. doi: 10.1016/j.molcel.2012.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Breaker RR. Kinetics of RNA Degradation by Specific Base Catalysis of Transesterification Involving the 2'-Hydroxyl Group. Journal of the American Chemical Society. 1999;121(23):5364–5372. [Google Scholar]
- Nick McElhinny SA, et al. Genome instability due to ribonucleotide incorporation into DNA. Nature Chemical Biology. 2010;6(10):774–781. doi: 10.1038/nchembio.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JS, Kunkel TA. Ribonucleotides in DNA: origins, repair and consequences. DNA Repair (Amst) 2014;19:27–37. doi: 10.1016/j.dnarep.2014.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JS, Lujan SA, Kunkel TA. Processing ribonucleotides incorporated during eukaryotic DNA replication. Nature Reviews Molecular Cell Biology. 2016;17(6):350–363. doi: 10.1038/nrm.2016.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones ME, Thomas SM, Rogers A. Luria-Delbruck fluctuation experiments: design and analysis. Genetics. 1994;136(3):1209–1216. doi: 10.1093/genetics/136.3.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q. A new practical guide to the Luria-Delbruck protocol. Mutat Research. 2015;781:7–13. doi: 10.1016/j.mrfmmm.2015.08.005. [DOI] [PubMed] [Google Scholar]
- Sambrook J. Molecular Cloning: a Laboratory Manual. Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Miyabe I, Kunkel TA, Carr AM. The major roles of DNA polymerases epsilon and delta at the eukaryotic replication fork are evolutionarily conserved. PLoS Genetics. 2011;7(12):e1002407. doi: 10.1371/journal.pgen.1002407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan SA, Williams JS, Clausen AR, Clark AB, Kunkel TA. Ribonucleotides are signals for mismatch repair of leading-strand replication errors. Molecular Cell. 2013;50(3):437–443. doi: 10.1016/j.molcel.2013.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JS, et al. Topoisomerase 1-mediated removal of ribonucleotides from nascent leading-strand DNA. Molecular Cell. 2013;49(5):1010–1015. doi: 10.1016/j.molcel.2012.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen AR, et al. Tracking replication enzymology in vivo by genome-wide mapping of ribonucleotide incorporation. Nature Structural & Molecular Biology. 2015;22(3):185–191. doi: 10.1038/nsmb.2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL. Methods for determining spontaneous mutation rates. Methods Enzymology. 2006;409:195–213. doi: 10.1016/S0076-6879(05)09012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JS, et al. Evidence that processing of ribonucleotides in DNA by topoisomerase 1 is leading-strand specific. Nature Structural & Molecular Biology. 2015;22(4):291–297. doi: 10.1038/nsmb.2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov YI, Shcherbakova PV, Kunkel TA. In vivo consequences of putative active site mutations in yeast DNA polymerases alpha, epsilon, delta, and zeta. Genetics. 2001;159(1):47–64. doi: 10.1093/genetics/159.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nick McElhinny SA, Kissling GE, Kunkel TA. Differential correction of lagging-strand replication errors made by DNA polymerases {alpha} and {delta} Proceedings of the National Acaddemy of Sciences U S A. 2010;107(49):21070–21075. doi: 10.1073/pnas.1013048107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AB, Lujan SA, Kissling GE, Kunkel TA. Mismatch repair-independent tandem repeat sequence instability resulting from ribonucleotide incorporation by DNA polymerase epsilon. DNA Repair (Amst) 2011;10(5):476–482. doi: 10.1016/j.dnarep.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan SA, et al. Mismatch repair balances leading and lagging strand DNA replication fidelity. PLoS Genetics. 2012;8(10):e1003016. doi: 10.1371/journal.pgen.1003016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijns MA, et al. Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell. 2012;149(5):1008–1022. doi: 10.1016/j.cell.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan SA, et al. Heterogeneous polymerase fidelity and mismatch repair bias genome variation and composition. Genome Research. 2014;24(11):1751–1764. doi: 10.1101/gr.178335.114. [DOI] [PMC free article] [PubMed] [Google Scholar]