

Abstract
Bulk autophagy is characterized by the sequestration of large portions of cytoplasm into double/multi-membrane structures termed autophagosomes. Here a simple protocol to monitor this process is described. Moreover, typical results and experimental validation of the method under autophagy-inducing conditions in various types of cultured mammalian cells are provided. During bulk autophagy, autophagosomes sequester cytosol, and thereby also soluble cytosolic proteins, alongside other autophagic cargo. LDH is a stable and highly abundant, soluble cytosolic enzyme that is non-selectively sequestered into autophagosomes. The amount of LDH sequestration therefore reflects the amount of bulk autophagic sequestration. To efficiently and accurately determine LDH sequestration in cells, we employ an electrodisruption-based fractionation protocol that effectively separates sedimentable from cytosolic LDH, followed by measurement of enzymatic activity in sedimentable fractions versus whole-cell samples. Autophagic sequestration is determined by subtracting the proportion of sedimentable LDH in untreated cells from that in treated cells. The advantage of the LDH sequestration assay is that it gives a quantitative measure of the autophagic sequestration of endogenous cargo, as opposed to other methods that either involve ectopic expression of sequestration probes or semi-quantitative protease protection analyses of autophagy markers or receptors.
Keywords: Biology, Issue 137, LDH, lactate dehydrogenase, sequestration, autophagy, autophagic cargo, phagophore, autophagosome, Bafilomycin, A1, LC3
Introduction
Autophagy (Greek for "self-eating") is an evolutionary conserved process for vacuolar/lysosomal degradation of intracellular material. Upon discovery of the autophagy-related ("ATG") genes, which are important for autophagy in yeast and humans, and the realization that autophagy plays a significant role in human health and disease (acknowledged by the 2016 Nobel Prize in Medicine or Physiology to Yoshinori Ohsumi), autophagy has quickly become one of the most intensely studied processes in cell biology1,2.
Macroautophagy (hereafter referred to as "autophagy") is characterized by the expansion and folding of intracellular membrane cisternae ("phagophores") into sealed, double- or multi-membrane structures ("autophagosomes") that effectively sequester the enwrapped material from the rest of the cytoplasm. Upon fusion of autophagosomes with lysosomes, the inner autophagosomal membrane and the sequestered cargo is degraded and recycled. Autophagosomes can sequester cytoplasmic material in both random (non-selective autophagy) and selective (selective autophagy) manners. Bulk autophagy most likely represents a mix of non-selective and selective autophagy.
In the 1960's and 70's ("the morphological era" of autophagy research), autophagic sequestration was mainly assessed through ultrastructural analyses. In the 1980's and beginning of the 1990's ("the biochemical era") Per Seglen and co-workers — who studied autophagy in primary rat hepatocytes — developed the first methods to quantitatively measure autophagic sequestration activity3. Using these assays, Seglen defined and characterized different steps of the autophagic-lysosomal pathway4,5, discovered and coined the amphisome6 (the product of endosome-autophagosome fusion), and was the first to describe the role of protein phosphorylation in autophagy regulation7. However, after the discovery of the ATGs in the 1990's ("the molecular era") and the first characterization of a mammalian ATG8 protein, microtubule-associated protein 1A/1B-light chain 3 (LC3) in 20008, the use of ATG proteins as markers for the autophagic process quickly gained popularity, and the older and more laborious biochemical methods were left behind. In fact, over the last 18 years, western blot and fluorescence microscopy analyses of LC3 have become the by far most popular (and in many cases the only) means of studying autophagy in mammalian cells. The advantage is the relative ease by which these methods can be carried out. The disadvantage is that one is studying a cart component (LC3) rather than actual autophagic cargo. This is a rather serious disadvantage, because the relationship between the states and/or flux of LC3 through the pathway versus the sequestration and flux of cargo is highly unclear. In fact, we have shown that bulk cargo flux can be maintained at high levels under conditions where there is no LC3 flux, despite the presence of conjugated LC3 in the cells9. Moreover, we demonstrated that bulk autophagy is unaffected by efficient LC3 depletion, and thus likely is LC3-independent9. This finding has later been confirmed by LC3 knock-out studies10,11, which also indicate that Parkin-dependent mitophagy (the selective autophagy of mitochondria) is independent of LC310,11.
In summary, there is a clear need for cargo-based assays to monitor autophagic activity. Optimally such assays should be broadly applicable, well-defined, and easy to perform. Over the last few years we have taken a particular interest in the LDH sequestration assay, which was developed by Per Seglen in the 1980's12, and is based on measuring the transfer of cytosolic LDH to sedimentable, autophagic vacuole-containing cell fractions. LDH is a stable, soluble cytosolic protein that is readily co-sequestered when phagophores enwrap cytoplasmic cargo. Sequestration of LDH is therefore a general measure of autophagic sequestration. LDH is exclusively degraded by the autophagic-lysosomal pathway12. Thus, in the presence of lysosomal degradation inhibitors, e.g., bafilomycin A1 (Baf)13, experimental treatment effects directly reflect alterations in autophagic sequestration activity. In the absence of degradation inhibitors, the net effect of alterations in LDH sequestration and degradation can be measured.
The LDH sequestration assay is broadly applicable, since LDH is highly and ubiquitously expressed in all cell types, and LDH levels can be accurately quantified by an enzymatic assay14,15. However, the original protocol12 — established in primary rat hepatocytes — was rather time-consuming and required a high amount of starting material as well as a custom-made electric discharge capacitor. In a step-wise manner, we have gradually transformed the assay into an easy and versatile method. First, the original protocol was adapted for use in mammalian cell lines16. Second, the method was substantially downscaled3,9. Third, several steps in the protocol were eliminated, including a laborious density cushion step17. This simultaneously enabled an even further downscaling of the method, from the original starting point of using a 10 cm plate per sample16 to using a single well from a 12-well plate per sample (i.e., approximately 15-fold less starting material)17. Fourth, we identified a commercial electroporation apparatus that could replace the custom-made electric discharge capacitor17.
Here our most up-to-date protocol of the LDH sequestration assay, which includes some further simplifications of the method as compared to the previously published17 is presented. Furthermore, a set of typical results obtained in a number of different cell types is shown, and importantly, multiple lines of experimental validations of the method using pharmacological as well as genetic knockdown and knockout approaches are provided. For an overall flow scheme of the whole protocol, see Figure 1.
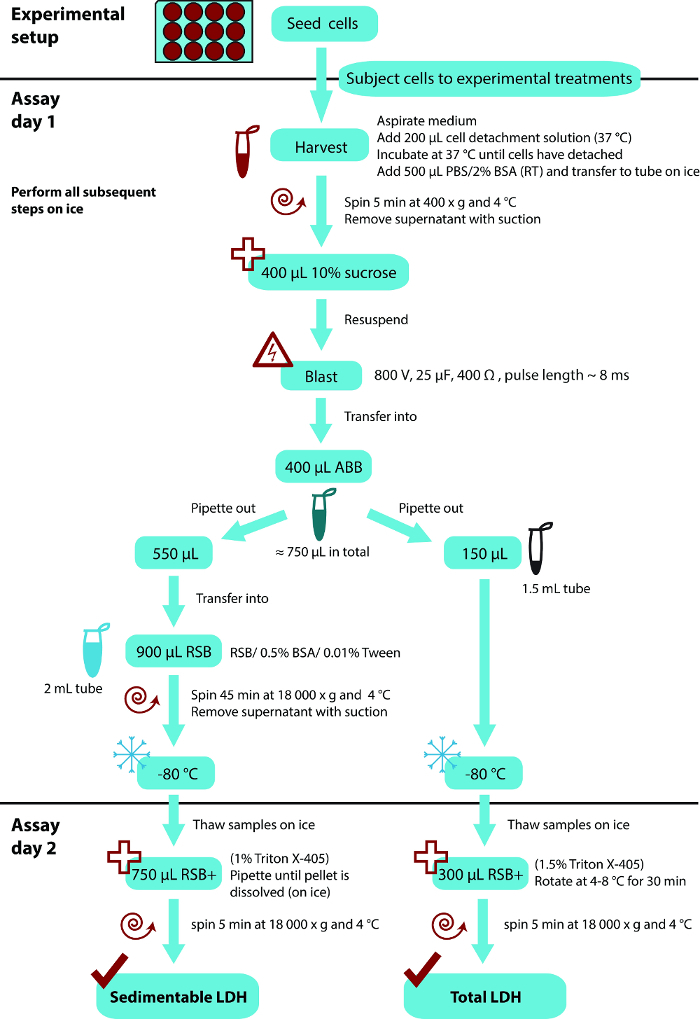
Protocol
1. Cell Seeding and Treatment
- Culture adherent cells in 75 cm2 tissue culture flasks in a humidified incubator with 5% CO2 at 37 °C, using the preferred culture medium for the cell type in question. Allow the cells to grow until they reach a near-confluent cell layer. NOTE: Use RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) for LNCaP, HEK293, mouse embryonic fibroblasts (MEFs), BJ, MCF-7, and RPE-1 cells.
- Wash the cells with 3 mL 37 °C phosphate-buffered saline (PBS), pH 7.4. Replace the PBS with 3 mL 0.25% (w/v) Trypsin-ethylenediaminetetraacetate (EDTA), and incubate the flask in a humidified incubator with 5% CO2 at 37 °C until the cells detach (2–5 min).
- Resuspend the detached cells with 7 mL culture medium containing 10% FBS. Mix a 10 µL cell suspension aliquot with 10 µL 0.4% Trypan Blue in a microcentrifuge tube, using a 0.5–20 µL pipette tip. Use the same pipette tip to immediately fill a counting chamber slide, and count the cells in an automated cell counter.
- Prepare a suitable dilution (see note below) of the cell suspension from step 1.1.2 using culture medium containing 10% FBS, and seed 1 mL of the diluted cell suspension in each well of a 12-well tissue culture plate (surface area ~3.8 cm2) using aseptic technique. Allow growth in a humidified incubator with 5% CO2 at 37 °C until the desired cell density has been reached, e.g., 60–90% confluence at harvest. NOTE: The appropriate dilution of the cell suspension that will give the desired cell confluency at harvest will vary from cell type to cell type, as well as according to the duration and type of experimental treatments. Thus, this must be empirically assessed in each case.
- For experiments that are to be both treated and harvested 2 days after seeding, seed 2.5 x 105 LNCaP, HEK293, or MCF-7 cells, 5 x 104 MEFs, 4 x 105 BJ, or 1.5 x 105 RPE-1 cells in each well of the 12-well plate.
- For cells that adhere loosely, coat the plates with the type of coating recommended for the cell type in question. For LNCaP (and HEK293) cells use plates coated with poly-D-lysine (PDL).
- To that end, add 500 µL PDL at 2.5 µg/mL in sterile H2O to each well, and incubate the plates in a sterile environment for 30 min at room temperature (20–25 °C). Remove the PDL with suction, and wash each well briefly with 1 mL sterile H2O. NOTE: Generally, step 1.2.1 is done without any experimental treatments. However, if performing RNAi, it may be convenient to start a reverse transfection with the seeding9.
- Perform experimental treatments in duplicate or triplicate wells per condition.
- For example, treat the cells with 50 nM of the mTOR-inhibitor Torin1, which generally is an efficient inducer of autophagic sequestration, or subject the cells to acute serum- and amino acid starvation by washing the cells with 1 mL of amino acid-free Earle's Balanced Salt Solution (EBSS) medium, and subsequently incubate the cells in 1 mL of EBSS in a humidified incubator with 5% CO2 at 37 °C.
- Leave one set of wells untreated in order to define background levels of sedimentable LDH.
- Add a saturating amount of the post-sequestration inhibitor bafilomycin A1 (Baf)3,13,16,18 in the absence or presence of the experimental treatments, 3–4 h before cell harvest. Incubate the cells in a humidified incubator with 5% CO2 at 37 °C.
- Use 100 nM Baf for LNCaP, HEK293, BJ, MCF-7 and RPE-1 cells, and 10 nM Baf for MEFs.
- For experimental treatments that have a duration of only 3–4 h (like those exemplified in step 1.3.1 typically have), add Baf simultaneously with the treatments. For longer experimental treatments, wait until 3-4 h before harvest, and add 2 µL of a 500x concentrated Baf stock directly into the medium.
- Mix by agitating the plate immediately after the addition of Baf. At this point it is also recommendable to add macroautophagic sequestration inhibitors as controls, e.g., 10 mM of the pan-phosphoinositide 3-kinase (PI3K) inhibitor 3-methyl adenine (3MA)19, or 10 µM of the selective PI3K class III inhibitor SAR-40520.
2. Cell Harvest and Preparation for Electrodisruption
At the end of the treatment period, aspirate the medium with suction and add 200 µL cell detachment solution (pre-heated to 37 °C) to each well. Incubate at 37 °C until the cells detach (typically around 5 min). NOTE: Whereas 0.25% (w/v) Trypsin-EDTA may be used instead of the cell detachment solution, the latter contains DNase, which helps reduce the viscosity of the detached cells. As long as the medium is thoroughly aspirated, it is not necessary to wash the cells before addition of Trypsin-EDTA or cell detachment solution.
Add 500 µL room temperature (20-25 °C) PBS, pH 7.4, containing 2% (w/v) bovine serum albumin (BSA) to each well, and resuspend with the pipette until no cell clumps are visible. Immediately transfer the cell suspension to 1.5 mL microcentrifuge tubes on ice. NOTE: Unless otherwise stated, perform all subsequent steps on ice.
Sediment the cells by centrifugation at 400 x g for 5 min at 4 °C.
Thoroughly aspirate the supernatant (with suction) to leave the cell pellets as dry as possible.
Add 400 µL 10% (w/v) sucrose (in ultrapure H2O) to each tube.
3. Plasma Membrane Electrodisruption and Separation of Sedimentable- and Total-cell Fractions
Resuspend the cell pellet with a pipette to obtain a near single-cell suspension, and transfer it to a 4 mm electroporation cuvette. NOTE: Pipetting up-and-down ~10–15 times, using a 100–1,000 µL pipette tip, is usually sufficient.
Place the cuvette in an exponential decay wave electroporator, and discharge a single electric pulse at 800 V, 25 µF, and 400 Ω; these settings produce a pulse of ~8 ms duration.
- Use a new pipette tip to transfer the cell disruptate to a 1.5 mL microcentrifuge tube containing 400 µL ice-cold phosphate-buffered sucrose solution (100 mM sodium monophosphate, 2 mM dithiothreitol (DTT), 2 mM EDTA, and 1.75% sucrose, pH 7.5), and mix briefly by pipetting.
- Optional: To verify efficient plasma membrane electrodisruption17, mix 10 µL of the diluted cell disruptate from step 3.3 with 10 µL 0.4% Trypan Blue in a 1.5 mL microcentrifuge tube. Transfer to a counting chamber and verify that the percentage of Trypan Blue positive cells is >99%.
- Leave the sample in the counting chamber for 30 min at room temperature (20–25 °C), and verify that the percentage of Trypan Blue positive cells has remained >99%.
- Optional: To verify that the electrodisruption has not been too harsh, that is, it has not disrupted intracellular organelles, perform steps 1.1–3.3 as described above, but use a larger starting material (a well from a 6-well plate with an ~80% confluent cell layer), and use 150 µL 10% sucrose in step 2.5 and 150 µL phosphate-buffered sucrose solution without DTT in step 3.3.
- Use a pipette to carefully layer 200 µL of the diluted cell disruptate solution on top of a 1.2 mL density cushion of phosphate-buffered 8% (w/v) density gradient medium (e.g, 8% Nycodenz, 50 mM sodium phosphate, 2.2% sucrose, 1 mM EDTA) in a 2 mL centrifuge tube. Centrifuge at 20,000 x g for 45 min at 4 °C in a microcentrifuge with soft-mode function (for gentle acceleration and deceleration), and carefully put the tubes on ice.
- Carefully remove 60 µL of the ~200 µL top fraction, making sure not to pick up any density gradient medium solution, and transfer to a fresh microcentrifuge tube. NOTE: This should contain cytosol of exceptional purity, termed "cell sap"21.
- Test the purity of the fraction obtained in the above step, by performing western blot analyses of organelle-contained proteins, using standard techniques and 4–20% gradient gels16.
- Perform for example immunoblotting for cathepsin B21, cytochrome c, and protein disulphide isomerase, to verify that the electric shock in step 3.2 has not disrupted lysosomes, mitochondria, or endoplasmic reticulum, respectively, and immunoblot for LDH to verify presence of a cytosolic protein in the cell sap.
- In parallel, perform immunoblotting on protein extracts made from the total cell disruptate solution16 to confirm that the antibodies used can detect the organelle-contained proteins that are being assessed.
Repeat steps 3.1–3.3 for each sample.
Remove 550 µL from each diluted cell disruptate solution (obtained at step 3.3) to 2 mL microcentrifuge tubes containing 900 µL ice-cold resuspension buffer (50 mM sodium monophosphate, 1 mM DTT, 1 mM EDTA, and 5.9% sucrose, pH 7.5) supplemented with 0.5% BSA and 0.01% Tween-20, and mix briefly by pipetting.
Centrifuge at 18,000 x g for 45 min at 4 °C to produce pellets containing "sedimented LDH". Thoroughly aspirate the supernatant (with suction) to leave the pellets as dry as possible. Place the samples in a -80 °C freezer.
Transfer 150 µL from each diluted cell disruptate solution (obtained at step 3.3) to new tubes, and place the samples in a -80 °C freezer. Use these samples to determine the "total LDH" levels in the cells. NOTE: At this point the experiment can be paused for as long as desired.
4. LDH Extraction and Measurement of LDH Enzymatic Activity
Thaw the "sedimented LDH" (from step 3.6) and "total LDH" samples (from step 3.7) on ice.
Add 300 µL of ice-cold resuspension buffer containing 1.5% Triton X-405 to the "total LDH" samples (yielding a final Triton X-405 concentration of 1%). Rotate the samples on a roller in a cold room (4–8 °C) for 30 min.
Add 750 µL of ice-cold resuspension buffer with 1% Triton X-405 to the "sedimented LDH" samples, and resuspend the pellets with a pipette until a homogenous solution is reached.
Centrifuge the samples from step 4.2 and 4.3 at 18,000 x g for 5 min at 4 °C to sediment undissolved cellular debris.
Mix 4 parts of cold 65 mM imidazole (pH 7.5)/0.75 mM pyruvate with one part of cold 65 mM imidazole (pH 7.5)/1.8 mM NADH to obtain a working solution that is stable for at least three weeks at 4 °C.
Mix 3–30 µL of the supernatants from step 4.4 with 200 µL of the step 4.5 working solution.
Determine the amount of LDH by measuring LDH enzymatic activity as the decline in nicotinamide adenine dinucleotide (reduced form) (NADH) absorbance at 340 nm at 37 °C compared to a standard with a known LDH concentration. Perform absorbance measurements until the reaction has approached completion, i.e. until the absorbance at 340 nm no longer changes with time. NOTE: This is the classical biochemical method to measure LDH activity. Although the current protocol performs the reaction at 37 °C, it can also be performed at room temperature (20–25 °C), which is advisable if doing manual spectrophotometry. The current protocol uses a robotic multianalyzer instrument, which in an automated fashion mixes samples with working solution in a 96-well plate, and measures the absorbance at 340 nm at 37 °C every 20 s for 3 min. Thereafter, the instrument software calculates the concentration of LDH, expressed as Units (U)/L, by comparing the slope of the absorbance measurements over time compared to a standard curve obtained through calibration with a standard of known LDH concentration. The linear range of detection by this approach is 30–1,500 U/L. As an alternative, a wide variety of commercially available kits to measure LDH exist. Some of them are based on coupling the enzymatic reaction to the generation of colorimetric or fluorescent products, enabling detection by other means than UV spectrophotometry, and with other linear ranges of detection.
5. Calculation of LDH Sequestration
Calculate the percentage of sedimented LDH to total LDH for each sample, taking the dilutions and sampling into account: Sedimented LDH (%) =
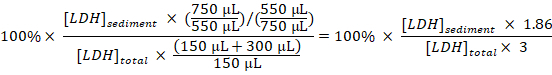 NOTE: During steps 3.1–3.3 approximately 50 µL is lost due to transfer into and out of the electroporation cuvette. Thus, calculate from having a total of 750 µL (instead of 800 µL) of diluted cell disruptate in step 3.3.
NOTE: During steps 3.1–3.3 approximately 50 µL is lost due to transfer into and out of the electroporation cuvette. Thus, calculate from having a total of 750 µL (instead of 800 µL) of diluted cell disruptate in step 3.3.Subtract the percentage of sedimented LDH obtained in the samples from untreated cells (step 1.3.2) from the percentage of sedimented LDH obtained in samples from experimentally treated cells, and divide by the treatment time with Baf to obtain the percentage of sequestered LDH per hour in the sampling period: Sequestered LDH (%/h) =
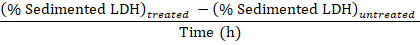
Representative Results
Using the protocol described here, bulk autophagic sequestration activity in a number of different mammalian cell lines, including LAPC4, DU145, Huh7, PNT2A, HeLa, VCaP, H3122, Hec1A, MCF-7, T47D, U2OS, PC3, G361, mouse embryonic fibroblasts (MEFs), RPE-1, HEK293, BJ, and LNCaP cells was measured. Sequestration was assessed under basal conditions (in complete, nutrient-rich medium), or in cells acutely starved for serum and amino acids (a bona fide autophagy-inducing condition22). The results indicated that LDH sequestration activity under starvation conditions varies extensively across different cell lines, ranging from barely detectable levels in LAPC4, DU145, Huh7 and PNT2 cells (data not shown) to ~1.6%/h in LNCaP cells (Figure 2A). The rate observed in starved primary rat hepatocytes is higher than in the cell lines mentioned above, and typically ranges from 2.5–4%/h23. Under basal conditions, LDH sequestration was virtually undetectable in half of the cell lines tested, and ranged from ~0.2%/h to ~0.5%/h in the other half (data not shown). In the cell lines that show detectable basal autophagic sequestration activity, acute serum and amino acid starvation typically induces a 3–4 fold increase in LDH sequestration rate (see for example the LNCaP experiment shown in Figure 2A). In the tested cell lines (mentioned above), the background percentage of sedimented LDH obtained in samples from untreated cells (step 1.3.2) is typically around 2–3%. From 22 independent experiments performed in various cell lines, the intra-experimental coefficient of variation (CV) between sedimented LDH values (% sedimented LDH) of treatment replicates was 5.8% ± 1.7% (average% CV ± standard deviation), ranging from 3.0–9.0%. Together, these numbers give an indication of what to expect when performing the LDH sequestration assay in mammalian cells.
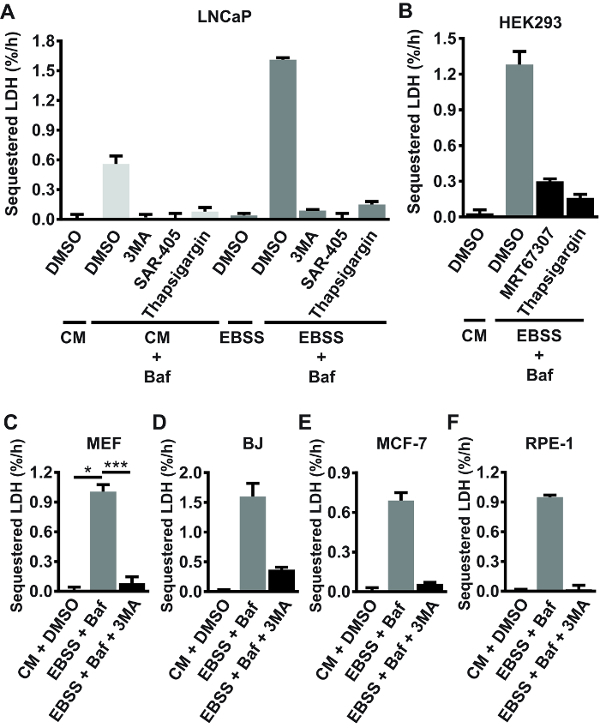
Use of chemical inhibitors as well as genetic knockdown and knockout approaches, extensively verifies that the LDH sequestration assay reliably measures autophagic sequestration activity. Autophagosome formation requires active PI3K class III (PIK3C3) and Unc-51 like autophagy activating kinase (ULK)24, as well as balance in intracellular Ca2+ homeostasis16. As shown in Figure 2A, both basal and starvation-induced LDH sequestration is completely abolished by the pan-PI3K inhibitor 3MA, the selective PIK3C3 inhibitor SAR-40520, or the ER Ca2+ pump inhibitor thapsigargin (TG)25 in LNCaP cells. LDH sequestration under starvation conditions (switch to amino acid-free Earle's Balanced Salt Solution (EBSS) medium) is also strongly reduced by TG, or by the ULK-inhibitor MRT67307 in HEK293 cells (Figure 2B). Moreover, starvation-induced LDH sequestration is consistently inhibited by 3MA in MEFs (Figure 2C), BJ (Figure 2D), MCF-7 (Figure 2E), and RPE-1 (Figure 2F) cells. Generally, incubation of cells in EBSS medium alone (i.e., in the absence of Baf or other post-sequestration inhibitors) does not lead to any measurable accumulation of sequestered LDH (see for example the LNCaP experiment shown in Figure 2A). This is likely because acute amino acid starvation leads to accelerated autophagic-lysosomal flux, resulting in rapid and continuous degradation of the sequestered LDH.
Next, various ATG gene knockout (KO) MEFs were employed to test whether LDH sequestration requires autophagy-related genes reported to be essential for autophagosome formation. Indeed, starvation-induced LDH sequestration is abolished in ATG5 KO MEFs26 (Figure 3A), confirming our previous findings9. Moreover, as shown in Figure 3B and 3C, starvation-induced LDH sequestration is blunted also in ATG7 KO MEFs27 and ATG9A KO MEFs28.
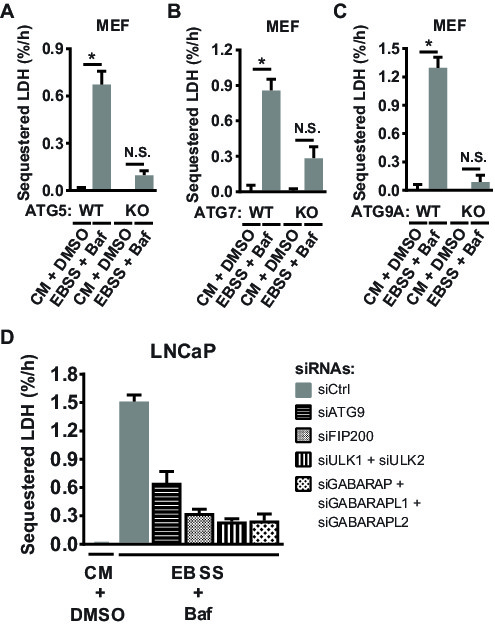
Finally, the LDH sequestration assay was tested in relation to whether RNAi-mediated silencing of key ATG gene transcripts would inhibit starvation-induced sequestration activity. Indeed, transfection with an ATG9A-targeting siRNA, or combined targeting of ULK1 and ULK2, strongly reduced LDH sequestration under starvation conditions in LNCaP cells (Figure 3D). Moreover, we confirmed our previous findings3,9 that targeting of focal adhesion kinase family interacting protein of 200 kDa (FIP200) or combined targeting of the γ-aminobutyric acid type A (GABAA) receptor-associated protein (GABARAP) family members inhibits starvation-induced LDH sequestration (Figure 3D).
Figure 1: Overall flow scheme of the LDH sequestration protocol. The protocol is based on a 12-well tissue culture plate format, using the indicated volumes per sample (from one well). The assay protocol can conveniently be divided into two separate working days, as indicated. However, it is also possible to perform the whole procedure in one day. Abbreviations: ABB, after blast buffer (see step 3.3 in the protocol for ingredients); RSB, resuspension buffer (see step 3.5 in the protocol for ingredients). Please click here to view a larger version of this figure.
Figure 2: Validation of the LDH sequestration assay using autophagy-inhibiting compounds. (A) LNCaP cells were either left untreated (for the purpose of background subtraction: see step 1.3.2) in complete growth medium (CM; RPMI 1640 + 10% fetal bovine serum (FBS)), or they were treated with DMSO vehicle (0.1%) or 100 nM Baf in CM or in serum- and amino acid free medium (EBSS). Additionally, some of the cells were treated with 3MA (10 mM), SAR-405 (10 µM), or Thapsigargin (300 nM), as indicated. After 3 h of treatment, the cells were harvested, and LDH sequestration rates were determined as detailed in the current protocol. (B) HEK293 cells were treated with DMSO vehicle (0.1%) in CM, or with 100 nM Baf in EBSS, as indicated. Additionally, some of the cells received 10 µM MRT67307 (an ULK inhibitor) or 300 nM Thapsigargin. LDH sequestration rates were determined after 3 h of treatment. (C-F) MEF (C), BJ (D), MCF-7 (E), or RPE-1 (F) cells were treated with DMSO vehicle (0.1%) in CM, or with Baf (10 nM in C, 100 nM in D-F) in EBSS with or without 10 mM 3MA, as indicated. LDH sequestration rates were determined after 3 h of treatment. A, B, D, E, and F show mean values from three biological replicates (triplicate wells) from one experiment (n = 1), with error bars representing standard deviation. C shows the mean values from three independent experiments (n = 3), with error bars representing standard error of the mean. *p <0.05, ***p <0.001, repeated samples one-way ANOVA.
Figure 3: Validation of the LDH sequestration assay by knockout (A-C) or knockdown (D) approaches. (A-C) ATG5 wild type (WT) or knockout (KO) MEFs (A), ATG7 WT or KO MEFs (B), or ATG9A WT or KO MEFs (C) were treated with DMSO vehicle (0.01%) in CM, or with 10 nM Baf in EBSS, as indicated. LDH sequestration rates were determined after 3 h of treatment. Mean values from four (A; n = 4) or three (B and C; n = 3) independent experiments, with error bars representing standard error of the mean. *p <0.05, repeated samples two-way ANOVA. N.S., not significant. (D) LNCaP cells were reverse transfected with 5 nM of a nontargeting control siRNA (siCtrl), or with 5 nM each of ATG9A-, FIP200-, ULK1-, ULK2-, GABARAP-, GABARAPL1-, or GABARAPL2-targeting siRNA oligoes, as indicated. After 48 h, the cells were either treated with DMSO vehicle (0.1%) in CM, or with 100 nM Baf in EBSS, as indicated. LDH sequestration rates were determined after 3 h of treatment. Shown are mean values from three biological replicates (triplicate wells) from one experiment (n = 1), with error bars representing standard deviation.
Discussion
In summary, the protocol described here represents a reliable and widely applicable method to monitor bulk autophagic sequestration activity in mammalian cells. Compared to the original method12,16, we have removed a number of unnecessary steps, simplified several of the remaining steps, and introduced a substantial downscaling. As a result, the protocol is greatly improved in relation to cost- and time-efficiency, and the same amount of samples can now be handled in less than half the time as compared to the original protocol. For 24 samples, steps 2–3 (day 1) requires approximately ½ h of preparation plus 3 h of efficient work, whereas step 4 (day 2) can be performed in ~2 h (time estimates given that all the necessary buffers are prepared beforehand). Since the assay on day 1 usually is preceded by cell treatments, including a 3–4 h incubation with Baf or other inhibitors of autolysosomal LDH degradation, it is convenient to divide the assay in two consecutive days. However, it is also possible to perform the whole assay in one and the same day. In that case, it would save time to snap-freeze the samples in liquid nitrogen in step 3.6 and 3.7. Although not tested with the current protocol, it is likely that the freeze-thaw step may be skipped altogether, since the more elaborate version of the assay in primary rat hepatocytes has been done without this step23.
The protocol presented here can be performed with relative ease. Of note, it is important to be accurate in the pipetting, since the assay includes several sampling and dilution steps. Moreover, the supernatant aspirations should be done thoroughly, so that minimal amounts of ions are present in the sucrose cell suspension in step 2.5, and so that as little cytosolic LDH as possible is contaminating the sedimented material in step 3.6. The assay as described here is flexible for a wide range of starting material. For example, in LNCaP cells, we have successfully used the assay with a range of different amounts of cells at harvest, spanning up to a 10-fold difference (from 2.5 x 105 cells to 2.5 x 106 cells). The minimum starting material needed is defined by how sensitive the detection method of LDH enzymatic activity is. The protocol can very likely be scaled substantially further down, by scaling down the volumes used at steps 2.5, 3.3, 3.5, 3.7, 4.2, and 4.3.
The most critical factor and technical challenge with the assay is the electrodisruption step. The strong, but brief, electric shock of cells in ion-free, isotonic solution results in a uniform and selective disruption of the plasma membrane, while leaving intracellular structure and organelles (including autophagic vacuoles) intact12,29. When using adherent cells, it is essential that they tolerate the enzymatic and mechanical treatments in steps 2.1–3.1 (cell detachment, centrifugation, and resuspension). For step 3.1, it is not critical to obtain a perfectly single-cell suspension. For example, this is very hard to achieve with LNCaP cells. Nevertheless, successful electrodisruption in near 100% of the cells is always obtained, even within cell clumps containing several dozens of physically attached cells. When testing a new cell type, it is advisable to verify the efficiency of the electrodisruption (see step 3.3.1)17. Close to 100% of the cells should be permanently Trypan Blue positive after the electrodisruption step17. If this is not the case, the electroporator settings must most likely be altered. Using the assay in 20 different mammalian cell types, we have never had to change the electroporator settings. Thus, once the right settings have been found for one mammalian cell line, it is likely to work for all other types of mammalian cells. To verify that the electrodisruption conditions are not too harsh, i.e.,that only the plasma membrane, and not the membranes of intracellular organelles have been disrupted, follow step 3.3.2.
Bulk autophagy is performed by autophagosomes that co-sequester substantial amounts of cytosol along with other cargo. This may happen via purely non-selective autophagy of portions of the cytoplasm, or in a manner that represents a mix of selective and non-selective autophagy. To date it is not clear whether or to which degree selective and non-selective autophagy co-exist as two different modes of autophagy, or whether autophagosomes as a rule simultaneously sequester cargo in both selective and non-selective manners. Importantly, however, a recent study reported that activators of selective autophagy induce a similar increase in bulk cytosolic cargo sequestration as in the sequestration of the specific cargo30. Results from our own laboratory are in agreement with this (our unpublished results). Analyzing the sequestration of soluble cytosolic proteins (e.g., LDH) therefore likely has the potential to detect alterations in macroautophagic sequestration activity under many, if not all conditions. However, although it still remains to be proven, the possibility cannot be excluded that certain types of conditions uniquely induce a type of selective-exclusive autophagy where the cargo is so tightly wrapped by the phagophore that even cytosol is excluded from being sequestered into the autophagosome. To probe for whether such condition may exist, bulk autophagy assays like the LDH sequestration assay should be run in parallel with selective autophagy assays.
One of the main advantages of the LDH sequestration assay is that it measures the sequestration of endogenous cargo, thus making it a broadly applicable method. Moreover, the assay measures sequestration activity in a highly quantitative manner. The LDH sequestration assay is an important tool for studying the mechanisms and regulation of autophagosome formation, since open phagophores cannot sequester LDH. It is very cumbersome and difficult, if not impossible, to assess whether autophagosome-like structures are sealed entities or not by electron microscopy. Another method that is being used to analyze whether autophagosomes are closed is to test the sensitivity of autophagy markers (e.g., LC3) or receptors (e.g., sequestosome 1 (p62/SQSTM1) or nuclear dot protein 52 (NDP52)) to proteases10,31,32. The disadvantage of this assay is that one is studying protease protection of cart components rather than autophagic cargo. Moreover, since the assay is based on western blotting, it is only semi-quantitative.
Whereas the ability to analyze autophagy of endogenous cargo is an advantage, an inherent limitation with the LDH sequestration assay, and any other assays that assess sequestration of endogenous cargo, is that a degradation inhibitor must be included in order to discern specific effects on autophagic sequestration rather than net effects of sequestration and degradation. Efficient inhibitors of LDH degradation include proton pump inhibitors like Baf or concanamycin A3,16,18, and lysosomotropic agents like chloroquine and ammonium chloride33. The protease inhibitor leupeptin works well in primary rat hepatocytes23, but is generally inefficient in the mammalian cell lines we have tested. We routinely use Baf13, which acts rapidly and is highly efficient in both nonmalignant and malignant cells. However, it is important to keep in mind that none of the LDH degradation inhibitors are entirely specific, and putative effects of the inhibitors on autophagy cannot be excluded. To minimize the risk of non-specific influence, it is recommendable to use concentrations of the inhibitor that are just at saturating levels, and to include the inhibitor only for the last few hours (3–4 h) of the experiment. The saturating concentration of Baf should be determined for each cell type. As a guide, 50–100 nM is saturating for most of the cultured mammalian cell lines we have tested, whereas some cell types such as MEFs require only 10 nM Baf for a full block in LDH degradation.
An obvious limitation with the LDH sequestration assay is that it can only be performed on living cells, thus excluding its use for analysis of autophagy in fixed cells and tissues. On the other hand, there are currently no assays established that can measure functional autophagic activity in fixed cells. Although not tested with the current protocol, it should be entirely possible to use the LDH sequestration assay to assess in vivo autophagic sequestration activity in experimental organisms. The limitation would be that the organism must tolerate treatment with an inhibitor of lysosomal LDH degradation, and that the time period between such treatment and performance of the assay should be relatively short, to minimize potential non-specific effects of the inhibitor. Interestingly, an early study by Kominami et al., indicated that 3–6 h treatment of rats with leupeptin (intraperitoneal injection of 2 mg/100 g of body weight) is appropriate to observe efficient accumulation of autophagically sequestered LDH in liver subcellular fractions enriched for autophagic vacuoles34.
Ectopic expression of pH-sensitive fluorescent sequestration probes such as Rosella35 or Keima36 can be used to visualize autophagic sequestration without the need of including degradation inhibitors. Moreover, unlike the LDH sequestration assay, such approaches can be used to visualize autophagic sequestration in single cells. Additionally, fusion of the probe to organelle-targeting sequences can be utilized to monitor sequestration of specific organelles. Powerful microscopic platforms may also allow for high-throughput screening analyses. The disadvantages are that ectopic probe expression, as well as accumulation of the probe in the lysosomal system, may influence the autophagic pathway. Moreover, unlike the LDH sequestration assay, one is dependent on cells that can be efficiently transfected. Finally, whereas the LDH sequestration assay provides a straight-forward quantitative output of percentage sequestered cytosol, the image-based methods generally cannot provide this type of absolute quantitative output. It would be advisable to make use of both types of approaches in a complementary manner. Another excellent method to study autophagic sequestration without a need for degradation inhibitors is to introduce radiolabeled raffinose into cells by reversible electro-permeabilization3,23,37,38. Raffinose is a membrane-impermeant and metabolically inert sugar that is resistant to lysosomal enzymes. Therefore, its autophagic sequestration can be followed by separation of cytosolic from sedimentable cell fractions3,23,37,38. This approach is, however, more time-consuming than the LDH sequestration assay, and requires additional safety precautions due to the use of radioactivity. Finally, the end-result of bulk autophagic sequestration and unobstructed autophagic cargo flux, the degradation of long-lived proteins, can be accurately measured by a well-established protein labelling-based method39,40,41,42,43. However, unlike the LDH sequestration assay, this method does not provide a specific read-out for autophagic activity, since long-lived proteins are degraded also by other mechanisms than autophagy, most notably by the proteasomal system (whereas LDH sequestration only occurs as a result of autophagy). Therefore, a number of control treatments need to be routinely included, e.g., chemical inhibitors of autophagic-lysosomal or proteasomal degradation, and genetic interference with autophagy3,16.
The LDH sequestration assay can be compared with the commonly used LC3 flux assays, which measure LC3-II levels by western blotting or LC3 puncta formation by fluorescence imaging in the absence or presence of inhibitors of lysosomal LC3 degradation (Baf is most frequently used), or the transition of fluorescently tagged LC3 variants to acidic environments22. Although these LC3-based assays may provide useful information, they can be difficult to interpret, in particular because the relationship between the degree of LC3 flux and the amount and type of cargo flux is unknown. As mentioned in the introduction, bulk autophagy and Parkin-dependent mitophagy do not require LC3 family proteins9,10,11,44. Moreover, in starved rat hepatocytes, LDH sequestration and degradation proceeds uninterrupted during time periods where there is no autophagic-lysosomal LC3 flux9. Thus, in this case, cargo flux can be completely separated from LC3 flux. More research comparing LC3 flux with different kinds of cargo flux is needed to better interpret results obtained with LC3 flux assays.
In its present form, the LDH sequestration assay is comparable to western blotting in terms of the required hands-on time and costs. In terms of measuring bulk autophagy, it is at least as efficient as the raffinose-based sequestration assay or the long-lived protein degradation assay described above. For specific measurement of bulk autophagic sequestration activity and formation of closed autophagosomes, it is much more efficient and straight-forward than LC3 flux assays or protease protection assays of LC3 or autophagy receptors, since the latter assays measure cart components rather than actual cargo. Even though we have substantially improved the throughput of the LDH sequestration assay, it is far from a high-throughput assay, and it cannot compete with the efficiency of the image-based assays described above. However, both the image-based assays and the LDH sequestration assay have their advantages and disadvantages, and thus both types of assays have their own values. It is likely possible via future adjustments to make the LDH sequestration assay semi high-throughput. For example, it should be possible to perform electrodisruption in a 96-well format, and it is conceivable that cytosolic LDH could be separated from sequestered LDH by filtration instead of centrifugation, or that the centrifugation step can be performed in a 96-well format. Moreover, assays to measure LDH activity that are much more sensitive than that used in the current protocol have been developed, and are commercially available. Other interesting future potentials of the assay are its putative use in other cell types than mammalian cells, for instance in yeast or other unicellular organisms, or even plant cells, as well as its use for in vivo measurements of autophagic sequestration activities in tissues of experimental organisms.
In conclusion, we believe that the LDH sequestration assay in its improved and revived form will be an important tool in future autophagy-related research.
Disclosures
The authors have no conflict of interest.
Acknowledgments
This work was financially supported by the Research Council of Norway, the University of Oslo, the Anders Jahre Foundation, the Nansen Foundation, and the Legacy in the memory of Henrik Homan. We thank Dr. Noboru Mizushima for the ATG5+/+ MEFs and ATG5-/- MEFs, Dr. Masaaki Komatsu for the ATG7+/+ MEFs and ATG7-/- MEFs, and Dr. Shizuo Akira for the ATG9A+/+ MEFs and ATG9A-/- MEFs. We thank Frank Sætre for technical assistance, and Dr. Per O. Seglen for constructive methodological discussions.
References
- Rubinsztein DC, Frake RA. Yoshinori Ohsumi's Nobel Prize for mechanisms of autophagy: from basic yeast biology to therapeutic potential. J R Coll Physicians Edinb. 2016;46(4):228–233. doi: 10.4997/jrcpe.2016.403. [DOI] [PubMed] [Google Scholar]
- Mizushima N. The exponential growth of autophagy-related research: from the humble yeast to the Nobel Prize. FEBS Lett. 2017;591(5):681–689. doi: 10.1002/1873-3468.12594. [DOI] [PubMed] [Google Scholar]
- Seglen PO, et al. Macroautophagic cargo sequestration assays. Methods. 2015;75:25–36. doi: 10.1016/j.ymeth.2014.12.021. [DOI] [PubMed] [Google Scholar]
- Hoyvik H, Gordon PB, Seglen PO. Use of a hydrolysable probe, [14C]lactose, to distinguish between pre-lysosomal and lysosomal steps in the autophagic pathway. Exp Cell Res. 1986;166(1):1–14. doi: 10.1016/0014-4827(86)90503-3. [DOI] [PubMed] [Google Scholar]
- Plomp PJ, Gordon PB, Meijer AJ, Hoyvik H, Seglen PO. Energy dependence of different steps in the autophagic-lysosomal pathway. J Biol Chem. 1989;264(12):6699–6704. [PubMed] [Google Scholar]
- Gordon PB, Seglen PO. Prelysosomal convergence of autophagic and endocytic pathways. Biochem Biophys Res Commun. 1988;151(1):40–47. doi: 10.1016/0006-291x(88)90556-6. [DOI] [PubMed] [Google Scholar]
- Holen I, Gordon PB, Seglen PO. Protein kinase-dependent effects of okadaic acid on hepatocytic autophagy and cytoskeletal integrity. Biochem J. 1992;284:633–636. doi: 10.1042/bj2840633. Pt 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo j. 2000;19(21):5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szalai P, et al. Autophagic bulk sequestration of cytosolic cargo is independent of LC3, but requires GABARAPs. Exp Cell Res. 2015;333(1):21–38. doi: 10.1016/j.yexcr.2015.02.003. [DOI] [PubMed] [Google Scholar]
- Nguyen TN, et al. Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J Cell Biol. 2016. [DOI] [PMC free article] [PubMed]
- Pontano Vaites L, Paulo JA, Huttlin EL, Harper JW. Systematic analysis of human cells lacking ATG8 proteins uncovers roles for GABARAPs and the CCZ1/MON1 regulator C18orf8/RMC1 in macro and selective autophagic flux. Mol Cell Biol. 2017. [DOI] [PMC free article] [PubMed]
- Kopitz J, Kisen GO, Gordon PB, Bohley P, Seglen PO. Nonselective autophagy of cytosolic enzymes by isolated rat hepatocytes. J Cell Biol. 1990;111(3):941–953. doi: 10.1083/jcb.111.3.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman EJ, Siebers A, Altendorf K. Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc Natl Acad Sci U S A. 1988;85(21):7972–7976. doi: 10.1073/pnas.85.21.7972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjivassiliou AG, Rieder SV. The enzymatic assay of pyruvic and lactic acids. A definitive procedure. Clin Chim Acta. 1968;19(3):357–361. doi: 10.1016/0009-8981(68)90258-1. [DOI] [PubMed] [Google Scholar]
- Bergmeyer HU, Bernt E. In: Methods of enzymatic analysis (2nd English ed) Bergmeyer HU, editor. Vol. 2. Verlag Chemie; 1974. pp. 574–579. [Google Scholar]
- Engedal N, et al. Modulation of intracellular calcium homeostasis blocks autophagosome formation. Autophagy. 2013;9(10):1475–1490. doi: 10.4161/auto.25900. [DOI] [PubMed] [Google Scholar]
- Luhr M, et al. A Simple Cargo Sequestration Assay for Quantitative Measurement of Nonselective Autophagy in Cultured Cells. Methods Enzymol. 2017;587:351–364. doi: 10.1016/bs.mie.2016.09.064. [DOI] [PubMed] [Google Scholar]
- Mousavi SA, et al. Effects of inhibitors of the vacuolar proton pump on hepatic heterophagy and autophagy. Biochim Biophys Acta. 2001;1510(1-2):243–257. doi: 10.1016/s0005-2736(00)00354-0. [DOI] [PubMed] [Google Scholar]
- Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A. 1982;79(6):1889–1892. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronan B, et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat Chem Biol. 2014;10(12):1013–1019. doi: 10.1038/nchembio.1681. [DOI] [PubMed] [Google Scholar]
- Saetre F, Hagen LK, Engedal N, Seglen PO. Novel steps in the autophagic-lysosomal pathway. Febs j. 2015;282(11):2202–2214. doi: 10.1111/febs.13268. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12(1):1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seglen PO, Overbye A, Saetre F. Sequestration assays for mammalian autophagy. Methods Enzymol. 2009;452:63–83. doi: 10.1016/S0076-6879(08)03605-7. [DOI] [PubMed] [Google Scholar]
- Hurley JH, Young LN. Mechanisms of Autophagy Initiation. Annu Rev Biochem. 2017;86:225–244. doi: 10.1146/annurev-biochem-061516-044820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase. Proc Natl Acad Sci U S A. 1990;87(7):2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuma A, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432(7020):1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- Komatsu M, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169(3):425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T, et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci U S A. 2009;106(49):20842–20846. doi: 10.1073/pnas.0911267106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon PB, Seglen PO. Autophagic sequestration of [14C]sucrose, introduced into rat hepatocytes by reversible electro-permeabilization. Exp Cell Res. 1982;142(1):1–14. doi: 10.1016/0014-4827(82)90402-5. [DOI] [PubMed] [Google Scholar]
- An H, Harper JW. Systematic analysis of ribophagy in human cells reveals bystander flux during selective autophagy. Nat Cell Biol. 2018;20(2):135–143. doi: 10.1038/s41556-017-0007-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fass E, Shvets E, Degani I, Hirschberg K, Elazar Z. Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with lysosomes. J Biol Chem. 2006;281(47):36303–36316. doi: 10.1074/jbc.M607031200. [DOI] [PubMed] [Google Scholar]
- Velikkakath AK, Nishimura T, Oita E, Ishihara N, Mizushima N. Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol Biol Cell. 2012;23(5):896–909. doi: 10.1091/mbc.E11-09-0785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Øverbye A, Sætre F, Hagen LK, Johansen HT, Seglen PO. Autophagic activity measured in whole rat hepatocytes as the accumulation of a novel BHMT fragment (p10), generated in amphisomes by the asparaginyl proteinase, legumain. Autophagy. 2011;7(9):1011–1027. doi: 10.4161/auto.7.9.16436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kominami E, Hashida S, Khairallah EA, Katunuma N. Sequestration of cytoplasmic enzymes in an autophagic vacuole-lysosomal system induced by injection of leupeptin. J Biol Chem. 1983;258(10):6093–6100. [PubMed] [Google Scholar]
- Rosado CJ, Mijaljica D, Hatzinisiriou I, Prescott M, Devenish RJ. Rosella: a fluorescent pH-biosensor for reporting vacuolar turnover of cytosol and organelles in yeast. Autophagy. 2008;4(2):205–213. doi: 10.4161/auto.5331. [DOI] [PubMed] [Google Scholar]
- Katayama H, Kogure T, Mizushima N, Yoshimori T, Miyawaki A. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol. 2011;18(8):1042–1052. doi: 10.1016/j.chembiol.2011.05.013. [DOI] [PubMed] [Google Scholar]
- Ogier-Denis E, et al. A heterotrimeric Gi3-protein controls autophagic sequestration in the human colon cancer cell line HT-29. J Biol Chem. 1995;270(1):13–16. doi: 10.1074/jbc.270.1.13. [DOI] [PubMed] [Google Scholar]
- Seglen PO, Gordon PB, Tolleshaug H, Hoyvik H. Use of [3H]raffinose as a specific probe of autophagic sequestration. Exp Cell Res. 1986;162(1):273–277. doi: 10.1016/0014-4827(86)90446-5. [DOI] [PubMed] [Google Scholar]
- Luhr M, Sætre F, Engedal N. The Long-lived Protein Degradation Assay: an Efficient Method for Quantitative Determination of the Autophagic Flux of Endogenous Proteins in Adherent Cell Lines. Bio-protocol. 2018;8(9):e2836. doi: 10.21769/BioProtoc.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronning OW, Pettersen EO, Seglen PO. Protein synthesis and protein degradation through the cell cycle of human NHIK 3025 cells in vitro. Exp Cell Res. 1979;123(1):63–72. doi: 10.1016/0014-4827(79)90421-x. [DOI] [PubMed] [Google Scholar]
- Seglen PO, Grinde B, Solheim AE. Inhibition of the lysosomal pathway of protein degradation in isolated rat hepatocytes by ammonia, methylamine, chloroquine and leupeptin. Eur J Biochem. 1979;95(2):215–225. doi: 10.1111/j.1432-1033.1979.tb12956.x. [DOI] [PubMed] [Google Scholar]
- Seglen PO, Solheim AE. Valine uptake and incorporation into protein in isolated rat hepatocytes. Nature of the precursor pool for protein synthesis. Eur J Biochem. 1978;85(1):15–25. doi: 10.1111/j.1432-1033.1978.tb12208.x. [DOI] [PubMed] [Google Scholar]
- Bauvy C, Meijer AJ, Codogno P. Assaying of autophagic protein degradation. Methods Enzymol. 2009;452:47–61. doi: 10.1016/S0076-6879(08)03604-5. [DOI] [PubMed] [Google Scholar]
- Engedal N, Seglen PO. Autophagy of cytoplasmic bulk cargo does not require LC3. Autophagy. 2016;12(2):1–3. doi: 10.1080/15548627.2015.1076606. [DOI] [PMC free article] [PubMed] [Google Scholar]