

Abstract
Due to recent technological progress, cryo-electron microscopy (cryo-EM) is rapidly becoming a standard method for the structural analysis of protein complexes to atomic resolution. However, protein isolation techniques and sample preparation methods for EM remain a bottleneck. A relatively small number (100,000 to a few million) of individual protein particles need to be imaged for the high-resolution analysis of proteins by the single particle EM approach, making miniaturized sample handling techniques and microfluidic principles feasible.
A miniaturized, paper-blotting-free EM grid preparation method for sample pre-conditioning, EM grid priming and post processing that only consumes nanoliter-volumes of sample is presented. The method uses a dispensing system with sub-nanoliter precision to control liquid uptake and EM grid priming, a platform to control the grid temperature thereby determining the relative humidity above the EM grid, and a pick-and-plunge-mechanism for sample vitrification. For cryo-EM, an EM grid is placed on the temperature-controlled stage and the sample is aspirated into a capillary. The capillary tip is positioned in proximity to the grid surface, the grid is loaded with the sample and excess is re-aspirated into the microcapillary. Subsequently, the sample film is stabilized and slightly thinned by controlled water evaporation regulated by the offset of the platform temperature relative to the dew-point. At a given point the pick-and-plunge mechanism is triggered, rapidly transferring the primed EM grid into liquid ethane for sample vitrification. Alternatively, sample-conditioning methods are available to prepare nanoliter-sized sample volumes for negative stain (NS) EM.
The methodologies greatly reduce sample consumption and avoid approaches potentially harmful to proteins, such as the filter paper blotting used in conventional methods. Furthermore, the minuscule amount of sample required allows novel experimental strategies, such as fast sample conditioning, combination with single-cell lysis for "visual proteomics," or "lossless" total sample preparation for quantitative analysis of complex samples.
Keywords: Biology, Issue 137, Electron microscopy, cryo, negative stain, microfluidics, sample conditioning, visual proteomics, single-cell lysis
Introduction
Hardware and software for the structural analysis of protein complexes by transmission electron microscopy (TEM) has massively advanced during recent years. The improvements made paved the way to a "resolution revolution"1,2 and fundamentally changed structural research. The revolution started with the advent of cryo-electron microscopy (cryo-EM)3,4, allowing the preparation of biological samples under close to physiological conditions while decreasing radiation sensitivity and preventing sample evaporation in the high vacuum of the transmission electron microscope5. In the following years, incremental technological progress gradually increased the resolution achievable. Among these innovations were the application of field-emission guns6,7, and, more recently, improved data analysis algorithms, such as maximum likelihood methods8,9. Direct electron detector cameras10,11,12,13, movie-mode imaging and the accompanying software developments14,15,16,17, provided the final breakthrough required to achieve atomic resolution for biological samples by single particle analysis (for a review see Cheng, Grigorieff, et al.18). The importance of cryo-EM was recently recognized by the award of the Nobel prize for chemistry to three of the pioneers.
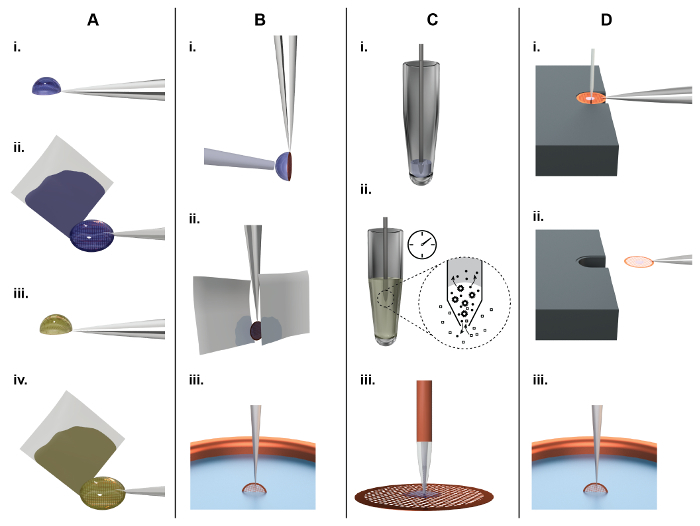
To image a biological sample by TEM, the method used to load the EM grid with sample (subsequently referred to as "grid preparation") must ensure that the resulting sample layer (i) is thin enough (<100 nm) to avoid extensive noise by inelastic or multiply scattered electrons, (ii) withstands the high vacuum of the electron microscope, and (iii) protects the biomolecules from radiation damage. Two main methods are used to fulfill these perquisites: Negative stain(NS)19,20 procedures (Figure 1A) adsorb the sample to a thin carbon film, embed the biomolecules in amorphous heavy metal and then allow the assembly to dry in air. This is simple and quick, and the loaded EM grids (subsequently referred to as "sample grids") are easy to store and can be kept for extended periods of time (generally years). In TEM, the preparations exhibit high contrast due to the NS and tolerate higher electron doses than cryo-preparations, but the resolution is limited to approximately 20 Å. Cryo-EM procedures (Figure 1B) employ holey carbon supports. A thin film of the sample solution is spanned across the holes and the EM grid is plunged into a cryogen, usually liquefied ethane, to rapidly cool it below -150°C. The result is an amorphous, vitrified, 50 to 100 nm-thick film of the solution in the support holes. This thin, amorphous film withstands the high vacuum in the electron microscope and, in the ideal case, preserves biological structures in their native state. The procedure allows biological samples to be imaged at high-resolution. However, the sample grid must be kept at temperatures below -150 °C at all times to avoid devitrification. It can be imaged using relatively high electron doses due to the low temperature, but the contrast and signal-to-noise ratio is nevertheless low. Therefore, averaging techniques are employed to increase contrast and, provided the sample is imaged from different angles, a high-resolution three-dimensional (3D) map can be reconstructed. The most commonly used and highly successful method for 3D reconstruction as of now is the single particle approach. For a recent review, see Cheng at al.18.
Negative stain TEM (NS-EM) is important for screening and quality control, when high-contrast is needed or when only limited amounts of sample are available (adsorption to the carbon film generally concentrates the sample). Single particle cryo-EM is the gold-standard method if high-resolution 3D reconstructions of the protein structure are aimed for.
Figure 1: Principles of TEM grid preparation and comparison between the classical (panel A, B) and a microfluidic approach (panel C, D). A) Classical NS-EM grid preparation: About 3 µL of sample are pipetted by hand onto an EM grid covered with a continuous carbon film (subsequently referred to as an 'NS-EM grid') (i). After incubation for approx. 10 s, filter paper is used to blot away the excess liquid from the side (ii), leaving the adsorbed biomolecules in a thin water film. Subsequently, the protein is incubated in a heavy metal salt solution, e.g., 2% uranyl acetate, for 20 s (iii), and again the liquid is removed by blotting from the side using filter paper (iv). Finally, the EM-grid is left to dry in air. B) Classical cryo-EM grid preparation: About 3 µL of sample are pipetted by a hand onto a holey carbon film. To form a thin sample film, the surplus liquid is removed by paper-blotting face-on from one or both sides (ii). Finally, the grid is rapidly plunged into liquid ethane for vitrification (iii). C) NS-EM grid preparation using the cryoWriter setup: A 5 nL volume is aspirated from the sample stock using a microcapillary (i). For sample conditioning, the microcapillary tip is immersed into the conditioning solution, e.g., 2% ammonium acetate. Ions and small molecules are exchanged by diffusion (ii). Note that the dimensions of the microcapillary ensure that the whole process is diffusion driven. Proteins have much lower diffusion constants than salt ions and are not significantly lost24. Finally, the sample is dispensed onto the grid and allowed to dry (iii). D) Principles of cryo-EM grid preparation using the cryoWriter-based method: An EM grid covered with a holey carbon film is placed on the surface of a temperature-controlled platform and held by tweezers. The temperature of the platform is controlled at an offset from the dew point temperature of the grid environment. The grid is moved relative to the microcapillary containing the sample and the microcapillary is lowered until it is a few micrometers above the grid. Subsequently, a few nanoliters of sample are dispensed from it while the stage is moved in a spiral pattern; excess liquid is re-aspirated (i). After EM grid priming, the microcapillary is withdrawn and the grid remains on the temperature controlled platform (subsequently referred as dew point (DP) stage) for a short time to allow a controlled amount of sample to evaporate. For plunge freezing, the grid is rapidly withdrawn from the stage using the tweezers (ii), flipped by 90° into the vertical position, and plunged into a cryogen bath (iii) (subsequently referred to as 'pick-and-plunge' mechanism). Please click here to view a larger version of this figure.
Unfortunately, the grid preparation methods used for NS and cryo-EM have not improved significantly since they were invented. Current drawbacks are the high sample consumption (approx. 3 µL of 1 mg/mL protein) and the large amount (> 99%) of sample lost (Figure 1A,B). Furthermore, the classical method used to prepare grids for cryo-EM is a harsh procedure for proteins: First, it involves an extensive face-on paper-blotting step (Figure 1B, ii), and, second, the protein is exposed to the air-water interface for a significant amount of time21. Here, an alternative method for sample pre-conditioning, sample grid preparation and post-processing (grid drying or vitrification) for NS-EM (Figure 1C) or cryo-EM (Figure 1D) is presented. The in-house built setup, called "cryoWriter", uses miniaturized sample handling technology and microfluidic principles to aspirate, condition and dispense sample, avoiding paper blotting completely and providing alternative methods to thin samples for cryo-EM. It significantly reduces sample consumption and improves user-control over sample preparation as a whole. Furthermore, the method allows novel experimental applications; such as the preparation of isolated biological components of individual cells in an approach called "single cell visual proteomics"22,23,24,25.
Protocol
A "cryoWriter" (Figure 2; for details see previous work24,26,27) or equivalent instrumentation is required for the following protocols. A list of suppliers for the main parts and consumables is given in the Table of Materials.

1. Negative Stain (NS) Grid Preparation
Turn on the instrument and start up the software. Initialize all necessary modules (syringe pump controller, motorized stages, surveillance cameras, and dew point stage).
Cool the sample support and the dew-point stage. If required, make sure that the dew-point stage temperature is regulated 1 - 2 °C above the dew point. NOTE: The stage is cooled by a commercial Peltier device with a PID controller.
Prepare NS by filling a 100 or 200 µL PCR tube with 100 - 150 µL of NS (e.g., commercially available 2% methylamine tungstate). Place the tube on the cooled sample support of the instrument.
- Position the sample.
- Put the sample (0.5 - 1 µM) into a 100 or 200 µL PCR tube. If less than 50 µL of sample is available, cut off the bottom of a PCR tube with a razor blade and use it as a sample well. This will ensure that the microcapillary can easily reach the sample. NOTE: It is easiest to aspirate samples from 100 or 200 µL PCR tubes, because the microcapillary used later to aspirate the sample is slightly tilted and the travel-height z-axis direction is limited.
- Place the PCR tube/container on the cooled sample support in the instrument to prevent evaporation. Alternatively, samples can be aspirated from well plates in the microscope stage top incubator at room temperature; cooling is not implemented for well plates.
- Define positions. Use the cryoWriter joystick that controls the motorized xy-stage and software control buttons for the linear x-, y-, and z-axis stages to position the microcapillary. Use the camera to check the position of the capillary.
- Move the microcapillary to the sample reservoir. Immerse the tip into the sample liquid and save this position as "sample".
- Move the microcapillary to the NS PCR tube, immerse the tip into the NS solution and save this position as "stain".
- Place the microcapillary roughly 100 µm above the center of the slot where the EM grid will be positioned and save this position as "grid_save".
- Aspirate sample and condition it for NS-EM.
- If not already installed, mount a 10-µL syringe (0.46 mm inner diameter) on a precision syringe pump.
- Glue one end of a 30 cm long fused silica microcapillary (outer diameter 360 µm, inner diameter 150 µm) to the syringe outlet.
- Connect the other end of the microcapillary to a short (5 cm) long tapered microcapillary via press fit connector. The tapered tip of the short microcapillary forms the dispensing tip.
- Fill the syringe with degassed double-distilled water (ddH2O; system liquid) and avoid the formation of air bubbles.
- Dispense a few tens of nanoliters of system liquid and remove any drops from the microcapillary with a lint-free tissue.
- Double-click on the saved sample position. This positions the microcapillary in the sample well. While the capillary is moving, dispense 3 x 0.5 nL of system liquid just before the microcapillary tip is immersed into the sample to prevent air bubble from being trapped there (see note 2 below).
- Aspirate 5 nL of sample.
- Double-click on the saved NS position. The microcapillary is withdrawn from the sample container and moved to the NS reservoir automatically. While the capillary is moving, manually dispense 3 x 0.5 nL of sample liquid just before the tip is immersed into the reservoir to make sure that there are no air bubbles (see note 2 below). NOTE: IMPORTANT: (1) When switching from aspiration to dispensing mode or vice versa, there is a small loss in piston stroke due to backlash in the gears of the syringe pump. According to the manufacturer, the backlash of a new unit lies between 7 and 12 µm. For our syringe with a barrel diameter of 0.46 mm, this translates to 1-2 nL. Therefore, 1-2 nL can be "dispensed", before sample is actually dispensed. Usually, a tiny droplet starts to exit the microcapillary tip after the third 0.5 nL dispense step. (2) An air bubble trapped above/below the sample would make dispensing less accurate and prevent sample conditioning by diffusion.
- Leave the microcapillary immersed for 3-12 min, depending on the sample buffer and nozzle geometry. NOTE: The higher the salt and/or phosphate concentration in the buffer, the longer the required immersion time. NS (relatively quickly) diffuses into the sample plug while buffer salts (relatively fast) and protein (much slower) diffuse out. This lowers the concentration of buffer salts in the sample preventing them from crystallizing when the loaded grid dries. Further, phosphate tends to form a precipitate in combination with NS.
- Prepare a grid and deposit a spot of the conditioned sample.
- While the sample is being conditioned, take a piece of adhesive tape and a PDMS block and clean the top side of the PDMS by applying and removing the adhesive tape to ensure that there is no dust. Put the PDMS block in a Petri dish. NOTE: New PDMS blocks are taken from the clean room.
- Carefully pick up a grid (e.g., Cu, 200 or 400 mesh coated with Parlodion/C film). Make sure to touch only the edge of the grid with the tweezers. Place it on the clean PDMS block with the carbon film facing upwards.
- Place the PDMS block with the grid in an air glow-discharge unit and glow discharge it for 20 s with 100 W power at 0.4 mbar. Store the grid in a closed Petri dish. Longer glow discharge times generally lead to a larger spreading of the deposited sample volume on the grid. As a result, the stain layer becomes thinner (weaker stain).
- 1 min before the immersion time is up, grasp the glow-discharged grid with the cryoWriter tweezers. Make sure the electromagnet is turned ON; otherwise turn it on in the software. Mount the tweezers on the electromagnet and use the manual micromanipulator screw to align the grid flat on the dew-point stage, carbon film side up. Make sure that the dew-point stage temperature is regulated 1-2 °C above the dew point to reduce the rate of evaporation after sample is loaded.
- When the immersion time is up, double-click on "grid_save". The microcapillary tip will be placed safely above the grid surface. Manually bring the microcapillary tip into contact with the grid. Lift the nozzle by 10 µm and position it above the center. CAUTION: Some samples containing detergents tend to move up along the outer surface of the microcapillary when liquid is dispensed due to the lower surface tension. It is important to be very close to the grid surface to prevent such losses.
- Dispense 5 nL of sample onto the grid. On a dry day, when rapid evaporation takes place at the microcapillary tip, one can also slowly dispense until the sample is at the very tip, and then quickly dispense 5 nL.
- Withdraw the microcapillary and let the conditioned sample dry slowly on the dew point stage (DP-stage).
- Once the sample spot has dried, remove the grid and store it at room temperature in a grid box or Petri dish.
- Dispense 500 nL of system liquid from the microcapillary and remove it with a lint-free tissue. Flush the capillary 5 times with either ethanol, detergent or 1 M NaOH. This cleans the microcapillary allowing it to be used with a different sample.
2. Cryo Grid Preparation
To prepare the instrument and sample, follow steps 1.1 to 1.5 described above. If NS is not required, omit steps 1.3 and 1.5.2. To exchange the sample buffer or condition the sample for cryo-EM by dialysis, e.g., to reduce the concentration of buffer salts or to introduce additives (e.g., trehalose, detergents) use the desired buffer instead of NS in steps 1.3 and 1.5.2.
- Prepare liquid ethane in a standard cryo container.
- Assemble the ethane cup, cryo box holder, and spider and fill the cryogen container to the brim with liquid nitrogen; usually requires about 200 mL. Wait a few minutes until the ethane cup has cooled down and is free of liquid nitrogen.
- Open the ethane gas bottle and slowly let the gas stream into the ethane cup. Let it fill with liquid ethane until the level is 2-3 mm below the top; this takes a few minutes and requires about 5 mL of liquid ethane.
- Take a cryo box and place it in a free slot in the cryogen container.
- Remove the spider, place the polystyrol lid on top, and place the cryogen container on the mounting in the cryoWriter.
- Glow discharge an EM grid.
- Take a piece of adhesive tape and a polydimethylsiloxane (PDMS) block and clean the top side PDMS by applying and removing the adhesive tape (removes any dust). Put the PDMS block in a Petri dish.
- Carefully pick a grid from the grid box. Make sure to touch only the edge of the grid with the tweezers. Place it on a clean PDMS block with the holey carbon film facing upwards.
- Place the PDMS block with the grid in a plasma cleaner and plasma-clean the grid surface (e.g. use 75% Ar/25% H2, power 50 W, pressure 25 mTorr). Place the PDMS block with the glow-discharged grid in a Petri dish.
- Position the grid in the instrument
- Grasp the glow-discharged grid with the cryoWriter tweezers. Make sure the electromagnet is turned ON; otherwise turn it on in the software. Mount the tweezers on the electromagnet and use the manual micromanipulator screw to align the grid flat on the stage, carbon film side up.
- Double-click on "grid_save". Adjust the microcapillary position so that the tip is approx. 10 µm above the grid surface. Make sure that the microcapillary can move freely across the grid without touching it anywhere, if necessary withdraw the microcapillary a few micrometers.
- Go back to the center of the grid and save the new position as "grid". CAUTION: Correct naming is mandatory for the macro script to work.
- Write sample and plunge-freeze grid.
- Flush the microcapillary with a few tens of nanoliters of system liquid and remove any drops from the microcapillary with a lint-free tissue.
- Start macro script. The macro will perform the following steps:
- Dispense 5 nL (to remove any air bubbles at the tip) and go to sample position.
- Aspirate 65 nL of sample. Infuse 5 nL back into the sample tube. This accounts for system backlash and allow synchronized writing, i.e., to ensure that dispensing and stage movement start at the same time.
- Move to "grid" position.
- Initiate 'writing pattern', which will cause the microcapillary to move across the grid, simultaneously dispensing 45 nL of sample.
- Afterwards, move the microcapillary back to the center of the grid, lower it another 10 µm, and withdraw excess sample liquid.
- Withdraw the microcapillary and turn off the electromagnet. This releases the tweezers and initiates plunge freezing.
Grip the tweezers and carefully release the magnetic adapter from the plunger. Quickly transfer the grid from the ethane cup into the cryogen container containing the cryo box and place the grid in a free slot.
3. Single-cell Lysate Preparation
- Prepare the microcapillaries for electroporation. NOTE: The tapered (laser-pulled and inspected) fused silica microcapillaries have a protecting polyimide coating on the outside, except for the tip, where the coating is burnt off during the tapering process.
- To coat the microcapillaries with a conductive layer, mount them at an angle of 45° on a metal rail, with the tips pointing upwards. Use an aluminum foil to shield the lower end (2 cm) of the tips, as uncoated polyimide forms the best seal with the press-fit connectors employed later.
- Sputter deposit a sticky layer of 20 nm Ti/W, and then a 200 nm-thick layer of Pt.
- Make the microcapillary tip hydrophobic.
- Prepare a 1 M solution of 1-dodecanethiol in EtOH.
- Glow-discharge the microcapillary in air for 1 min with 100 W power at 0.4 mbar.
- Immerse the tip of the microcapillary in the 1 M solution of 1-dodecanethiol for a few hours (ideally overnight). NOTE: It is best to prepare tips freshly one day before use.
- Install a new microcapillary.
- Remove the microcapillary from the 1-dodecanethiol solution, rinse the outside with ethanol and flush the inside with ethanol using a syringe. NOTE: If no functionalized microcapillaries are available, quickly glow discharge the tip of a microcapillary and dip it in a drop of commercial car window treatment, which is a mixture of PDMS and sulphuric acid. The resulting hydrophobic functionalization is not as good as with 1-dodecanethiol, but generally sufficient.
- Cleave its uncoated end with a capillary cutter. NOTE: A clean cut is important for a good seal with the press-fit connector.
- Use a toothpick to apply silver paste to the sharp boarder formed between glass and the polyimide coating when the polyimide coating was burnt off by the laser during capillary pulling. NOTE: This reinforcement is necessary as the Pt coating is very weak in this region, and electrical conduction can easily break down.
- Wash the polyimide end of the microcapillary with acetone. Leave a small drop of acetone at the end and insert it into the orifice of the press-fit connector. Apply light pressure to form a good seal.
- Calibrate the microcapillary position.
- Turn on the instrument and start the software as described in step 1.1. Additionally, initialize the microscope camera and the function generator.
- Place a glass microscope slide into the slide holder on the microscope.
- Lower the microcapillary close to the glass slide and center it over the microscope objective until it appears in the microscope camera view. Use the x- and y- axis linear stages to position the tip of the microcapillary at the center of the image. Then slowly lower the tip until it slightly touches the glass slide. CAUTION: Choose very small steps (5 µm) towards the final approach. Otherwise, the tip can be damaged.
- Press the "Calibrate Nozzle" button. This retracts the microcapillary 40 mm, and sets this position as home position.
- Preparation of stamped PDMS pieces and ITO slides
- Mix PDMS and crosslinker in 10:1 ratio, pour into a large Petri dish to a depth of about 2-3 mm. Bake at 60° C for a few hours in a hybridization incubator or similar device.
- With a hammer and a stamp (12 mm diameter), cut holes in the PDMS layer. Afterwards, use a scalpel to cut out 2 cm long squares or rectangles including the holes to obtain stamped pieces that fit on a microscope slide. Usually, two stamped pieces are mounted on one microscope slide.
- Wash the PDMS pieces and the ITO slides first with detergent and then with 70% ethanol. Place them, wet, in a Petri dish and let the ethanol fully evaporate in an oven at 60° C.
- Glow-discharge the ITO slides for 1 min with 100 W power at 0.4 mbar and then apply 1 mL of coating solution (e.g., poly-L-lysine (PLL)). Incubate for 5 min, remove the solution with a pipette, and apply 1 mL of ddH2O. Slightly agitate for 1 min, and then remove the ddH2O using a pipette. Let the slides dry at 60° C.
- Prepare the cell culture. Follow standard adherent-cell culturing protocol (e.g., Arnold, et al.24,25). Seed cells on ITO during normal splitting/passaging runs.
- Take a freshly PLL-coated ITO slide and add a PDMS piece. Apply some pressure to form a watertight seal. This produces small wells with the slide as their base.
- Add about 300 µL of cells suspended in fresh medium to the PDMS wells. The seeding density should be around 75,000 cells per well.
- Incubate the ITO slides for 1-2 days under standard conditions.
- Prepare for cell lysis experiment:
- Pre-warm a few milliliters of the electroporation buffer (e.g., phosphate buffered saline (PBS)).
- Prepare set-up for NS-EM grid preparation NOTE: Details for preparing the set-up are shown in steps 1.1-1.5 (for NS) or step 2.1-2.4 (for cryo). Here we describe the most important steps for NS-preparation.
- Prepare NS by filling a 100 or 200 µL PCR tube with 100-150 µL of NS (e.g., commercially available 2% methylamine tungstate). Place the tube on the cooled sample support of the instrument.
- Move the microcapillary to the NS PCR tube, immerse the tip into the NS solution and save this position as "stain".
- Load the standard lysis parameters, e.g., the lysis voltage.
- Take the ITO slide from the incubator and mount it on the microscope insert. Use two screws to fix the slide on the aluminum insert and to ensure electrical contact between the ITO coating of the slide and the electrically grounded aluminum frame.
- Remove the cell culture medium and wash twice with 300 µL of electroporation buffer. Keep the cells in electroporation buffer.
- Place the aluminum insert holding the ITO slide in the live-cell incubator stage on the setup.
- Locate the cell culture in the microscope view and choose an area with no cells. Approach the tip to the ITO surface and gently touch it, then withdraw the tip 100 µm and save the position as "cells".
- Quickly leave the cell culture and flush the microcapillary tip with a few tens of nanoliters of system liquid then put it into the cell culture again. Dispense a few nanoliters during immersion into the PDMS well to ensure that no air bubbles are trapped at the tip.
- Gently approach the ITO surface (initially, you must be at position "cells", i.e., 100 µm above the surface). Upon contact, retract tip 10 µm.
- Select a nearby cell for lysis. Place the tip of the microcapillary above the targeted cell.
- Start the macro script for single-cell lysis.
- Name the position the microcapillary should be moved to after a cell has been successfully lysed. This will be the NS reservoir (NS-EM), a desalting buffer (cryo-EM) or the EM grid (cryo-EM). Avoid spelling errors and press "ok".
- The macro proceeds without user intervention:
- i) The microscope stage and cell culture 100 µm are moved to the left, a snapshot of the targeted cell is taken, and 50 nL of ddH2O system liquid are dispensed from the microcapillary. This displaces and dilutes the high salt buffer and applies osmotic pressure to the cell.
- ii) The stage is moved back to position the tip above the targeted cell again. The predefined voltage burst is applied, and after 500 ms the pump system starts to aspirate 3 nL of sample at a flow rate of 2 µL/min.
- iii) The stage is moved to the left again, allowing the cell to be inspected. A window appears, asking for user input.
- Say whether the lysis step was successful or not.
- If the answer is no, take over, flush the microcapillary and target a new cell.
- If the answer is yes, a snapshot of the removed (lysed) cell is taken, and afterwards the microcapillary is moved to the location specified in 3.8.1. If this is NS or a buffer reservoir, use the standard user interface to dispense 3 x 0.5 nL of liquid while the microcapillary is moving to ensure that there are no air bubbles. The tip is immersed in the reservoir liquid by the macro. NOTE: You can continue to condition the sample and prepare grids as explained in sections 1.6.9 - 1.7.9 for NS-EM or section 2.2 - 2.6 for cryo-EM. Below, the necessary steps for NS-grid preparation are explained.
- Leave the microcapillary immersed for 8-12 min, depending on the nozzle geometry NOTE: The high phosphate concentration in the buffer requires a longer immersion time for conditioning.
- Dispense 5 nL of the conditioned cell lysate onto the grid.
- Withdraw the microcapillary and let the conditioned sample dry slowly on the dew point stage (DP-stage) regulated 1 - 2°C above the dew point temperature.
- Remove the grid and store it at room temperature in a grid box or Petri dish.
Representative Results
The "cryoWriter" setup was developed (depicted in Figure 2) in order to test the miniaturized EM grid preparation procedures proposed in Figure 1C,D. Figure 2A shows an overview of the various components mounted to an inverse fluorescence microscope. A cell-culturing module is installed on the left side of the microscope; a module for EM grid preparation is located on the right. The cell-culturing module (Figure 2B) allows the growth of adherent eukaryotic cells and live-cell imaging of the cell culture by the light microscope. Individual cells are lysed by the combined action of osmotic shock, electroporation and aspiration of the cell content into a microcapillary (Figure 2B, Figure 6A)24,25. The aspirated lysate sample can then be used to prepare grids for NS- or cryo-EM. Alternatively, a stock protein solution in a PCR tube can be the sample source. The microcapillary (Figure 2B) employed is connected to a high-precision pump system allowing sample volumes to be aspirated and dispensed with sub-nL precision. As detailed in the protocols, all sample processing is performed within this microcapillary or on the EM grid itself without significant sample transport. For example, the same microcapillary is used to lyse individual eukaryotic cells, aspirate the lysate, condition it, and finally dispense aliquots onto EM grids. The grid preparation module consists of a movable DP-stage that allows the temperature of the EM grid placed on it to be precisely controlled (Figure 2C). For NS-TEM, the prepared sample grid can then simply be removed from the cold stage and allowed to dry in air at room temperature. However, the so-called coffee-ring effects that can then result need to be avoided for quantitative TEM where protein 'particles' are counted. To do so, grids are dried slowly on the DP-stage using a gradually increasing temperature gradient to slow down liquid evaporation. For cryo-EM, the temperature of the grid is kept close to the dew point; a positive offset of approximately 8 °C is chosen, allowing the controlled evaporation of sample liquid for thin film stabilization and thinning, which can be monitored by a sensor if necessary26. After the selected thinning time, a pick-and-plunge mechanism is activated and the sample is vitrified (Figure 2C). Note that this plunging mechanism is not needed for NS-EM grids, which are stored at room temperature.
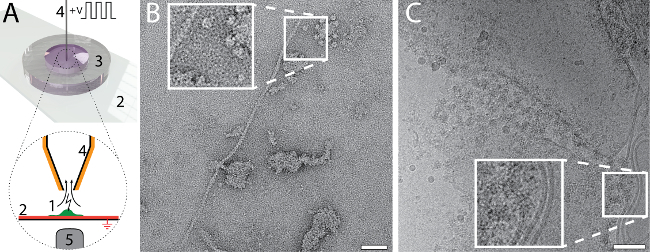
Figure 2: Overview of the cryoWriter setup. A) Overview of the cryoWriter setup mounted on an inverse light-microscope (1). B) Inset of area indicated on the left side in panel A. Cell culturing compartment (2), with a microcapillary (3) for sample manipulation and cell lysis positioned above a miniaturized PDMS-based cell culture plate (4). C) Inset of area indicated on the right side in panel A. 'Pick-and-plunge' mechanism. A holey carbon film EM grid (5) is mounted between the tips of tweezers (6) and positioned horizontally in direct contact with the temperature-controlled stage (7), referred as the dew-point stage (DP-stage) in the main text. The stage temperature is tightly controlled via a PID controller and a water-cooled Peltier element, keeping it at or close to the dew point temperature, depending on the ambient environment. The DP-stage (7) is mounted on a motorized xy axis to move the grid relative to the microcapillary. The microcapillary itself is mounted on a z-stage and can be lowered until it is very close to the surface of the EM grid and used to dispense nanoliter-sized volumes onto the sample support covering it (a continuous thin carbon layer for NS-EM or a holey carbon film for cryo-EM). Note that liquid uptake and dispensing is performed using a high-precision pump system (8). The dispensed liquid can be distributed by moving the grid relative to the microcapillary in a spiral pattern. For cryo-EM preparation, the pick-and-plunge freezing mechanism (9) rapidly transfers the sample-loaded grid into liquid ethane (10) for rapid cooling and sample vitrification. Please click here to view a larger version of this figure.
Figure 3 shows representative results obtained for NS-EM grids prepared using the cryoWriter setup. The tip of the microcapillary was loaded with 5 nL of sample from a stock solution and dipped into a reservoir of NS solution (2% methylamine tungstate) for several minutes to allow diffusive exchange of NS and salt ions (for a theoretical discussion see Arnold, et al.24). Afterwards, the conditioned sample was dispensed onto the thin carbon film of a NS-EM grid and dried. Figure 3A shows the use of a slot grid in the same way to visualize the complete droplet, as required for quantitative TEM. In order to avoid the coffee ring effect, the freshly glow-discharged grid was initially held at the dew-point temperature (no water evaporation) and then slowly warmed up on the DP-stage. Note, that for most applications (e.g., quality control of the sample or structural analysis) this slow-drying process is not needed. High-quality NS-preparations are obtained without it, as shown in Figure 3B,C. Conditioning times for phosphate-free low salt buffers are around 3 min, e.g., with low-salt Tris-buffer (20 mM Tris-HCl pH 7.4 with 50 mM NaCl), as shown in Figure 3B using tobacco mosaic virus (TMV) as sample. Figure 3C presents a worst-case scenario as the TMV was in PBS buffer (2.7 mM KCl, 1.5 mM KH2PO4, 136.9 mM NaCl, 8.9 mM Na2HPO4·7H2O, pH 7.4). Phosphate ions form transient crystals with the heavy-metal ions of NS (see Figure 5C), lengthening the conditioning time required (7 min). Other heavy metal salts can also be used with the grid preparation module, e.g., 2% methylamine vanadate or ammonium molybdate (see also Arnold, et al.24). However, uranyl acetate is not suitable; the crosslinking effect of this stain leads to aggregates if the protein sample is conditioned in solution, before adsorption to a carbon film (see Figure 5E)23.

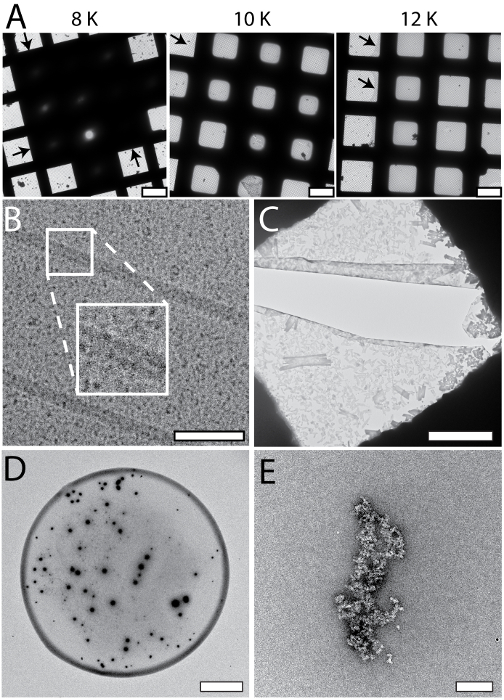
Figure 3: Typical results for NS grids prepared using the cryoWriter setup as indicated in Figure 1C. A) Overview image of a 3 nL droplet dispensed on a slot grid after conditioning with 2% methylamine tungstate. B) TMV in 20 mM TRIS buffer. The inset shows a 3x enlargement of the indicated region. Adapted from Arnold, et al.24 (further permissions related to the material excerpted should be directed to the ACS). C) Tobacco mosaic virus (TMV) in PBS buffer. The inset shows a 3x enlargement of the indicated region. Adapted from Arnold, et al.24 (further permissions related to the material excerpted should be directed to the ACS). Scale bars: A, 100 µm; B, 50 nm; C, 80 nm. Please click here to view a larger version of this figure.
Typical results obtained for cryo-EM grids prepared using the cryoWriter setup are depicted in Figure 4. Panel 4A shows a grid atlas of the area covered by vitrified sample. Panel 4B shows the homogeneity of the vitreous ice in a selected grid slot. In both cases, the sample was in 25 mM HEPES-KOH pH 7.5, 50 mM NaCl buffer containing 0.05% Fos 14 detergent. Many samples and buffers were tested and a comparable high quality vitreous ice was obtained, but the conditions required are buffer dependent (see also discussion of Figure 5). Panel 4C shows apoferritin particles and a bacteriophage in Tris-HCl buffer (20 mM Tris-HCl, 50 mM NaCl; pH 7.4) imaged at high defocus to increase contrast. Panel 4D shows a 200 kDa membrane protein stabilized by amphipoles.

Figure 4: Typical results for cryo-EM grids prepared using the cryoWriter setup as indicated in Figure 1D. The samples and the buffers vary in the examples shown. All samples were loaded on holey carbon films. A) Collage of overview images ("grid atlas") of a sample containing a 150 kDa membrane protein, the periphery of the vitreous ice is indicated by white arrows. B) Enlarged grid slot from a grid prepared with the same buffer, showing the holey carbon film with vitreous ice. Some holes are not filled with sample buffer as indicated by white arrows. C) Carbon hole with vitrified sample containing apoferritin protein complexes and bacteriophages. Inset: twofold enlargement showing the tail of a bacteriophage. The white asterisk indicates the carbon film. Note that the image was recorded with high defocus to increase contrast. D) A 200 kDa membrane protein reconstituted in amphipols. Inset: a 2x enlargement of the indicated region shown with increased contrast. Scale bars: A, 100 µm; B, 10 µm; C and D, 80 nm. Please click here to view a larger version of this figure.
The cryoWriter setup allows systematic screening for optimal EM-grid preparation conditions; an example is shown in Figure 5A (apoferritin in 25 mM HEPES-KOH pH 7.5, 50 mM NaCl, 0.05% Fos 14). In this experiment, the vitreous ice "thinning" temperature was varied, but the thinning time (i.e., the time gap between sample application and plunge freezing) remained constant (1 s). At low offset temperatures (e.g., 8 K), the sample layer was too thick. At higher offset temperatures, the vitreous ice in the holes was thinner (10 K, 12 K), until at some stage (above 18 K) the grid became completely dry (not shown). In the results presented here, an offset of 12 K lead to a large homogeneous area of vitreous ice as indicated by the black arrows. Such optimization experiments can be performed with the buffer of the target sample using "test" proteins (such as apoferritin). The best conditions found are then applied to the target sample. Furthermore, grids with parameters far from the optimum can often be recognized during the preparation procedure and do not need to be screened in the electron microscope, saving significant time. Figure 5 also shows a gallery of typical cryo-EM (Panel B) and NS-EM (Panels C to E) artifacts specific to the cryoWriter setup. The cryo-EM grid shown in Panel 5B with TMV in PBS containing 0.1% decyl-β-D-maltopyranoside (2.7 mM KCl, 1.5 mM KH2PO4, 136.9 mM NaCl, 8.9 mM Na2HPO4·7H2O, pH 7.4, 0.1%DM) was excessively thinned. The background of the image is grainy because the salt concentration became too high. In general, the visual appearance of a sample does not seem to be a linear function of the salt concentration; grains suddenly become prominent when a threshold concentration is reached during the thinning process. Note that unwanted substances can be removed by a conditioning step prior to grid preparation, as described for NS-EM in protocol section 1.6. NS can cause other artifacts. In the example shown in Panel 5C, PBS buffer (2.7 mM KCl, 1.5 mM KH2PO4, 136.9 mM NaCl, 8.9 mM Na2HPO4·7H2O, pH 7.4) without sample was conditioned in 2% methylamine tungstate for 3 min. Precipitates and crystals are evident and exert extensive forces on the carbon surface leading to cracks. The precipitates only form in a certain PBS and NS concentration range and can be avoided by conditioning the sample for longer (compare Figure 3C). Panel 5D shows the periphery of a dispensed NS sample droplet exhibiting a "coffee ring". This would disturb quantitative, total sample analysis and can be avoided by slowing down the drying process, i.e., by keeping the EM grid at the dew point temperature during sample application, and then gradually increasing the temperature to dry it (see Figure 3A). Panel 5E (apoferritin in 20 mM HEPES, pH 7.0, conditioned 3 min) shows the crosslinking activity of 2% uranyl acetate stain, which cannot be used to condition protein samples before they are adsorbed to carbon film supports.
Figure 5: Systematic changes and artifacts observed when the cryoWriter set-up was used to prepare grids for NS- and cryo-EM. A) Systematic vitreous ice thickness variation; optimization of grid preparation for cryo-EM. The offset temperature of the DP-stage was varied (8 K to 12 K) keeping the thinning time constant (1 s). The arrows indicate the periphery of the sample layer. B) Salt effects; too highly concentrated, i.e., the thinning step was too long. The inset depicts a 2x enlargement of the indicated region. C) Salt precipitates formed by PBS buffer in the presence of heavy metal salts. Samples containing PBS buffer must be conditioned longer than samples in other buffers. Here, PBS buffer without sample was conditioned in 2% methylamine tungstate for 3 min, a typical time for other sample buffers. Note the crack in the carbon film most probably due to the strong forces from the precipitates acting on the thin support during the drying process. D) 'Coffee ring' effect. E) Apoferritin conditioned in 2% uranyl acetate for 3 min. Uranyl ions exhibit significant crosslinking activity and the apoferritin clusters form large aggregates. Scale bars: A, 80 µm; B, 80 nm C, 12 µm; D, 80 µm; E, 200 nm. Please click here to view a larger version of this figure.
The small amount and volume required for EM grid preparation using the cryoWriter setup enables new types of experiments. For example, the total contents of a single cell can be collected and prepared for NS- and cryo-EM. The procedure is indicated in Figure 6A. An adherent, eukaryotic cell (HEK 293) is lysed by simultaneous electroporation and sample aspiration (Panel 6A)25. A total volume of 3 nL is aspirated, which contains the cell lysate, and is retained in the microcapillary for further processing. For the NS-EM shown in Panel 6B, the cell-culturing medium was exchanged with PBS buffer (2.7 mM KCl, 1.5 mM KH2PO4, 136.9 mM NaCl, 8.9 mM Na2HPO4·7H2O, pH 7.4) prior to cell lysis. The cell contents were aspirated in 3 nL of buffer and conditioned in a reservoir of NS as indicated in Figure 1C for 10 min. Afterwards, a 5 nL volume was dispensed onto the continuous carbon film of a NS-EM grid. Individual proteins, e.g., filamentous actin, and membrane patches with attached proteins can be recognized in the image. For the cryo-EM, shown in Figure6C, a volume of 3 nL was dispensed on a holey carbon EM grid without re-aspiration to remove liquid. The relatively thick film of sample formed was extensively thinned before vitrification. To do this, the DP-stage temperature was gradually increased, starting at the dew-point temperature. The thinning process was monitored by a real-time sensor-system until a pre-specified threshold was reached triggering the 'pick-and-plunge' mechanism and sample vitrification (for details see Arnold, et al.26). Membrane structures and proteins can be recognized in the image.
Figure 6: Single cell visual proteomics using the cryoWriter set-up. A) Lysis of a single adherent eukaryotic cell. The cell is grown (1, green) on an functionalized, ITO coated (red) glass-slide (2) in a miniaturized petri-dish (3)25. The ITO layer is electrically grounded. The cell is approached by the microcapillary (4), which is coated with platinum. An initial osmotic shock (not indicated) is given to facilitate lysis, which is performed by a series of electric pulses and by the shear forces exerted during the aspiration of the cell lysate. The process can be monitored by light microscopy; the objective lens of the microscope is indicated (5). For details, see our previous work24,25. B) NS-EM image of lysate from an individual HEK 293 cell. Filamentous actin and membrane patches with attached proteins are visible. Panel adapted from Arnold, et al.24 (further permissions related to the material excerpted should be directed to the ACS). C) Cryo-EM image of lysate from an individual HEK 293 cell. The inset shows a 2x enlargement of the indicated region where typical membrane structures with associated proteins are visible. Panel C adapted from Arnold, et al. 26 Scale bars: B, 50 nm; C, 80 nm. Please click here to view a larger version of this figure.
Discussion
The 'cryoWriter' instrument and the protocols required to prepare sample grids for NS- and cryo-EM from nL sized total sample volumes and completely avoid the classical paper-blotting step are presented. Microfluidic principles and a micromechanical system are combined in the cryoWriter to make this possible.
Our experience shows that when the miniaturized methods presented in this manuscript are used, the parameter space for EM-grid preparation is larger than for classical methods, and under tighter user control. Importantly, the increased reproducibility achieved makes it possible to pre-screen with sample buffer system, complemented with a readily available test protein, to determine the optimum parameters before the actual experiment is performed. This keeps consumption of the sample of interest to the absolute minimum and is highly recommended. Critical steps for both NS- and cryo-EM grid preparation are: (i) Priming of the pump system; for sub-nanoliter volume dispensing, the system liquid (water) must be degassed and bubble free. (ii) Precise control of the glow discharge or plasma cleaning step; the surface characteristics of the EM grid are crucial for reproducible results. (iii) Sample conditioning; the time required for conditioning, e.g., with NS, depends on the buffer type, salt content and concentration (Figure 3), as well as on the nozzle geometry of the microcapillary24. (iv) Evaporation rates for NS-EM preparations for quantitative EM; the coffee-ring effect can prohibit the quantitative analysis of NS-EM preparations, and must be suppressed by slow evaporation rates controlled by the DP-stage.
Different aspects of the presented grid preparation methods can be freely combined allowing the development of versatile protocols for specific samples. Typical examples would be the removal of a substance that prevents high-resolution EM, e.g., glycerol, by a conditioning step prior to grid preparation for cryo-EM; the introduction of mediator molecules, such as ligands, by conditioning before the grids are prepared; or the examination of single cell lysate by NS- or cryo-EM (Figure 6).
The use of microfluidics and minimal sample amounts in the presented methods completely removes the need for paper-blotting steps. This is a great advantage, because paper blotting is a harsh treatment for proteins, potentially contaminating the sample with unwanted ions and inherently leading to massive sample loss. On the other hand, effects potentially caused by the air-water interface of the thin sample film formed when cryo-EM samples are prepared in the classical manner are not avoided when the cryoWriter is used. Grids suitable for cryo-EM can be prepared with a less than 0.2 s wait time between sample application and vitrification (data not shown). However, as proteins travel a few tenths of a nanometer in a few nanoseconds by diffusion, there is still enough time for them to collide with the air-water interface of a 100 nm thick sample film several times. However, the amount of proteins sticking to the air-water interface might be significantly reduced by these short time gaps and might prevent protein denaturation or restricted particle orientation. Another promising approach that might protect sensitive proteins from the air-water interface is to cover the sample film by low molecular weight surface-active substances. These compounds could be rapidly introduced by a conditioning step in the cryoWriter before grid preparation. The high surface-to-volume ratio of microfluidic systems is a further limitation of the cryoWriter, as sample can potentially be lost by unspecific adsorption to the microcapillary surface and disturb quantitative analysis by particle counting. The problem is addressed in two ways: First, the sample does not travel long distances within the microcapillary. Indeed, the nanoliter sample volume remains at the capillary tip throughout the processing. Second, the surface to volume ratio is further reduced by using microcapillaries with relatively large inner diameters, e.g., 180 µm. Third, the surfaces of the microcapillaries can be easily passivated if necessary, e.g., by treating them with commercially available polylysine ethanol glycols (PLL-PEG).
The high-resolution analysis of proteins by the single particle approach used in EM only requires 100,000 to a few million images of individual protein particles. This means that microfluidic techniques can provide enough protein complexes for the structural investigation. A miniaturized immuno-precipitation method for the fast isolation of protein complexes (around 1 hour) from minimal cell amounts (approx. 40,000 cells) was developed earlier28. This method will now be directly linked to the miniaturized sample preparation stage of the cryoWriter. The final goal is to develop an integrated microfluidic pipeline for ultra-fast protein isolation and cryo-EM grid preparation that requires less than two hours in all. Furthermore, as demonstrated by Figure 6, the minute amount and volume of material needed for sample preparation and the almost lossless conditioning and grid preparation procedure achieved using the cryoWriter, make it possible to study the protein complexes of individual cells. Together, the miniaturized immuno-precipitation method and the cryoWriter lay the foundation of a new proteomics method called "single cell visual proteomics", as we recently demonstrated for heat-shock experiments24. Data analysis algorithms geared to the analysis of "visual proteomics" images are currently being tested.
Disclosures
The authors Stefan A. Arnold, Henning Stahlberg and Thomas Braun declare the following competing financial interest: The cryoWriter concept is part of patent application PCT/ EP2015/065398 and EP16194230.
Acknowledgments
The authors would like to thank the workshop of the Biozentrum of the University Basel for their support, S. A. Müller for critical discussions and for carefully reading the manuscript, A. Fecteau-LeFebvre for technical assistance with EM, Ricardo Adaixo, Frank Lehmann for membrane protein test samples (all from the C-CINA, Biozentrum, University of Basel) and A. Engel, emeritus University Basel for his inspiring conversations. Test samples were kindly provided by P. Ringler, M.-A. Mahi, and T. Schwede (Biozentrum, University of Basel), P. Leiman (Laboratory of Structural Biology and Biophysics, EPFL) and R. Diaz- Avalos (New York Structural Biology Center, USA). The project was supported by the Swiss Nanoscience Institute (SNI, project P1401, ARGOVIA project MiPIS) and the Swiss National Science Foundation (SNF, project 200021_162521).
References
- Kuhlbrandt W. Biochemistry: The resolution revolution. Science. 2014;343(6178):1443–1444. doi: 10.1126/science.1251652. [DOI] [PubMed] [Google Scholar]
- Bai X-c, McMullan G, Scheres SHW. How cryo-EM is revolutionizing structural biology. Trends Biochem Sci. 2015;40(1):49–57. doi: 10.1016/j.tibs.2014.10.005. [DOI] [PubMed] [Google Scholar]
- Dubochet J, Adrian M, Chang JJ, Homo JC, Lepault J, McDowall AW, Schultz P. Cryo-electron microscopy of vitrified specimens. Q Rev Biophys. 1988;21(2):129–228. doi: 10.1017/s0033583500004297. [DOI] [PubMed] [Google Scholar]
- Lepault J, Booy FP, Dubochet J. Electron microscopy of frozen biological suspensions. J Microsc. 1983;129:89–102. doi: 10.1111/j.1365-2818.1983.tb04163.x. Pt 1. [DOI] [PubMed] [Google Scholar]
- Baker LA, Rubinstein JL. Radiation damage in electron cryomicroscopy. Methods Enzymol. 2010. [DOI] [PubMed]
- Crewe AV, Eggenberger DN, Wall J, Welter LM. Electron gun using a field emission source. Rev Sci Instrum. 2003.
- Zemlin F. Expected contribution of the field-emission gun to high-resolution transmission electron microscopy. Micron. 1994;25(3):223–226. [Google Scholar]
- Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012;180(3):519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigorieff N. FREALIGN: high-resolution refinement of single particle structures. J Struct Biol. 2007;157(1):117–125. doi: 10.1016/j.jsb.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Li X, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S, Agard Da, Cheng Y. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat methods. 2013;10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milazzo A-C, Cheng A, Moeller A, Lyumkis D, Jacovetty E, Polukas J, Ellisman MH, Xuong N-H, Carragher B, Potter CS. Initial evaluation of a direct detection device detector for single particle cryo-electron microscopy. J Struct Biol. 2011;176(3):404–408. doi: 10.1016/j.jsb.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruskin RS, Yu Z, Grigorieff N. Quantitative characterization of electron detectors for transmission electron microscopy. J Struct Biol. 2013;184(3):385–393. doi: 10.1016/j.jsb.2013.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veesler D, Campbell MG, Cheng A, Fu C-y, Murez Z, Johnson JE, Potter CS, Carragher B. Maximizing the potential of electron cryomicroscopy data collected using direct detectors. J Struct Biol. 2013;184(2):193–202. doi: 10.1016/j.jsb.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MG, Cheng A, Brilot AF, Moeller A, Lyumkis D, Veesler D, Pan J, Harrison SC, Potter CS, Carragher B, Grigorieff N. Movies of ice-embedded particles enhance resolution in electron Cryo-microscopy. Structure. 2012;20(11):1823–1828. doi: 10.1016/j.str.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripstein ZA, Rubinstein JL. Processing of Cryo-EM movie data. Methods in Enzymology. 2016;579:103–124. doi: 10.1016/bs.mie.2016.04.009. [DOI] [PubMed] [Google Scholar]
- Li X, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S, Agard DA, Cheng Y. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nature Methods. 2013;10(6):584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLeod RA, Kowal J, Ringler P, Stahlberg H. Robust image alignment for cryogenic transmission electron microscopy. J Struct Biol. 2017;197(3):279–293. doi: 10.1016/j.jsb.2016.12.006. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Grigorieff N, Penczek PA, Walz T. A primer to single-particle cryo-electron microscopy. Cell. 2015;161(3):438–449. doi: 10.1016/j.cell.2015.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S, Horne RW. A negative staining method for high resolution electron microscopy of viruses. Biochim Biophys Acta. 1959;34:103–110. doi: 10.1016/0006-3002(59)90237-9. [DOI] [PubMed] [Google Scholar]
- De Carlo S, Harris JR. Negative staining and cryo-negative staining of macromolecules and viruses for TEM. Micron. 2011;42(2):117–131. doi: 10.1016/j.micron.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaeser RM. How good can cryo-EM become. Nat Methods. 2016;13:28–32. doi: 10.1038/nmeth.3695. [DOI] [PubMed] [Google Scholar]
- Engel A. in. In: Graslund A, Rigler R, Widengren J, editors. Single Molecule Spectroscopy in Chemistry, Physics and Biology. Vol. 96. Heidelberg, Berlin: Springer Berlin; 2010. pp. 417–431. [Google Scholar]
- Kemmerling S, Ziegler J, Schweighauser G, Arnold SA, Giss D, Müller SA, Ringler P, Goldie KN, Goedecke N, Hierlemann A, Stahlberg H, Engel A, Braun T. Connecting µ-fluidics to electron microscopy. J Struct Biol. 2012;177(1):128–134. doi: 10.1016/j.jsb.2011.11.001. [DOI] [PubMed] [Google Scholar]
- Arnold SA, Albiez S, Opara N, Chami M, Schmidli C, Bieri A, Padeste C, Stahlberg H, Braun T. Total sample conditioning and preparation of nanoliter volumes for electron microscopy. ACS nano. 2016;10(5):4981–4988. doi: 10.1021/acsnano.6b01328. [DOI] [PubMed] [Google Scholar]
- Kemmerling S, Arnold SA, Bircher BA, Sauter N, Escobedo C, Dernick G, Hierlemann A, Stahlberg H, Braun T. Single-cell lysis for visual analysis by electron microscopy. J Struct Biol. 2013;183:467–473. doi: 10.1016/j.jsb.2013.06.012. [DOI] [PubMed] [Google Scholar]
- Arnold SA, Albiez S, Bieri A, Syntychaki A, Adaixo R, McLeod RA, Goldie KN, Stahlberg H, Braun T. Blotting-free and lossless cryo-electron microscopy grid preparation from nanoliter-sized protein samples and single-cell extracts. J Struct Biol. 2017;197(3):220–226. doi: 10.1016/j.jsb.2016.11.002. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan C, Bieri A, Sauter N, Roizard S, Ringler P, Müller SA, Goldie KN, Enimanev K, Stahlberg H, Rinn B, Braun T. openBEB: open biological experiment browser for correlative measurements. BMC Bioinformatics. 2014;15:84. doi: 10.1186/1471-2105-15-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giss D, Kemmerling S, Dandey V, Stahlberg H, Braun T. Exploring the interactome: microfluidic isolation of proteins and interacting partners for quantitative analysis by electron microscopy. Anal Chem. 2014;86:4680–4687. doi: 10.1021/ac4027803. [DOI] [PubMed] [Google Scholar]