

Abstract
Transforming Growth Factor β (TGF-β) signaling regulates many important functions required for cellular homeostasis and is commonly found overexpressed in many diseases, including cancer. TGF-β is strongly implicated in metastasis during late stage cancer progression, activating a subset of migratory and invasive tumor cells. Current methods for signaling pathway analysis focus on endpoint models, which often attempt to measure signaling post-hoc of the biological event and do not reflect the progressive nature of the disease. Here, we demonstrate a novel adenovirus reporter system specific for the TGF-β/Smad3 signaling pathway that can detect transcriptional activation in live cells. Utilizing an Ad-CAGA12-Td-Tom reporter, we can achieve a 100% infection rate of MDA-MB-231 cells within 24 h in vitro. The use of a fluorescent reporter allows for imaging of live single cells in real-time with direct identification of transcriptionally active cells. Stimulation of infected cells with TGF-β displays only a subset of cells that are transcriptionally active and involved in specific biological functions. This approach allows for high specificity and sensitivity at a single cell level to enhance understanding of biological functions related to TGF-β signaling in vitro. Smad3 transcriptional activity can also be reported in vivo in real-time through the application of an Ad-CAGA12-Luc reporter. Ad-CAGA12-Luc can be measured in the same manner as traditional stably transfected luciferase cell lines. Smad3 transcriptional activity of cells implanted in vivo can be analyzed through conventional IVIS imaging and monitored live during tumor progression, providing unique insight into the dynamics of the TGF-β signaling pathway. Our protocol describes an advantageous reporter delivery system allowing for quick high-throughput imaging of live cell signaling pathways both in vitro and in vivo. This method can be expanded to a range of image based assays and presents as a sensitive and reproducible approach for both basic biology and therapeutic development.
Keywords: Cancer Research, Issue 137, TGF-β, live cell imaging, adenovirus, cell signalling, breast cancer, Smad3, transcriptional activation
Introduction
Transforming Growth Factor β (TGF-β) is an essential cytokine implicated in human development that signals through a heterodimeric complex consisting of type II and type I receptors1. Binding to type II receptor results in the recruitment and phosphorylation of type I receptor, which in turn phosphorylates downstream Smad2/3 proteins2,3. These activated Smad2/3 proteins bind to Smad4, forming a complex that translocates into the nucleus and regulates gene transcription4. Under homeostatic conditions TGF-β/Smad signaling is tightly regulated; however, in many diseases, the signaling pathway is deregulated and often overexpressed leading to progression of disease5,6,7. Recent studies have demonstrated that cellular response to TGF-β is heterogenous and subpopulations of TGF-β/Smad active cells are responsible for biological function in a time-dependent manner8,9. Common cellular analysis of TGF-β/Smad signaling involves the use of fixed endpoint assays that provide only a snapshot of cellular activity and often quantitate the average TGF-β/Smad effect10. These methods, however, may not accurately represent the molecular behaviors of TGF-β/Smad signaling in the physiological state during disease progression. Image-based analysis of live cells capture the dynamics of cellular and biological processes with both a spatial and temporal understanding.
Our goal was to develop a sensitive high-throughput method for live cell imaging of TGF-β/Smad signaling using adenovirus-based reagents. Here, we infected the human breast cancer cell line MDA-MB-231 with an adenovirus expressing the Smad3 CAGA motif binding sequence and a luciferase (Luc) or Td-Tomato (Td-Tom) reporter gene. Adenoviral reporter systems provide a quick and cheap method for plasmid introduction that can result in a 100% infection rate in cancer cell lines. Adenoviral reporter systems have also been successfully applied to cell lines that are difficult to transfect with conventional plasmid11. In this protocol we will describe a reproducible and noninvasive process to achieve live cell imaging of the TGFβ/Smad signaling pathway both in vivo and in vitro.
Protocol
All animal experiments were approved by the University of Melbourne Animal Ethics Committee.
Note: The sequence, construction and generation protocol for the adenoviral vectors pAd-CMV-Td-Tom, pAd-CMV-GFP and pAd-CAGA12-Luc/Td-Tom have been previously described11,12,13. All vectors are commercially available.
1. Virus Titer Determination Using 50% Tissue-culture Infectious Dose (TCID50)
Prepare 1 x 106 HEK293A cells in 10 mL of DMEM + 10% FBS media.
Seed 100 µL of cell suspension into each well of a 96-well flat-bottom cell culture plate.
Prepare 1 mL of a 1:100 dilution of virus stock in complete medium. Add 11 µL of diluted virus to each well of column 1.
Perform serial 10-fold dilutions directly in the 96 well plate by mixing 11 µL of diluted virus with 100 µL of cell suspension until a final dilution of 1 x1012 is reached. Although cells are non-adherent at this stage, the number of cells transferred between wells remains constant.
Pipette 11 µL of each dilution into each column starting with the highest dilution of 104. Replace pipette tips with each dilution to limit cross-over of viral particles.
Add 11 µL of virus-free complete media to column 12 as a negative control.
Incubate plate for 7 - 10 days at 37 °C with 10% CO2.
Using a microscope, score the number of wells in each column displaying signs of cytopathic or cytotoxic effects. Consider a well positive if any sign of infection is present.
Calculate the fraction of positive wells for each dilution using the Reed and Muench (1938) statistical method14.
Calculate the proportional distance (PD) using: (A-50)/(A-B), where A is the percentage response above or equal to 50% and B is the percentage response below 50%.
Calculate TCID50 using: 10X-PD, where X is the dilution giving a response greater than 50%.
Calculate viral titer from using the following: 0.69/(0.011 x TCID50) PFU/mL
2. Adenovirus MOI Determination
Seed 1.0 - 1.5 x 105 MDA-MB-231 cells with 500 μL of DMEM culture media containing 5% FBS in each well of a 12 well plate (35% cell confluency, 6 wells in total).
Calculate the volumes of Ad-CMV-Td-Tom stock required for 0, 250, 500, 2500, and 5000 MOI using following formula: MOI = (volume [μL] x PFU/μL)/number of cells. For Ad-CMV-Td-Tom with a titer of 1 x 1010 (PFU/mL), the volumes of virus infection required MOIs are 0, 3.75, 7.5, 37.5, and 75 μL respectively.
Infect the cells with the calculated volumes of Ad-CMV-Td-Tom in Section 2.2 by directly pipetting viral stock into the wells with thorough mixing.
Place the plate in the incubator at 37 °C with 10% CO2 for 24 h.
Remove the media from the wells and immediately fix the cells with 500 μL of 10% formalin for 5 min.
Aspirate the formalin from wells and wash wells with 500 μL of PBS once.
Stain the cell nucleus with 500 μL of Hoechst solution for 5 min.
Remove the Hoechst solution and wash the wells with 500 μL of PBS 3 times.
Image the Td-Tomato protein and cell nucleus signal using 200X magnification on a fluorescence microscope. Excite Td-Tomato protein at 554 nm and the Hoechst dye at 352 nm.
Analyze images using ImageJ to determine the working MOI required for 100% infection of Td-Tomato15.
3. Dual-adenovirus Transduction in Live Single Cells
Seed 1.0 - 1.5 x 105 MDA-MB-231 cells with 500 μL of DMEM culture media containing 5% FBS in each well of a 12 well plate (35% cell confluency, 2 wells in total).
Infect cells with 75 μL of Ad-CMV-GFP (titer = 5 x 109 PFU/mL) and 7.5 μL of Ad-CAGA12-Td-Tom (titer = 5 x 1010 PFU/mL) to obtain a 2,500 MOI, which can achieve 100% infection positivity.
Treat cells with or without 0.25 μL of TGF-β per well (10 mg/mL in stock, 5 ng/μL as the final concentration) immediately after adenovirus infection.
Place the plate in the incubator at 37 °C with 10% CO2 for 24 h.
Image live cells with phase contrast fluorescence microscope. Excite GFP at 470 nm and Td-Tomato protein at 554 nm.
Fix cells and stain cell nucleus as described in Steps 1.6 - 1.9.
Image the Td-Tomato protein and Hoechst stain using a fluorescence microscope. Excite the Hoechst dye at 352 nm.
Using ImageJ, calculate the percentage of GFP and Td-Tomato positive cells15.
4. Live Single Cell Signaling in Wound Healing Assay
Seed 4 - 5 x 105 MDA-MB-231 cells with 1 mL of DMEM culture media containing 5% FBS in each well of a 6 well plate (50% cell confluency, 2 wells in total).
Infect the cells with 22.5 μL of Ad-CAGA12-Td-Tom (titer = 5 x 1010 PFU/mL, MOI = 2,500).
Place the plate in the incubator at 37 °C with 10% CO2 for 24 h.
Scratch two crossed lines on the cell layer of each well using a P200 pipette tip.
Remove the old media from wells and replace with 2 mL of fresh 5% FBS media with or without 0.5 μL of TGF-β per well (10 mg/mL in stock, 5 ng/μL as final concentration).
Place the plate in the incubator at 37 °C with 10% CO2 for 5 min and take images of selected wound areas using 100X magnification on a phase contrast microscope.
Place the plate in the incubator at 37 °C with 10% CO2 for 24 h.
Take images of the wound closure at the same areas as before.
Fix the cells, stain cell nucleus as described in Steps 1.6 - 1.9 and visualize Td-Tomato signal in the wound area and non-wound area using 200X magnification on a fluorescence microscope.
Quantify the percentage of Td-Tomato positive cells in the wound and non-wound area using ImageJ software15.
5. In Vitro Luciferase Assay
Prepare a cell suspension of 3 - 5 x 104 MDA-MB-231 cells in 1 mL of DMEM media containing 5% FBS.
Infect cell suspension with 2.5 μL of Ad-CAGA12-Luc (titer = 5 x 1010 PFU/mL, MOI = 2,500).
Seed the infected cells into a 96 well plate at 3,000 cells/100 µL/well.
Place the plate in the incubator at 37 °C with 10% CO2 for 24 h.
Remove old media from wells and replace with 100 µL of fresh 5% FBS media with or without 0.1 μL of TGF-β per well (1 mg/mL in stock, 1 ng/μL as the final concentration) and in the presence or absence of 0.17 μL of TGF-β inhibitor per well (7 mg/mL in stock, 12 ng/μL as final concentration) (a novel TGF-β ligand trap protein, Chen et al., unpublished work).
Place the plate in the incubator at 37 °C with 10% CO2 for 24 h.
Thaw out the luciferase assay system substrate and prepare 1x cell culture lysis reagent in double distilled water (DDW).
Remove media from the 96 wells and lyse wells with 50 μL of 1x cell culture lysis buffer on ice using an orbital shaker with gentle agitation for 30 min.
Transfer 30 μL of lysate from each well into an opaque luciferase plate and allow the plate to warm to room temperature.
On the luminometer software, create a new protocol. Select for 1 injector with measurements before and after injection. Set measurements before injection at 1 s for both Delay in Measurement and Integration Time. For after injection measurement, set the Injection Volume to 40 µL with a Delay in Measurement of 1 s after injection and 5 s Integration Time. Confirm settings by selecting OK.
Place the injector tube into DDW and click the Injector 1 button under the Prime settings to clean the injector tube before use. Next remove the injector tube from the DDW and place in air followed by two further Primes of injector 1 to flush all solution from the tube. Place the injector tube into Firefly substrate and again Prime twice to prepare the substrate.
Place the opaque luciferase plate with the samples into the luminometer and select Options on the software. Highlight the wells of interest and click Apply. Press Start to measure luciferase signal.
Once measurements are complete, remove and rinse the plate with water. Recover any remaining substrate into the injector tube by placing the tube into air and Prime injector 1 back into firefly substrate tube. Place the injector tube into DDW and perform two further Primes to clean the injector tube of any remaining substrate.
6. Cell Preparation for Live Signaling Imaging In Vivo
48 h before tumor implantation, seed 2 - 3x106 MDA-MB-231 cells into 175 cm2 flasks (10 flasks in total). Culture cells in 20 mL of DMEM media containing 5% FBS at 37 °C, 10% CO2.
24 h later, add 300 μL of Ad-CAGA12-Luc (titer = 5 x 1010 PFU/mL, MOI = 2,500) into each flask. Keep cells in culture for another 24 h.
On the day of implantation, remove the old media and wash the adenovirus infected cells once with 20 mL of PBS, pH 7.4. Add 2 mL of 0.05% trypsin-EDTA into the flask and shake the flask slightly to allow trypsin to cover all the flask surface. Place the flask in to the incubator 37 °C, 10% CO2 for 3 min.
Quench the trypsin by adding 8 mL of FBS-containing DMEM media into flasks. Transfer all cell suspensions into a glass reagent bottle.
Count the cell number using a hemocytometer and transfer 4.8 x 107 cells in two 50ml centrifuge tubes.
Centrifuge the cells at 400 x g for 5 min at RT to remove trypsin and resuspend cells in 720 mL of fresh FBS-DMEM media.
Transfer the cell suspensions into a sterile 5 mL tube and add 160 μL Matrigel (10% total matrigel suspension).
Keep cells on ice until implantation.
7. Orthotopic Implantation
Weigh 12 SCID mice and randomly allocate mice into control and treatment groups (6 mice/group).
Perform the implantation in a sterile bio-cabinet to maintain a sterile environment. Anaesthetize the mouse using 2.5 - 4.5% isoflurane with an oxygen flow rate of 1 L/min. Place the mouse on a heat pad and dispense a drop of lubricating eye ointment onto both eyes to avoid corneal damage. Fix the mouse to the heat pad in a supine position while maintaining anaesthesia by placing the snout into a nose cone connected to the isoflurane.
Confirm the success of anaesthesia by the lack of reaction to toe pinch. Reduce isoflurane to a maintenance dose of 1.5 - 2.5% with 1 L/min oxygen for the reminder of implantation.
Clean the skin from the fourth nipple on both sides to the midline by using the cotton swab dipped into 80% ethanol.
Find the 4th mammary fat pad through palpation and squeeze the fat pad to further expose the tissue.
Mix the cell suspension well by pipetting up and down. Gently aspirate 50 µL of cell mixture into a 27G insulin syringe and inject into the mammary fat pad on both sides of the mouse.
Confirm a successful injection by feeling for swelling of the fat pad. Release the fat pad gently and place the mouse back into the cage.
Leave the cages on the heat pad for at least 30 min to allow the mice to gain consciousness and then place the cages back in the holding room for 3 days.
8. IVIS Imaging
Open the Living Image software.
Initialize the IVIS system by clicking the Initialize IVIS System button on the control panel. Wait until the temperature sign turns green.
Select the Luminescent Imaging Mode on the control panel.
Turn on isoflurane anaesthesia to the chamber and IVIS system.
Anaesthetize the mice by using 2.5 - 4.5% isoflurane with 1 L/min oxygen. Once anaesthetized, set isoflurane at 1.5 - 2.5% for maintenance. Inject 150 mg/kg body weight of D-luciferin into mice by intraperitoneal (i.p.) injection.
Immediately transfer mice into the IVIS system, place them onto the temperature-controlled imaging platform and place their nose into the nose cone.
Click on the Sequence Setup button on the control panel and select Medium Binning.
Set up the imaging exposure time as follows: 10 s, 20 s, 30 s, 60 s, and 120 s. Start imaging approximately 3 min after the luciferin injection and continue to image until the signal starts to drop (normally this will take around 20 min).
Remove the mice from IVIS while maintaining isoflurane anaesthetic through a nose cone and treat the mice with 50 µL PBS or 50 µL TGF-β signaling inhibitor (50 μg/tumor) via intra-tumor injection.
Put the mice back in the cage and leave them on the heat pad for at least 30 min. Then place the cage back in the holding room.
Repeat IVIS imaging procedure as described above the day following treatment.
9. Data Analysis
Open saved files in Living Image software.
Find the image information using View | Image Information.
Select photos taken at a similar time point with the same exposure time. Load photos as a group.
Uncheck the Individual box in Image Adjust to normalize the intensity.
Select Photon mode to analyze the intensity. Quantify the intensity of the signal by selecting the Region of Interest (ROI) button in ROI Tools.
Drag the ROI circle to the region containing the bioluminescent signal and click the Measure button to acquire the signal intensity.
Representative Results
Live single cell imaging in vitro
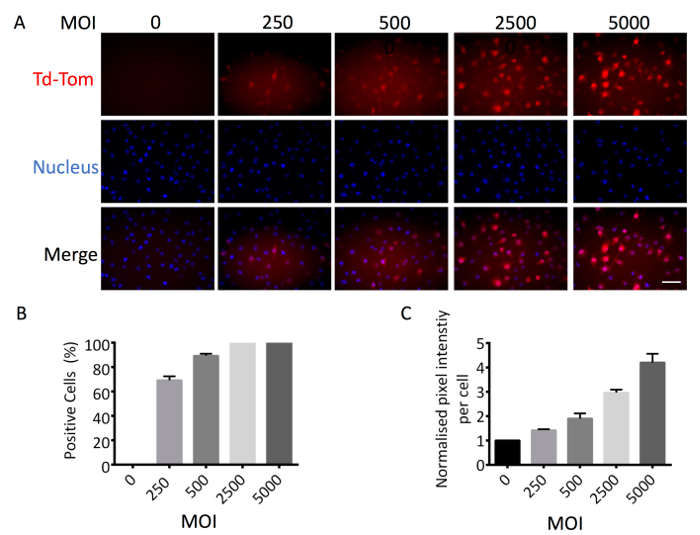
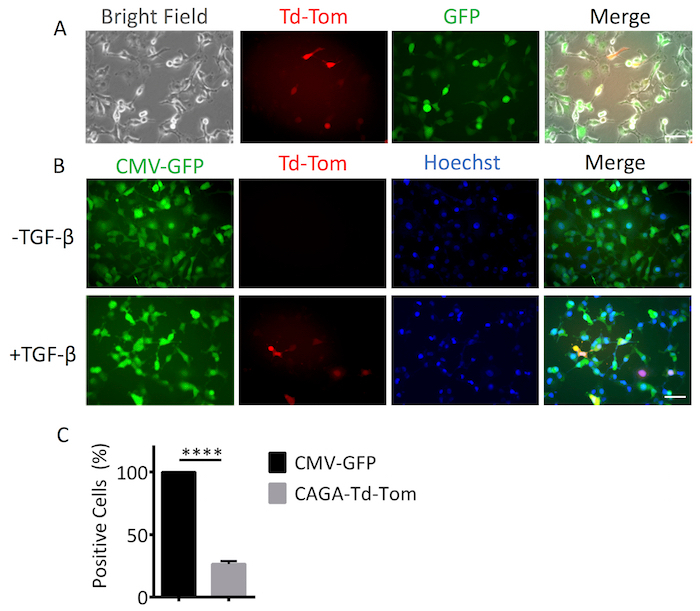
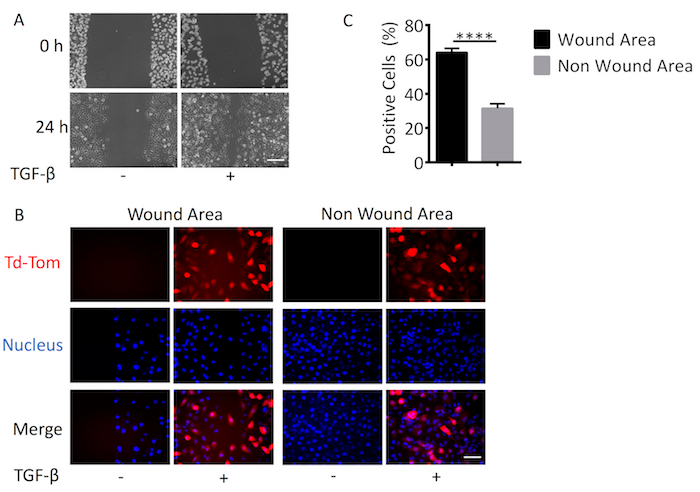
To accurately assess the activation of TGF-β/Smad signaling in single cells using adenovirus based reagents, it is important to first determine the optimal Multiplicity of Infection (MOI) for each cell line. The optimal MOI is determined when 100% of cells are positive for adenoviral infection with no present cytopathic or cytotoxic effects. To determine this, we used a constitutively active Ad-CMV-Td-Tom reporter that acted as a marker of positive infection. The percentage of adenovirus infected cells was increased in a MOI dependent manner from 0 to 5,000 MOI. At 2,500 MOI, we observed 100% Td-Tom expression in MDA-MB-231 cells (Figure 1A, 1B). Pixel intensity/cell also increased with MOI, providing enhanced sensitivity and detection of transcriptional output (Figure 1C). Therefore, a working MOI of 2,500 was used to perform a dual infection where cells were infected with Ad-CMV-GFP (served as positive control for cell infection) and Ad-CAGA12-Td-Tom simultaneously. Single cells presented varying levels of Td-Tom expression indicating heterogenous activation of TGF-β/Smad3 transcription across cells in both live and fixed cells. (Figure 2A, 2B). 100% of MDA-MB-231 cells presented with GFP expression while only around 26% of cells displayed detectable TGF-β/Smad3 transcriptional activity 24 h after TGF-β stimulation, compared to 0% positivity without TGF-β treatment (Figure 2C). To use the adenovirus based reagent to investigate the TGF-β/Smad3 transcriptional activity during biological processes, MDA-MB-231 cells were infected with Ad-CAGA12-Td-Tom at 2500 MOI and subjected to a wound healing assay. Infected MDA-MB-231 cells exhibited normal migration behavior and cells treated with 5 ng/mL TGF-β showed enhanced wound closure compared to non-TGF-β treated cells (Figure 3A). Notably, about 62% cells in the wound area presented positive TGF-β/Smad3 transcriptional activity, which is significantly higher than observed in non-wound area (32%) after TGF-β treatment (Figure3B). As such, the adenovirus-based reagents validated their application for imaging of the TGF-β signaling pathway in live single cell in vitro.
Live TGF-β signaling imaging in vivo
The sensitivity of the Ad-CAGA12- Luc reporter was first tested in vitro in response to TGF-β stimulation and the presence of a TGF-β inhibitor. When infected MDA-MB-231 cells were stimulated with 1 ng/mL TGF-β, luciferase reporter activity increased 1,200-fold compared to basal untreated MDA-MB-231 levels. This response was remarkably blocked by 12 µg/mL of TGF-β inhibitor (Figure 4A). The Ad-CAGA12-Luc reagent was subsequently tested in a breast tumor orthotopic implantation animal model. Both control and the TGF-β inhibitor treated groups demonstrated similar luciferase reporter activity before treatment. However, for the control group, the luciferase signal did not change after PBS treatment, whereas, mice treated with the TGF-β inhibitor showed around 3-fold signal reduction (Figure 4B, 4C). Collectively, these results demonstrate the potential for live and real-time monitoring of TGF-β signaling activity in vivo without the need for animal sacrifice.
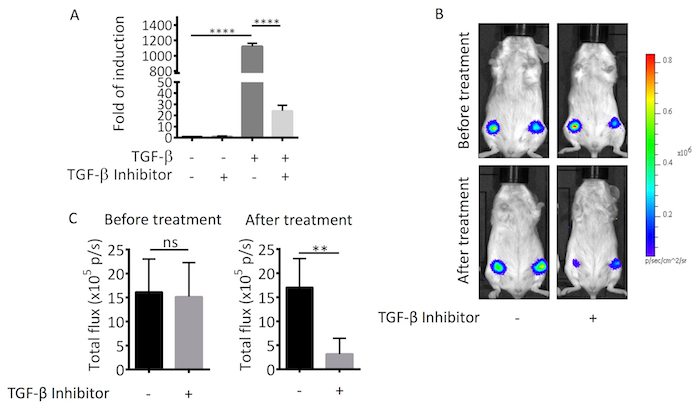
Figure 1. Determination of the MOI of adenovirus-based reagent. MDA-MB-231 cells were infected with Ad-CMV-Td-Tom at different MOI and seeded into a 12 wells plate. 24 h after infection, cells were fixed and nuclear stained by Hoechst. (A) Images were taken to visualize Td-Tom positive cells (red) and nucleus (blue) and quantified by ImageJ. (B) Percentage of Td-Tom positive cells. (C) Td-Tom pixel intensity/cell normalized to background. These figures are representative of at least 3 independent experiments. Data shown are means of triplicates ± SD. Scale bar = 50 µm (200X). Please click here to view a larger version of this figure.
Figure 2. Single cell imaging for dual infection. MDA-MB-231 cells were dual infected with Ad-CMV-GFP and Ad-CAGA12-Td-Tom at MOI (2,500) and seeded into a 12 wells plate. 24 h after infection, cells were treated with or without 5 ng/mL TGF-β. (A)24 h after treatment, cells were live imaged to visualize GFP positive (green) and Td-Tom positive (red) cells. Cells were then fixed and nuclear stained by Hoechst for quantification. (B) Images were taken to visualize GFP positive (green), Td-Tom positive (red) cells and nucleus (blue). (C) The GFP or Td-Tom positive cells were quantified by ImageJ and the percentage of positive cells was calculated. These figures are representative of at least 3 independent experiments. Data shown are means of triplicates ± SD. Statistical significance was determined by Student's t-test (**** p ≤0.0001). Scale bar = 50 µm (200X). Please click here to view a larger version of this figure.
Figure 3. Live cell TGF-β signaling promotes cell migration. MDA-MB-231 cells were infected with Ad-CAGA12-Td-Tom at MOI (2,500) and seeded onto a 6 wells plate. 24 h after infection, cells were scratched using a P200 pipette tip to create a wound and media replaced with or without 5 ng/mL TGF-β. After another 24 h, cells were fixed and nuclear stained by Hoechst for visual representation. (A) Phase contrast of wound closure, Scale bar = 100 µm (100X). (B) Td-Tom positive (red) cells and nucleus (blue). Scale bar = 50 µm (200X) (C) Td-Tom positive cells were quantified by ImageJ and the percentage of positive cells was calculated. These figures are representative of at least 3 independent experiments. Data shown are means of triplicates ± SD. Statistical significance was determined by Student's t-test (**** p ≤0.0001). Please click here to view a larger version of this figure.
Figure 4. In vivo live TGF-β signaling imaging. (A) MDA-MB-231 cells were infected with Ad-CAGA12-Luc and then seeded into 96 wells plate for 24 h. Thereafter, cells were treated with or without 1 ng/mL TGF-β in the presence or absence of 12 µg/mL TGF-β inhibitor overnight. The luciferase activity was measured based on the whole cell lysate. Luciferase activities were presented as the fold of induction normalized to no treatment control. Data shown are means of triplicates ± SD. (B) MDA-MB-231 cells were infected with Ad-CAGA12-Luc 24 h before implantation. Cells were implanted into mammary fat pad on both sides of SCID mice. 72 h later, IVIS imaging was performed before and after 24 h treatment (PBS for control group and 50 µg/tumor TGF-β inhibitor for treatment group). (C) Photon flux was quantified by Living Image software. Data shown are means of each treatment group (n = 12) ± SD. Statistical significance was determined by Student's t-test (** p ≤0.01, **** p ≤0.0001). p/s refers to photons/s. Please click here to view a larger version of this figure.
Discussion
We have developed a technique to allow for real-time imaging of TGF-β/Smad3 signaling in single live cells. Using this novel method, we have previously identified a sub-population of cells with dynamic TGF-β/Smad3 transcriptional activity that was associated with enhanced invasion and migration8. This method improves on traditional assays for TGF-β signaling, such as western blots of Smad3 phosphorylation and TGF-β targeted gene expression, by capturing the heterogeneity of TGF-β/Smad3 signaling within the tumor population and highlighting the importance of single cell biology. Additionally, alternate methods of single cell imaging such as nuclear translocation can be time consuming and expensive if performed in live cells and may not always translate to gene transcription16,17. While these methods are important to examine the upstream pathways of signal transduction, the adenovirus reporter system provides a unique perspective on TGF-β/Smad3 signaling in individual cells in relation to biological function.
Significantly, our method provides a sensitive and reproducible method for in vivo detection of the TGF-β/Smad3 signaling pathway in real-time during tumor progression and treatment. Similar to traditional stable luciferase in vivo imaging, detection of TGF-β/Smad3 signaling within the mouse is dependent on the number of cells and level of promoter activity18. While subcutaneous tissue provides little interference for analysis, increased depth of cells will reduce the sensitivity of signal detection. It is possible to expand the method for use in other organs, such as lung and bone, and other cancers; however, signal optimization or ex vivo imaging is recommended to achieve the sensitivity required for the cancer model of interest.
An additional benefit of using the adenovirus reporter system is that its application is likely to expand to analysis of multiple signaling pathways simultaneously in live cells. In Figure 2A, we demonstrated that MDA-MB-231 cells could be transduced with 2 separate adenovirus vectors, CMV-GFP and CAGA-TOM. This dual-infection system can be further expanded to report two different signaling pathways via both fluorescence reporter proteins and luciferase reporters (using Firefly and Guassia luciferase reporter proteins). Our lab has achieved simultaneous signaling of BMP/Smad1 and TGF-β/Smad3 signaling pathways in a number of live cancer cell lines (data not shown).
Adenovirus reporter systems provide an easy and convenient method for live cell imaging of the TGF-β/Smad signaling pathway both in vitro and in vivo. This system has distinct advantages over other commonly used reporter systems. Firstly, adenovirus vectors are reported to have among the highest viral transduction efficacy, making them an optimal system for cell lines that are difficult to transduce19,20,21,22. Additionally with current advancements in viral technologies, 'gutless' adenovirus vectors can be generated by removing most of the viral genome allowing for increased exogenous DNA capacity23. In comparison to non-viral delivery systems, adenovirus vectors represent a cost-effective, quick and easy application for cell transduction that results in far greater delivery efficiency19,20,21,22.
This protocol uses a second-generation adenovirus expression system, which is replication-incompetent in mammalian cells that do not express the E1a and E1b proteins, minimizing biosafety risk24. However, despite these enhanced biosafety features, adenovirus particles can still transduce primary human cells with high efficacy25,26. US Biosafety Level 2 and containment of adenoviral vectors is recommended when handling and disposing of adenovirus contaminated equipment. Large-scale amplification of adenovirus in HEK293A cells can result in a rare generation of replication-competent "wild-type" adenovirus27. PCR screening should be performed to identify wild-type contamination27.
Replication-incompetent adenovirus vectors are transient in expression and do not integrate with host DNA20,21. It is recommended that the experimenter identify the stability in expression of their reporter protein when using the adenovirus reporter system to confirm the validity of long-term experiments. The expression time of the adenovirus vector within a cell is proportional to the division time of the cell and viral MOI. To ensure reproducible results, it is critical that the viral titer is accurately determined for each viral batch. Additionally, excessively high volumes of the adenovirus can result in cytotoxic or cytopathic effects and it is important that an optimal MOI is empirically determined for each cell lined used to ensure best results. Further, in vivo use of adenovirus vectors can induce immune responses; where immune response is not the primary observation of the experiment, immune-compromised animals are recommended20,21. Additional quality control is required when using the adenoviral vectors to target immune responses, though the use of the adenovirus has been validated as an adjuvant to vaccine therapies28,29.
The adenovirus reporter system can be used on a broad range of primary and cultured cell lines of both dividing and non-dividing nature20,22. Using the adenovirus reporter system, we have achieved 100% infection rates in a number of cancer cell lines including brain, breast, colorectal as well Primary Mouse Embryological Fibroblast (MEF) and Canine Kidney Epithelial (MDCK) cells. Additionally, adenoviral vectors can be further modified to increase the affinity for cell-type specific receptors, providing improved organ targeting and efficacy for transduction30,31.
Applications of this method can be expanded to other signaling pathways and cell hosts. The principal advantages of the adenoviral reporter system include the cheap,high transduction efficacy, quick experimental setup and lack of integration with host DNA21,32. This method can be applied to many current assays and in vivo models, including in the evaluation of new targeted therapeutics33. We hope that the use of this technique will enhance our understanding of the relationship between signaling pathways and biological processes leading to new biological discoveries and the development of new therapies.
Disclosures
The authors report no conflicts of interest.
Acknowledgments
This work was supported by grants from the National Health and Medical Research Council (NHMRC) to H-JZ. TMBW is a recipient of an Australia Postgraduate Award from the Australian Government and the Ann Henderson Top-Up Scholarship from Australian Rotary Health in partner with Rotary of Templestowe and Dine for a Cure.
References
- Massague J, Like B. Cellular receptors for type beta transforming growth factor. Ligand binding and affinity labeling in human and rodent cell lines. The Journal of biological chemistry. 1985;260:2636–2645. [PubMed] [Google Scholar]
- Xu L, Chen YG, Massague J. The nuclear import function of Smad2 is masked by SARA and unmasked by TGFbeta-dependent phosphorylation. Nature Cell Biology. 2000;2:559–562. doi: 10.1038/35019649. [DOI] [PubMed] [Google Scholar]
- Massague J. TGFbeta signaling in context. Nature reviews. Molecular cell biology. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Development. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. The New England journal of medicine. 2000. pp. 1350–1358. [DOI] [PubMed]
- Zhu HJ, Burgess AW. Regulation of transforming growth factor-beta signaling. Molecular cell biology research communications : MCBRC. 2001;4:321–330. doi: 10.1006/mcbr.2001.0301. [DOI] [PubMed] [Google Scholar]
- Massagué J. TGFβ in Cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luwor RB, et al. Single live cell TGF-beta signaling imaging: breast cancer cell motility and migration is driven by sub-populations of cells with dynamic TGF-beta-Smad3 activity. Molecular cancer. 2015;14:50. doi: 10.1186/s12943-015-0309-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke DC, Liu X. Decoding the quantitative nature of TGF-beta/Smad signaling. Trends in Cell Biol.ogy. 2008;18:430–442. doi: 10.1016/j.tcb.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R, Li N, Xu J, Li W, Fang X. Quantitative single-molecule study of TGF-beta/Smad signaling. Acta Biochimica et Biophysica Sinica (Shanghai) 2017;50:51–59. doi: 10.1093/abbs/gmx121. [DOI] [PubMed] [Google Scholar]
- Luwor RB, et al. New reagents for improved in vitro and in vivo examination of TGF-beta signalling. Growth factors. 2011;29:211–218. doi: 10.3109/08977194.2011.615311. [DOI] [PubMed] [Google Scholar]
- Dennler S, et al. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998;17:3091–3100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luwor RB, et al. Targeting Stat3 and Smad7 to restore TGF-beta cytostatic regulation of tumor cells in vitro and in vivo. Oncogene. 2013;32:2433–2441. doi: 10.1038/onc.2012.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed LJ, Muench H. A Simple Method of Estimating Fifty Per Cent Endpoints. American Journal of Epidemiology. 1938;27:493–497. [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennler S, Prunier C, Ferrand N, Gauthier JM, Atfi A. c-Jun inhibits transforming growth factor beta-mediated transcription by repressing Smad3 transcriptional activity. The Journal of biological chemistry. 2000;275:28858–28865. doi: 10.1074/jbc.M910358199. [DOI] [PubMed] [Google Scholar]
- Fink SP, Mikkola D, Willson JK, Markowitz S. TGF-beta-induced nuclear localization of Smad2 and Smad3 in Smad4 null cancer cell lines. Oncogene. 2003;22:1317–1323. doi: 10.1038/sj.onc.1206128. [DOI] [PubMed] [Google Scholar]
- Zinn KR, et al. Noninvasive bioluminescence imaging in small animals. ILAR J. 2008;49:103–115. doi: 10.1093/ilar.49.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TK, Eberwine JH. Mammalian cell transfection: the present and the future. Analytical and Bioanalytical Chemistry. 2010;397:3173–3178. doi: 10.1007/s00216-010-3821-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell WC. Update on adenovirus and its vectors. Journal of General Virology. 2000;81:2573–2604. doi: 10.1099/0022-1317-81-11-2573. [DOI] [PubMed] [Google Scholar]
- Lee CS, et al. Adenovirus-Mediated Gene Delivery: Potential Applications for Gene and Cell-Based Therapies in the New Era of Personalized Medicine. Genes & Diseases. 2017;4:43–63. doi: 10.1016/j.gendis.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang II, Huang II. Adenovirus technology for gene manipulation and functional studies. Drug Discovery Today. 2000;5:10–16. doi: 10.1016/s1359-6446(99)01433-6. [DOI] [PubMed] [Google Scholar]
- Kochanek S, et al. A new adenoviral vector: Replacement of all viral coding sequences with 28 kb of DNA independently expressing both full-length dystrophin and beta-galactosidase. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:5731–5736. doi: 10.1073/pnas.93.12.5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Finer MH. Second-generation adenovirus vectors. Nature Medicine. 1996;2:714–716. doi: 10.1038/nm0696-714. [DOI] [PubMed] [Google Scholar]
- Durham HD, et al. Toxicity of replication-defective adenoviral recombinants in dissociated cultures of nervous tissue. Experimental Neurology. 1996;140:14–20. doi: 10.1006/exnr.1996.0110. [DOI] [PubMed] [Google Scholar]
- MacKenzie KL, Hackett NR, Crystal RG, Moore MA. Adenoviral vector-mediated gene transfer to primitive human hematopoietic progenitor cells: assessment of transduction and toxicity in long-term culture. Blood. 2000;96:100–108. [PubMed] [Google Scholar]
- Zhang WW, Koch PE, Roth JA. Detection of wild-type contamination in a recombinant adenoviral preparation by PCR. Biotechniques. 1995;18:444–447. [PubMed] [Google Scholar]
- Choi Y, Chang J. Viral vectors for vaccine applications. Clinical and Experimental Vaccine Research. 2013;2:97–105. doi: 10.7774/cevr.2013.2.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Cassan SC, et al. The requirement for potent adjuvants to enhance the immunogenicity and protective efficacy of protein vaccines can be overcome by prior immunization with a recombinant adenovirus. Journal of immunology. 2011;187:2602–2616. doi: 10.4049/jimmunol.1101004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasnykh VN, Mikheeva GV, Douglas JT, Curiel DT. Generation of recombinant adenovirus vectors with modified fibers for altering viral tropism. Journal of Virology. 1996;70:6839–6846. doi: 10.1128/jvi.70.10.6839-6846.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham TJ, et al. Increased in vitro and in vivo gene transfer by adenovirus vectors containing chimeric fiber proteins. Journal of Virology. 1997;71:8221–8229. doi: 10.1128/jvi.71.11.8221-8229.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith F, Jacoby D, Breakefield XO. Virus vectors for gene delivery to the nervous system. Restorative neurology and neuroscience. 1995;8:21–34. doi: 10.3233/RNN-1995-81207. [DOI] [PubMed] [Google Scholar]
- Zhou F, et al. Nuclear receptor NR4A1 promotes breast cancer invasion and metastasis by activating TGF-beta signalling. Nature communications. 2014;5:3388. doi: 10.1038/ncomms4388. [DOI] [PubMed] [Google Scholar]