

Abstract
The current understanding of the dynamics and structural features of T-cell synaptic interfaces has been largely determined through the use of glass-supported planar bilayers and in vitro-derived T-cell clones or lines1,2,3,4. How these findings apply to the primary human T cells isolated from blood or lymphoid tissues is not known, partly due to significant difficulties in obtaining a sufficient number of cells for analysis5. Here we address this through the development of a technique exploiting multichannel flow slides to build planar lipid bilayers containing activating and adhesion molecules. The low height of the flow slides promotes rapid cell sedimentation in order to synchronize cell:bilayer attachment, thereby allowing researchers to study the dynamic of the synaptic interface formation and the kinetics of the granules release. We apply this approach to analyze the synaptic interface of as few as 104 to 105 primary cryopreserved T cells isolated from lymph nodes (LN) and peripheral blood (PB). The results reveal that the novel planar lipid bilayer technique enables the study of the biophysical properties of primary human T cells derived from blood and tissues in the context of health and disease.
Keywords: Retraction, Issue 137, primary human T cells, peripheral blood, lymph nodes, planar lipid bilayers, structure, synaptic interface
Introduction
Scientific knowledge of the structural features of T-cell immune synapses and their link to the functional activity of T cells has been generated primarily from the study of cell lines and clones derived from PB. To what degree these findings relate to primary T cells obtained from blood or human lymphoid tissues remains unclear, as the synaptic interfaces of T cells residing in lymphoid and other tissues have not been analyzed thus far. Importantly, emerging data suggest that tissue-resident and lymphoid-organ-derived T cells may have significant differences in their phenotype and functional activity compared to those in PB6,7. This has further solidified the need to better understand the features of the T-cell synaptic interface in primary human T cells.
To this end, we have developed a novel mini-scale approach exploiting lipid bilayers built into multichannel flow slides enabling us to perform the imaging of T-cell/bilayer interfaces with less than 105 primary T cells isolated from human PB and LN. This novel technique allows the study of the biophysical properties of primary human T-cell synaptic interfaces in order to better model and understand in vivo cell-cell interactions.
Protocol
This study was conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from all participants, and blood and lymph node samples were acquired with the approval of the Institutional Review Board at the University of Pennsylvania (IRB#809316, IRB# 815056). All human subjects were adults. Cord blood samples were kindly provided by Labor and Delivery of the Department of Obstetrics & Gynecology at Thomas Jefferson University. All samples were de-identified.
1. Isolation of CD4+ T Cells for an Image Analysis
- Thaw a 1 mL aliquot containing 107 frozen peripheral blood mononuclear cells (PBMCs) or lymph node mononuclear cells (LNMCs) from collected samples. In a sterile hood, add the thawed cells to 9 mL of RPMI supplemented with penicillin/streptomycin and glutamine.
- Centrifugate the cells for 10 min at 300 x g at 4 °C, aspirate the supernatant, and resuspend the cells in 5 mL of supplemented RPMI containing 10% FBS (complete medium). Incubate the cells overnight in a CO2 incubator at 37 °C.
The next day, purify CD4+ T cells by negative immunomagnetic sorting using a commercially available kit according to the manufacturer's instruction.
To measure the number of freshly purified CD4+ T cells, mix 5 µL of the cell suspension with an equal volume of a trypan blue solution. Load a hemocytometer with a cell-trypan blue mixture and count the live cells inside the 5 sections of the hemocytometer.
Take the average of the cell number and determine the number of cells in the original cell suspension: the number of cells/1 mL = the average count x 2 x 104. If the total number of isolated cells is too small, use the cells as is without counting.
Centrifuge the cells at 300 x g for 10 min and resuspend them in an assay buffer (20 mM HEPES, pH 7.4, 137 mM NaCl, 2 mM Na2HPO4, 5 mM D-glucose, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, and 1% human serum albumin) at 105 cells/50 µL or less and keep the cells at 4 °C (for 1–2 h) until ready to use in the experiments.
Dispose of all biological waste according to the relevant institutional guidelines.
If desired as a control cell population, prepare activated CD8 T cells from cord blood PBMC, place 107 cells in 5 mL of complete medium in a T25 culture flask covered with a mixture of anti-CD3 and anti-CD28 antibodies at 10 µg/mL and 1 µg/mL, respectively.
The next day, remove the activated cord blood cells from the flask, wash them 1x with fresh complete medium and expand the cells in the presence of recombinant IL-2 (100 U/mL) for 2 weeks.
Purify the cord-blood CD8+ T cells by negative immunomagnetic sorting using the commercially available kit according to the manufacturer's instruction. Count the cells and exchange the media to the assay buffer as described in steps 1.3–1.6 for LN and PB CD8+ T cells.
2. Components for the Preparation of the Planar Lipid Bilayers
Prepare 3 kind of liposomes as described elsewhere5: (a) 0.4 mM DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine) liposomes, (b) 0.4 mM DOPC liposomes containing 33 mol% DOGS-NTA (1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl)iminodiacetic acid)succinyl] (ammonium salt)) lipids, (c) 0.4 mM DOPC liposomes containing 4 mol% Biotinyl-Cap-PE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(cap biotinyl) (sodium salt)).
- Prepare 5% casein solution as previously described5.
- Dissolve 5 g of casein powder in 100 mL of ultrapure water and add 350 µL of 10 M sodium hydroxide. Stir everything on a regular magnetic stirrer at a slow speed according to the available scale, at room temperature for 2 h, and then overnight at 4 °C. Adjust the pH to 7.3 and ultracentrifuge the solution for 2 h at 100,000 x g at 4 °C. Filter the supernatant with a 0.22 µm sterile filter and store the solution in aliquots at -80 °C. CAUTION: Sodium hydroxide solution can cause chemical burns and may induce permanent blindness upon contact with eyes. Use rubber gloves, safety clothing, and eye protection when handling this chemical or its solutions.
- Label anti-CD3 antibody with biotin to produce mono-bionylated antibody molecules with a previously described approach8.
- Prepare a solution of Biotin-PEO4-NHS in dimethyl sulfoxide (DMSO) at 0.1 mg/mL. Add 3.7 µL of the Biotin-PEO4-NHS solution to 1 mg of antibody in 0.5 mL of phosphate-buffered saline (PBS) containing 100 mM sodium bicarbonate.
- Incubate the mixture for 2 h at room temperature. Prepare a solution of Alexa Fluor 488 NHS ester at 10 mg/mL in DMSO. Add the Alexa Fluor 488 NHS ester solution to the antibody labeled by Biotin-PEO4-NHS at a 10-fold molar excess.
- Incubate the mixture for 1 h at room temperature with slow stirring on a regular magnetic stirrer. Separate the unbound dye using size-exclusion chromatography.
- Determine the antibody concentration by measuring the optical density of the antibody solution at 280 nm (A280). Measure the optical density of the labeled antibodies at 577 nm (A577).
- Determine the dye-to-antibody ratio using the following equation:
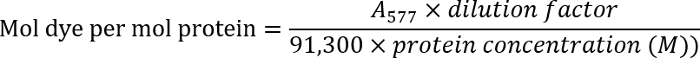 NOTE: Find additional details in the manufacturer's protocol.
NOTE: Find additional details in the manufacturer's protocol.
- Express a recombinant soluble ICAM-1 protein in a Drosophila expression system as described previously3,4,9,10.
- Clone, with cDNA encoding, the ectodomain of ICAM-1 into Drosophila expression vector pMT/V5-His with an inducible metallothionein promoter to produce a recombinant protein appended with a His6 tag on the C-terminal end.
- Co-transfect S2 cells with the resulting ICAM-1 containing plasmid and a G418 expression vector. Select stable transfectants using Schneider's Drosophila Media supplemented with 10% fetal calf serum (FCS) and 0.5 mg/mL of G418 for 3 weeks. Expand the cells in serum-free insect medium and induce a protein expression with 0.5 mM CuSO4 for 3 d.
- Concentrate the culture supernatant 10x and dialyze against PBS via a tangential flow concentrator as previously described11.
- Apply the concentrated culture supernatant to a column containing Sepharose with a covalently immobilized anti-ICAM-1 monoclonal antibody and elute the bound ICAM-1 with a 50 mM glycine buffer, pH 3.0. Immediately neutralize the eluted ICAM-1 protein with a 2 M Tris buffer, pH 8.0.
- Dialyze the eluted material against PBS, pH 8.0 and add the dialyzed material to a column containing Ni-NTA agarose. Elute soluble ICAM-1 with 200 mM imidazole, pH 8.0. Dialyze the eluted material against a PBS buffer, pH 8.0.
- Label the purified ICAM-1 with Cy5 NHS ester according to the manufacturer's instruction. NOTE: The best final dye-to-protein ratio is 1:1.
Produce Fab fragments from an anti-CD107a antibody by papain digestion and purify the Fab fragments by ion exchange chromatography as described previously3. Label the Fab fragments with Alexa Fluor 568 NHS ester according to the manufacturer's instruction.
3. Formation of Glass-supported Planar Lipid Bilayers
Prepare a fresh acidic piranha solution by mixing 140 mL of concentrated sulfuric acid and 60 mL of 30% hydrogen peroxide. Wash the glass coverslips for the flow slides by soaking them in the acid piranha solution for 30 min. Hold the glass coverslip with polypropylene scissor-type forceps. CAUTION: Piranha solution is an extremely strong oxidizer. Remember to wear safety glasses or goggles or a full-face shield together with thick rubber gloves at all times while handling the solution. Only work with piranha solution under a fume hood. Avoid heating, transporting, or shaking it at any time during use, as it may explode. Collect the piranha waste into a glass bottle with a lead-containing hole. Contact the Institutional Safety Committee about proper waste utilization.
- Rinse the washed coverslips 7x with ultrapure water by transferring them sequentially into beakers containing fresh water. Set the wet coverslips aside to let the remaining water roll off the clean glass, leaving the dry glass behind.
- Alternatively, use a pipette tip attached to a vacuum pump to carefully remove the remaining water droplets from the coverslips.
In the sterile hood, perform dilutions of various lipids to produce the liposome mix for making bilayers. First, combine 37 µL of DOPC liposomes and 3 µL of Biotinyl-Cap-PE liposomes. Second, mix 14 µL of DOPC liposomes and 15 µL of DOGS-NTA liposomes. Third, add 1 µL of the first mix to 29 µL of the second mix to fabricate the final liposome mixture.
- In the sterile hood, set up a workspace with dry coverslips close by. Aliquot 2 µL of the final liposome mixture (see step 3.3) precisely in the center of a self-adhesive slide channel. Immediately and very precisely align a clean and dry coverslip with the slide and gently lower the coverslip on the sticky side of the slide.
- If preparing more than one slide, work on one slide at a time since the liposome mixture evaporates rapidly. Turn the slide over and use the outer ring of polypropylene scissor-type forceps to apply a gentle pressure to the peripheral contact of the coverslip with the slide, making sure that the slip is tightly attached to the slide to preclude leakage. NOTE: Do not press against the channels of the slide to avoid breaking or cracking the coverslip.
- Turn the slide over again and mark the position of the formed bilayer, which looks like a drop between the coverslip and the channel slide, by drawing 4 dots with a permanent marker around the bilayer on the external slide side of the assembly.
Before the first injection of a liquid into the channel, designate one port of the channel as the entry port and the other as the exit port and maintain this designation throughout the experiment.
To avoid forming bubbles, insert the end of the pipet tip directly into the entry port of the channel.Slowly fill the channels of the slide with 50 µL of warm (at least room temperature) assay buffer (see step 1.5 for the buffer composition).
Prepare a 0.5 M nickel(II) chloride solution. Thaw a 2 mL aliquot of casein solution in a water bath at 37 °C for 30 min and supplement it with a nickel chloride solution at a final concentration of 200 µM.
Wash the bilayers by first injecting 100 µL of the casein solution in the entry port of the channel and then immediately removing 100 µL out of the exit port on the slide by pipetting. Block the bilayers with the same solution by injecting 100 µL of the casein solution into the entry port of each channel and incubating the slide for 45 min at room temperature.
Thaw aliquots of Cy5-ICAM-1-His6 and streptavidin proteins. Combine the proteins in the assay buffer at the final concentration of 2 µg/mL each. Centrifuge the solution for 30 min at 20,000 x g and 4 °C to remove any aggregates.
Remove the rest of the blocking solution from the exit port of the slide channel by pipetting. Inject 100 µL of the solution containing ICAM-1 and streptavidin into the entry port.
Incubate the slide for 45 min at room temperature. Remove any excess of the protein solution from the exit port. Wash the bilayer 2x by first injecting 100 µL of the assay buffer in the entry port of the channel and then immediately removing 100 µL out of the exit port.
Dilute an Alexa-Fluor-488-labelled anti-CD3 antibody with the assay buffer to a final concentration of 2 µg/mL. Inject 100 µL of the antibody solution into the entry port of the slide and incubate it for 45 min at room temperature. Remove any excess of the protein solution from the exit port. Wash the bilayer 2x with 100 µL of the assay buffer as in step 3.11.
4. Imaging of the T Cells Interaction with the Planar Bilayer
- Preheat the stage and the objective of a confocal or total internal reflection fluorescence (TIRF) microscope until the temperature is equilibrated at 37 °C. Set up the slide with the bilayer(s) on the heated stage. Move the stage to an appropriate position according to the ink marks and focus on the bilayer employing fluorescence of Cy-labelled ICAM-1 molecules.
- Use a 61X objective for the confocal microscope, or a 100X objective for the TIRF microscope, with appropriate filter settings.
- For granule release imaging by TIRF microscopy, add Alexa-Fluor-568-labeled anti-CD107a antibody Fab fragments to the cell suspension at a final concentration of 4 µg/mL before injecting the cells into the entry ports.
- Resuspend the prepared CD4+ T cells isolated from LN or PB or cord blood and inject 50 µL of the cell suspension into the entry port of the slide channel containing the bilayer.
Choose the desired numbers of fields and record images of each field 1x every 2 min for 30 min after the injection.
Exploit bright-field, reflected light, and fluorescent channels (Alexa 488 and Cy5) of the confocal microscope to acquire the images. Use TIRF mode for Alexa-Fluor-488 and Alexa-Fluor-568 fluorescence and widefield for Cy5 fluorescence, as well as bright-field imaging, on the TIRF microscope.
5. Image Analysis
Analyze the acquired images using appropriate software. Observe cell morphology in the transmitted light images and exclude clustered and visibly damaged or apoptotic cells from the analysis. Include in the analysis only those cells that productively interact with the bilayer surface (i.e., cells accumulating Alexa-Fluor-488 fluorescence (anti-CD3 antibodies) at the interface).
Determine the size of the cell adhesion area at 20 min after the initiation of the cell-bilayer interaction. NOTE: The adhesion area is the dark area developed at the cell-bilayer interface on the interference reflection microscopy (IRM) images.
Observe any accumulation of Cy5-ICAM-1 fluorescence and a formation of the ring junction by segregated Cy-ICAM-1 molecules at the cell-bilayer interface. If accumulated ICAM-1 molecules formed an adhesion ring junction on at least two consecutive images, designate such cells as cells developing a peripheral supramolecular activating cluster (pSMAC)12.
Evaluate the granule release by measuring the Alexa-Fluor-568 fluorescence intensity at the T cell-bilayer interface over the background fluorescence outside the contact area in close proximity to the cell. Designate cells with a ratio of Alexa-Fluor-568 signal-to-background of at least 1.3 as degranulating cells.
Representative Results
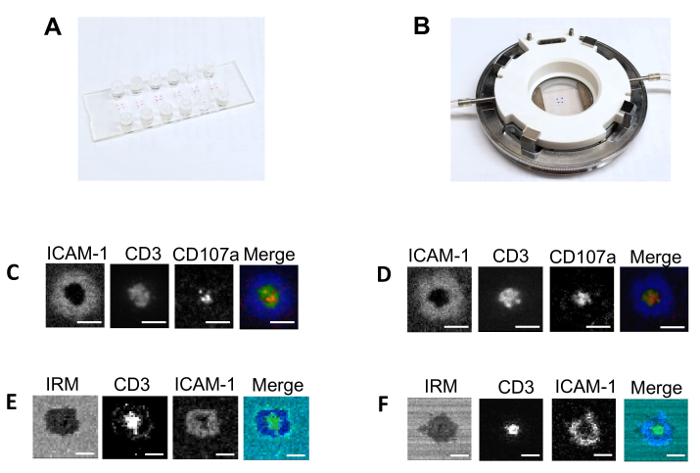
First, we compared the structure of the synaptic interface formed by activated cord-blood-derived CD8+ T cells exposed to lipid bilayers built either in traditional large-scale flow cell systems (see the Table of Materials for details)1,2,3,4 or in multichannel flow slides. The bilayers contained fluorescent-labeled anti-CD3 and ICAM-1 at 50 molecules/µm2 and 300 molecules/µm2, respectively. CD8+ T cells derived from human cord blood were activated by a stimulation with plate-bound anti-CD3 and anti-CD28 antibodies13,14. There was no difference between CD8+ T cells that formed classical immunological synapses on the lipid bilayers built in either the flow cell or the multichannel flow slide (Figure 1). All other experiments were performed with lipid bilayers made in the multichannel flow slides.
Next, we examined the ability of PB- and LN-derived human CD4+ T cells to form synaptic interfaces with the planar lipid bilayers and release granules. We exploited TIRF microscopy to maximize the vertical resolution at the T cell-bilayer interface to visualized degranulating cells and to evaluate the kinetics of the granule release. We observed 4 groups of cells for both the LN- and PBMC-derived CD4+ T cells: some T cells established mature immune synapses, but either did or did not release granules, other T cells released the granules without a formation of mature synapses, and still other T cells neither formed synapses nor released the granules (Figure 2)7. The difference between LN- and PBMC-derived CD4+ T cells became apparent when the kinetics of the granule release was evaluated. PBMC-derived CD4+ T cells were able to begin releasing granules almost twice as fast as LN-derived CD4+ cells (Figure 3)7. Overall, the data demonstrate that despite a heterogeneity of LN- and PBMC-derived CD4+ T cells, the latter contained a fraction of T cells capable of rapid degranulation.
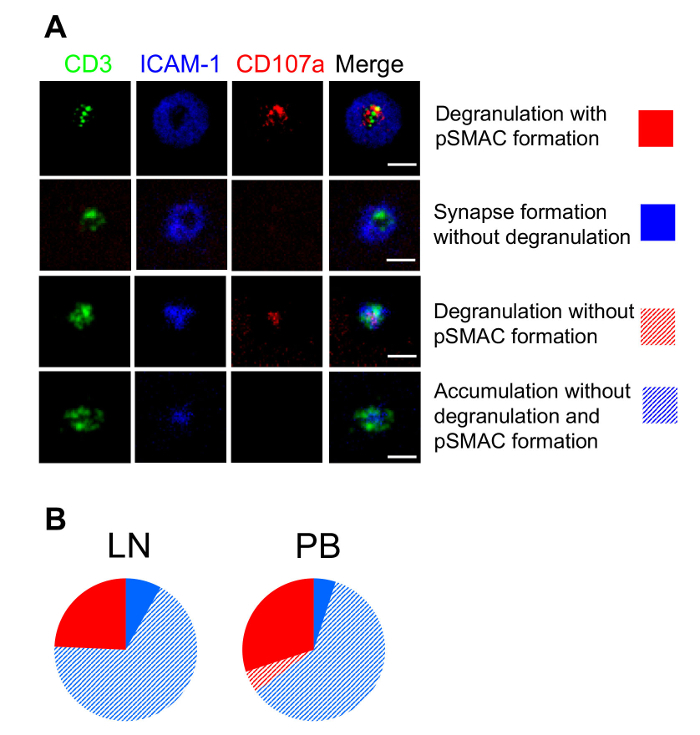
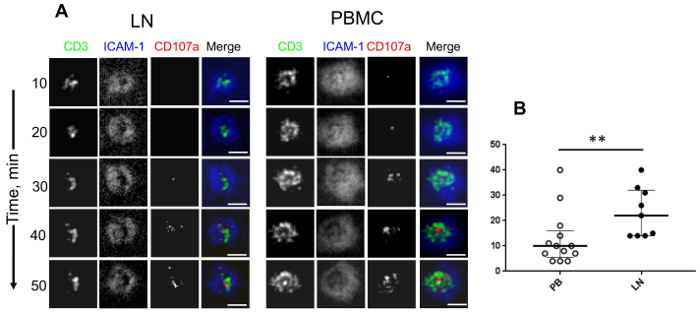
Figure 1: Comparison of the flow slide system and conventional flow chamber. (A) This panel shows the multichannel flow slide allowing an assembly of 6 glass-supported planar lipid bilayers. Each bilayer is connected to the delivery and the exit ports. Less than 1 x 105 cells are sufficient for the analysis. (B) This panel shows the conventional flow chamber system allowing an assembly of 1 glass-supported planar lipid bilayer. 2 x 106 cells are required for the analysis. The other panels show (C and D) representative TIRF microscopy and (E and F) confocal with interference reflection microscopy (IRM) images of the interface developed by activated cord-blood-derived CD8 T cells. The cells were exposed to a lipid bilayer containing ICAM-1 (300 molecules/µm2) and anti-CD3 antibodies (50 molecules/µm2) acquired (C and E) in flow slides or (D and F) in a conventional flow system under similar conditions. The TIRF microscopy and confocal with IRM images are taken using 100X and 60X magnification objectives, respectively. The percentage of T cells that form mature synapses on the bilayers built in either flow slide or conventional flow chamber was very similar but varies between experiments from 75% to 90%. In all images, Cy-5-labelled ICAM-1 molecules are shown in blue; Alexa-Fluor-488-labeled anti-CD3 antibodies are shown in green; Alexa-Fluor-568-labeled anti-CD107a antibodies bound to CD107a are shown in red; IRM images are shown in cyan; and the scale bars are 10 µm in length. Please click here to view a larger version of this figure.
Figure 2: Structure of a T cell–bilayer interface and the pattern of degranulation by lymph node and peripheral blood mononuclear CD4+ T cells. LNMC- or PBMC-derived cells were sorted to separate CD4+ T cells that were exposed to the bilayers containing 50 molecules/µm2 of anti-CD3 antibodies and 300 molecules/µm2 of ICAM-1 protein. The interface between the cells and the bilayers was imaged by TIRF microscopy. The scale bars are 5 µm. (A) These representative images of a T cell - bilayer interface demonstrate the structure of the T cell–bilayer interfaces and degranulation pattern. (B) These diagrams show a representation of LNMC- and PBMC-derived CD4+ T cells with a different structure of synaptic interfaces and patterns of granule release. Please click here to view a larger version of this figure.
Figure 3: Dynamic changes of the structure of a T cell–bilayer interface and the kinetics of a degranulation of lymph node and peripheral blood mononuclear CD4+ T cells. (A) This panel shows time-dependent changes of a T cell–bilayer interface and the appearance of released granules. The scale bars are 5 µm. (B) This panel shows a quantitation of the kinetics of a granule release by LN-derived CD4+ T cells (closed circles) and PBMC-derived CD4+ T cells (open circles). Each individual circle indicates the time of the first appearance of a detectable granule release by individual cells. The median and IQR (interquartile range) are shown for all scatter plots. Mann-Whitney tests were performed to compare differences between the indicated groups of T cells. * P <0.05, ** P <0.01. Please click here to view a larger version of this figure.
Discussion
The novel technique described here utilizes similar reagents required to build planar bilayers in the conventional flow cell5 and can be successfully applied to perform the imaging of primary human T cell–bilayer interfaces3,4,15. The technique offers a significant reduction in the fluorescent molecules usage and requires 10–20x fewer T cells as compared to a flow cell system5, creating the opportunity to analyze primary human T cells from blood and other tissues.
The number of injected cells used in this study allowed us to image about 50 cells per imaging field with a 60X objective. A larger concentration resulted in cell aggregation, reducing the number of cells suitable for analysis. We could further reduce the number of injected cells 10–50x, but the lower limit of the cell number depends on cell heterogeneity, on the number of imaging fields, and on the experimental design. Some investigators have previously used bilayers built in an open chamber with a glass bottom that required a similar number of cells for analysis16. However, the closed system described here, with a channel height of 0.1 - 0.4 mm, allows for rapid cell sedimentation to synchronize the cell attachment and analysis of the T-cell response without the risk of liquid evaporation. The sticky slide system further permits the formation of up to three bilayers per channel. Thus, the same cell sample could be used for the analysis of cell behavior on distinct bilayers with a different composition of stimulatory, adhesion, and costimulatory molecules.
Some precautions should be taken during the assembly of the sticky slides and coverslips for a successful bilayer formation. Particularly, the coverslips should be completely dry, as even a small amount of liquid left on the glass surface could result in leakage during the bilayer washing procedure. Importantly, it is essential to even the attached coverslip at the edges of the contact with the outer ring of polypropylene scissor-type forceps to avoid leakage (as described in step 3.4.1). It is also important to avoid any bubbling in the entry ports of the channels. If any bubbles enter a channel, they are almost impossible to remove and ruin the bilayer in the channel. Similarly, it is important to avoid any fast pipetting, since the turbulent flow of liquid may introduce bubbles into the channel and damage the bilayer.
A release of cytolytic granules is detected by the appearance of CD107a at the T cell-bilayer interface. Fab regions of Alexa-Fluor-568-labeled anti-CD107a antibodies are added to CD8 T cells prior to the loading on the bilayers. TIRF microscopy exploits an evanescent wave to illuminate the area about 100 nm above the bilayer in the volume defined as evanescent volume, allowing to increase the vertical resolution of the imaging. Labeled antibody Fabs, which diffuse very rapidly within the evanescent volume at 37 °C, do not generate a detectable fluorescent signal. During the membrane fusion of specialized lysosomes containing lytic molecules with the cell membrane, CD107a membrane protein appears on the T cells contact surface in distinct locations. The Fabs get bound to the CD107s protein at that time. Attached to the CD107a protein, fluorescent-labeled Fab(s) form immobile clusters that generate a bright fluorescence detectable by TIRF microscopy, indicative of T cell degranulation.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by the R01AI118694 NIH grant to Michael R. Betts, which includes sub-award 566950 to Yuri Sykulev. We thank the Sidney Kimmel Cancer Center Bioimaging Shared Resource for their excellent support.
References
- Grakoui A, et al. The immunological synapse: a molecular machine controlling T cell activation. Science. 1999;285:221–227. [PubMed] [Google Scholar]
- Somersalo K, et al. Cytotoxic T lymphocytes form an antigen-independent ring junction. Journal of Clinical Investigation. 2004;113:49–57. doi: 10.1172/JCI200419337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal AM, et al. Protein kinase C theta regulates stability of the peripheral adhesion ring junction and contributes to the sensitivity of target cell lysis by CTL. The Journal of Immunology. 2008;181:4815–4824. doi: 10.4049/jimmunol.181.7.4815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal AM, et al. Kinetics of early T cell receptor signaling regulate the pathway of lytic granule delivery to the secretory domain. Immunity. 2009;31:632–642. doi: 10.1016/j.immuni.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dustin ML, Starr T, Varma R, Thomas VK. Supported planar bilayers for study of the immunological synapse. Current Protocols in Immunology. 2007. Chapter 18, Unit 18 13. [DOI] [PubMed]
- Reuter MA, et al. HIV-Specific CD8(+) T Cells Exhibit Reduced and Differentially Regulated Cytolytic Activity in Lymphoid Tissue. Cell Reports. 2017;21:3458–3470. doi: 10.1016/j.celrep.2017.11.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buggert M, et al. Limited immune surveillance in lymphoid tissue by cytolytic CD4+ T cells during health and HIV disease. PLoS Pathogens. 2018;14:e1006973. doi: 10.1371/journal.ppat.1006973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco YR, Fleire SJ, Cameron T, Dustin ML, Batista FD. LFA-1/ICAM-1 interaction lowers the threshold of B cell activation by facilitating B cell adhesion and synapse formation. Immunity. 2004;20:589–599. doi: 10.1016/s1074-7613(04)00105-0. [DOI] [PubMed] [Google Scholar]
- Anikeeva N, et al. Distinct role of lymphocyte function-associated antigen-1 in mediating effective cytolytic activity by cytotoxic T lymphocytes. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:6437–6442. doi: 10.1073/pnas.0502467102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steblyanko M, Anikeeva N, Campbell KS, Keen JH, Sykulev Y. Integrins Influence the Size and Dynamics of Signaling Microclusters in a Pyk2-dependent Manner. The Journal of Biological Chemistry. 2015;290:11833–11842. doi: 10.1074/jbc.M114.614719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anikeeva N, Lebedeva T, Sumaroka M, Kalams SA, Sykulev Y. Soluble HIV-specific T-cell receptor: expression, purification and analysis of the specificity. Journal of Immunological Methods. 2003;277:75–86. doi: 10.1016/s0022-1759(03)00179-0. [DOI] [PubMed] [Google Scholar]
- Monks C, Freiberg B, Kupfer H, Sciaky N, Kupfer A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature. 1998;395:82–86. doi: 10.1038/25764. [DOI] [PubMed] [Google Scholar]
- Riddell SR, Greenberg PD. The use of anti-CD3 and anti-CD28 monoclonal antibodies to clone and expand human antigen-specific T cells. Journal of Immunological Methods. 1990;128:189–201. doi: 10.1016/0022-1759(90)90210-m. [DOI] [PubMed] [Google Scholar]
- Lin SJ, Yu JC, Cheng PJ, Hsiao SS, Kuo ML. Effect of interleukin-15 on anti-CD3/anti-CD28 induced apoptosis of umbilical cord blood CD4+ T cells. European Journal of Haematology. 2003;71:425–432. doi: 10.1046/j.0902-4441.2003.00162.x. [DOI] [PubMed] [Google Scholar]
- Anikeeva N, Sykulev Y. Mechanisms controlling granule-mediated cytolytic activity of cytotoxic T lymphocytes. Immunologic Research. 2011;51:183–194. doi: 10.1007/s12026-011-8252-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppa JB, et al. TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature. 2010;463:963–967. doi: 10.1038/nature08746. [DOI] [PMC free article] [PubMed] [Google Scholar]