

Abstract
The human genome encodes four bestrophin paralogs, namely BEST1, BEST2, BEST3, and BEST4. BEST1, encoded by the BEST1 gene, is a Ca2+-activated Cl- channel (CaCC) predominantly expressed in retinal pigment epithelium (RPE). The physiological and pathological significance of BEST1 is highlighted by the fact that over 200 distinct mutations in the BEST1 gene have been genetically linked to a spectrum of at least five retinal degenerative disorders, such as Best vitelliform macular dystrophy (Best disease). Therefore, understanding the biophysics of bestrophin channels at the single-molecule level holds tremendous significance. However, obtaining purified mammalian ion channels is often a challenging task. Here, we report a protocol for the expression of mammalian bestrophin proteins with the BacMam baculovirus gene transfer system and their purification by affinity and size-exclusion chromatography. The purified proteins have the potential to be utilized in subsequent functional and structural analyses, such as electrophysiological recording in lipid bilayers and crystallography. Importantly, this pipeline can be adapted to study the functions and structures of other ion channels.
Keywords: Retraction, Issue 138, Mammalian membrane protein, ion channels, retinal degenerative diseases, protein expression, protein purification, Bestrophin-1, BEST1, Ca2+-activated Cl- channel (CaCC)
Introduction
Bestrophins are a family of ion channels conserved through species varying from bacteria to humans1. In humans, the BEST1 gene, located on chromosome 11q12.3, encodes the membrane protein Bestrophin-1 (BEST1) that is predominantly expressed in the basolateral membrane of retinal pigment epithelium (RPE) cells of the eyes2,3,4. Comprised of 585 amino acids, the first ~350 of which are highly conserved among species and contain its transmembrane region, BEST1 acts as a CaCC in humans1,5,6. Furthermore, the BEST1 homologs in chickens and Klebsiella pneumoniae both function as homopentamers7,8, suggesting a high level of conservation throughout evolution.
In humans, over 200 mutations in the BEST1 gene have been clinically linked to a group of retinal degeneration diseases called bestrophinopathies1,9. Five specific bestrophinopathies have been reported, including Best disease, adult-onset vitelliform dystrophy, autosomal dominant vitreoretinochoroidopathy, autosomal recessive bestrophinopathy, and retinitis pigmentosa3,4,10,11,12,13,14. These diseases, which lead to decreased eyesight and even blindness, are currently untreatable. In order to develop therapeutic treatments and potentially personalized medicine, it is critical to understand how these BEST1 disease-causing mutations influence the function and structure of the BEST1 channel15. For these purposes, researchers need to obtain purified bestrophin (wild type and/or mutant) channels and conduct in vitro analyses5,8.
The first key step is the expression of bestrophin channels from higher species in mammalian cells. As baculovirus transduction of HEK293-F cells (the BacMam system) is a powerful method to heterologously express membrane proteins16,17, this protocol utilizes an optimized BacMam vector (pEG BacMam) for robust expression of the target protein18, which in this case is a mammalian bestrophin homolog. This vector has been used for the expression of various membrane proteins, including G-protein-coupled receptors, nuclear receptors, and other ion channels18. There is also evidence that the produced proteins are suitable for crystallography18. With high levels of expression in HEK293-F cells, the proteins can then be purified using chromatography; specifically, in the case of bestrophins, both affinity and size-exclusion chromatography can be used.
Once this protocol is fine-tuned for a bestrophin channel, the purified protein can then be analyzed for its function and structure through planar lipid bilayer and X-ray crystallography, respectively5,8. Altogether, these techniques provide a powerful pipeline for functional and structural investigations of bestrophins and other ion channels.
Protocol
1. Producing BacMam Expression Baculoviruses
Insert the coding sequence of a desired mammalian bestrophin protein into the pEG BacMam vector18 with a Tobacco Etch Virus (TEV) protease recognition sequence, followed by a GFP-10x His-tag at the C-terminus of the protein.
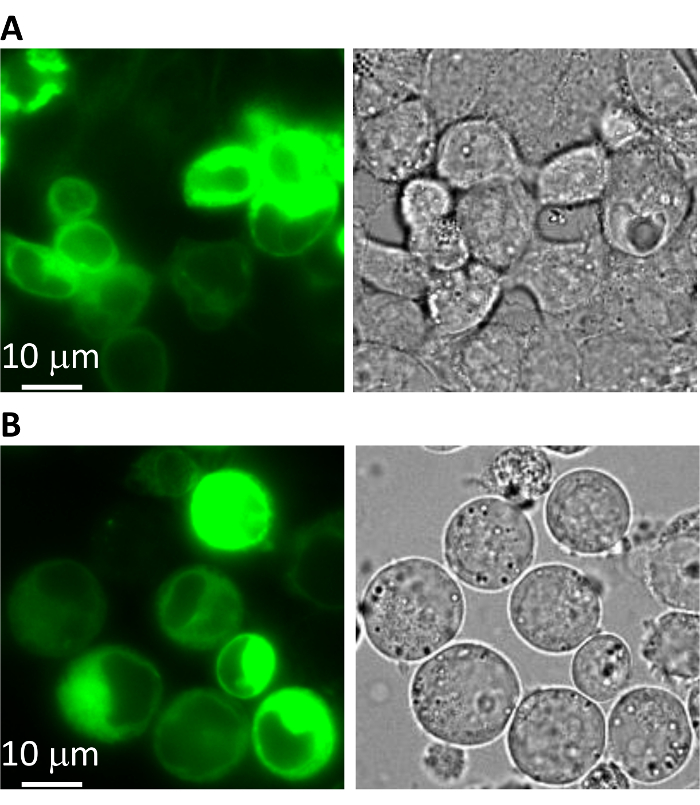
Transiently transfect the expression plasmid into adhesive HEK293 cells19,20,21,22,23. Check the expression of the GFP-fusion protein under a fluorescent microscope with 10X or 20X magnification, a 488 nm excitation source, and a 510 nm emission filter (Figure 1A). Note: Polymer-based transfection is routinely performed with 1 μg of plasmid DNA for a 35 mm dish of HEK293 cells.
Produce the baculoviruses in Sf9 insect cells as previously described16,17. Note: The passage 3 (P3) virus, which will be used to infect the HEK293-F cells for protein production, can be stored at 4 °C for 1 month.
Determine the titer (infectious units) of the P3 virus by the plaque formation assay16,17. Note: As the protein of interest is tagged with GFP, a faster method for checking infectious units is to infect Sf9 cells with serial dilutions of the virus and count the number of fluorescent cells after 48 h.
2. Protein Expression
Maintain the HEK293-F cells in a suspension culture with the HEK293-F expression medium in a 37 °C humidified incubator with 8% CO2. Use disposable culture flasks that are 2-2.5 times bigger than the culture volume and rotate the culture on an orbital platform at 135 rpm. Note: It is recommended to infect the cells when they are between passages 5 and 30 and to perform these actions under a cell culture hood.
24 h before viral infection, add 15 μL of Trypan blue to a 15 μL aliquot of cell culture, and check the cell density and viability using a hemocytometer under a microscope. Dilute the cells to 0.6 x 106 cells/mL in 2 x 2 L flasks each, with 500 mL of medium pre-warmed at 37 °C. Note: Dead cells are stained blue by Trypan blue. The desired cell viability is >95%.
On the day of infection, check the cell density and viability (step 2.2). Note: A high cell viability of >95% is essential for efficient infection and protein expression. The expected cell density is ~1.0 x 106 cells/mL (grown overnight from 2 x 500 mL of the 0.6 x 106 cells/mL cell culture). The expected total number of cells is ~1.0 x 109.
Add the P3 baculoviruses to the cells at a multiplicity of infection (MOI) of 5. To determine the amount of virus to inoculate, use the following equation:
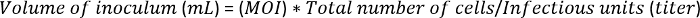
Shake the cells on the orbital platform at 135 rpm in a humidified 37 °C incubator with 8% CO2 for 24 h.
Add 10 mM of sodium butyrate to the cell culture and continue incubating for 48 h. Note: For some proteins, reducing the cell culture temperature to 30 °C after sodium butyrate addition may improve expression and stability.
Under a fluorescence microscope with 10X or 20X magnification, a 488 nm excitation source, and a 510 nm emission filter, check the percentage and brightness of green cells, which directly represent the protein (bestrophin-GFP-10xHis) yield (Figure 1B). Note: The percentage of green cells is calculated by: 100 x (Green cells/Total cells). Good protein expression level is indicated by strong green fluorescence under the microscope.
Harvest the cells by centrifuging at 1,000 x g in 4 °C for 20 min. Remove the supernatant and re-suspend the cell pellets with phosphate-buffered saline (PBS) to a final volume of ~80 mL. Split the cell suspension into 2 x 50 mL conical tubes and centrifuge at 1,000 x g in 4 °C for 20 min. Store the cell pellets at -80 °C.
3. Protein Purification
Incubate the conical tubes containing the cell pellets in a stirring water bath at room temperature for 10-15 min so that they thaw. Re-suspend the cells in 2x (w/v) volume of buffer A (Table 1) (e.g., 10 g of cell pellets in 20 mL of buffer) supplemented with proteinase inhibitors (Aprotinin, Leupeptin, Pepstatin A and phenylmethylsulfonyl fluoride) at 0.1-1.0 mM. Pipet up and down extensively to obtain a homogenous cell suspension.
Lyse the cells using a high-pressure homogenizer. Run the cell suspension through the homogenizer at 7-10 MPa for a total of 3-4 times to achieve complete homogenization. Keep the cell lysate on ice for 2-3 mins between rounds.
Add detergent [e.g., 2% w/v sol-grade n-Dodecyl-β-D-Maltopyranoside (DDM)] to the cell lysate. Incubate with agitation for 1 h at 20 °C to extract the membrane proteins. Note: The type of detergent needs to be optimized for each protein target18,24.
Centrifuge at 150,000 x g in an ultracentrifuge at 4 °C for 1 h. Note: All of the following steps are performed at 4 °C.
Collect the clear cell lysate and flow it through a 5 mL His Trap Ni2+-NTA affinity column pre-equilibrated with buffer A (Table 1). Note: After centrifugation, the clear cell lysate will be sandwiched between a pellet at the bottom and a cloudy layer on top. Use a 10 mL transfer pipet to carefully collect the clear lysate and then switch to a 1 mL pipet for the last few milliliters of lysate. Avoid the pellet, which can clog the Ni2+ column; however, a small amount of the top layer is fine or can be filtered out with a 1.5 μm filter.
Wash the column with 25 mL of buffer B and then 40 mL of buffer C (Tables 2 and 3, respectively). Note: This is a good place to take a break, as total purification may take up to 12 h and the protein can remain stable while attached to the column overnight.
Attach the column to a fast protein liquid chromatography (FPLC) system and elute the protein from the column with 13 mL of buffer D (Table 3) with fractionation. Collect the protein-enriched fractions according to the UV absorbance readout. Note: The FPLC conditions are as follows: a 1.0 mL fraction size, a pre-column pressure alarm set to 0.3 MPa, and a flow rate of 0.5 mL/min.
Measure the eluted protein product concentration on a microvolume spectrophotometer. Read the absorbance at 280 nm.
(Optional) To remove the GFP-10xHis tag, add the TEV protease at a 1:1 mass ratio and incubate at 4 °C for 30 min. Note: The tag may or may not affect the function or structure of the purified channel. Calculate the TEV amount from the volume and concentration of the collected protein from steps 3.7-3.8. For instance, if 10 mL of elution product is collected and measured at 0.1 mg/mL, the total protein mass is 10 x 0.1 = 1 mg, and 1 mg of TEV will be used for digestion.
Concentrate the protein with a 15 mL centrifugal (50 or 100 kDa) filter unit by spinning at 4,000 x g in 4 °C to a final volume of 400-500 μL (for a 2 mL sample-loading loop on FPLC). Note: The centrifuging time for concentration varies depending on the concentration and size of the protein. To avoid over-concentrating, which may cause protein precipitation, spin for 10 min at first, then observe the remaining volume and estimate the time for a subsequent spin (e.g., 2-5 min), and so on, to obtain the final volume. Pipet between spins to disperse the protein, which is pulled to the bottom of the filter.
Transfer the concentrated protein to a new 1.5 mL tube and remove any precipitate or bubbles from the concentrated product. Spin at >12,000 x g for 5-10 min at 4 °C and collect the supernatant in a new 1.5 mL tube.
Load the final product with a 1 mL syringe and a round-tip needle on a FPLC system for size-exclusion chromatography with a size-exclusion column pre-equilibrated with buffer E (Table 5). Note: The FPLC conditions are as follows: a 0.5 mL fraction size, a pre-column pressure alarm set to 2.50 MPa, a flow volume set to 30 mL of buffer E(Table 5), and a flow rate set to 0.5 mL/min.
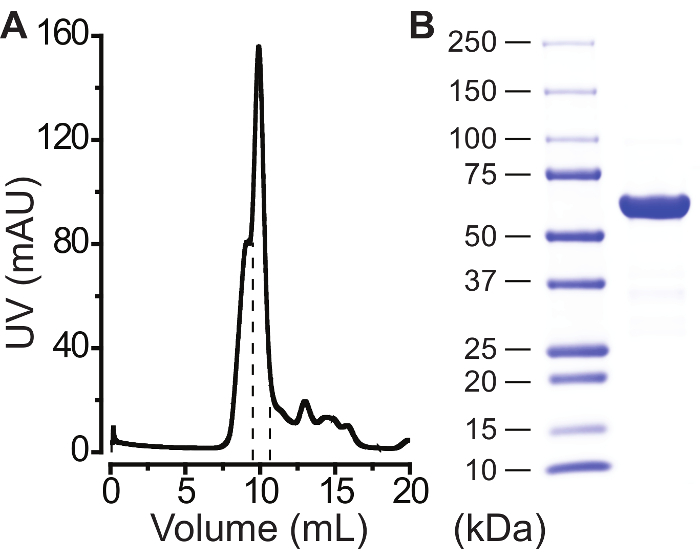
Note that well-behaved proteins run as a single peak (Figure 2A) and collect the protein fraction(s) corresponding to that peak (Figure 2A).
Concentrate the protein with a 4 mL or 0.5 mL centrifugal filter unit (of the same molecular weight cut-off as in step 3.10) and spin at 4,000 x g at 4 °C to a final concentration of 5-10 μg/μL. Use the spectrophotometer to check the final product concentration (step 3.8). Note: Centrifugation time varies, as the final product volume can differ between purification trials. Usually, centrifugation takes 10-20 min. It is recommended to pipet between spins to disperse the protein, which is pulled to the bottom of the filter.
Transfer the concentrated protein to a new 1.5 mL tube. Remove any precipitate by centrifuging at >12,000 x g for 5-10 min at 4 °C. Transfer the supernatant to a new 1.5 mL tube. Make 10 μL aliquots for storage at -80 °C and save one 2-5 μL small aliquot to run on a 4-15% gradient SDS-PAGE gel at 9 V/cm (Figure 2B).
Confirm the identity of the purified protein by mass spectrometry8. Note: In vitro analysis of the function and structure of the purified protein can be performed through planar lipid bilayer and X-ray crystallography, respectively5,8.
Representative Results
The fluorescence intensity in transiently-transfected adhesive HEK293 cells (Figure 1A) is a good indicator for the projected protein expression level in suspension HEK293-F cells (Figure 1B). If the target protein is not well-expressed or is mis-localized in HEK293 cells after transient transfection, it is recommended to consider modifying the expression construct (e.g., changing the position of the GFP tag or making mutations/truncations on the target protein). Small HEK293-F suspension cultures (e.g., 25 mL cultures) are used to optimize the conditions for protein expression, among which the infection MOI and culturing temperature have been the most important in previous experiences.
A successful purification is indicated by a single main peak at the expected elution volume in size-exclusion chromatography (Figure 2A) and a single dominant band on a denatured SDS-PAGE gel (Figure 2B). If multiple peaks appear in size-exclusion chromatography, it is important to collect each peak and run a native gel for determining which peak contains the functional channel pentamers. It should be noted that between two protein constructs, the expression level, indicated by fluorescence intensity at the time of harvest, does not nesessarily correlate with the final yield, as each protein construct behaves differently during purification. For a well-behaved bestrophin protein, 200-500 µg of the final purified product can typically be obtained from 1 L of HEK293-F suspension cells.
Figure 1: Expression of a mammalian bestrophin protein. Microscopic images of a GFP-tagged mammalian bestrophin expressed in (A) adhesive HEK293 cells by plasmid transfection, and in (B) suspension HEK293F cells by baculovirus infection. Left = green fluorescence; right = bright-field. Please click here to view a larger version of this figure.
Figure 2: Purification of a mammalian bestrophin protein. (A) Affinity-purified GFP-tagged proteins ran on a size exclusion gel-filtration column as one main peak. Fractions between dashed lines were collected. (B) Final protein product ran on a SDS-PAGE gel with ladders (left column). Please click here to view a larger version of this figure.
| Reagent | MW | Amount per 1 L | Final |
| HEPES | 238.3 | 11.9 g | 50 mM |
| NaCl | 58.44 | 17.529 g | 300 mM |
| Glycerol | 92.09 | 50 mL | 5% (v/v) |
| Imidazole | 68.1 | 1.3616 g | 20 mM |
| MgCl2 | 203 | 0.20331 g | 1 mM |
| tris(2-carboxyethyl)phosphine | 287 | 2.5 mL 200 mM stock | 0.5 mM |
Table 1: Protein Purification Buffer A (Resuspension Buffer)
| Reagent | MW | Amount per 1 L | Final |
| HEPES | 238.3 | 11.9 g | 50 mM |
| NaCl | 58.44 | 17.529 g | 300 mM |
| Glycerol | 92.09 | 50 mL | 5% (v/v) |
| Imidazole | 68.1 | 2.7232 g | 40 mM |
| MgCl2 | 203 | 1.01655 g | 5 mM |
| tris(2-carboxyethyl)phosphine | 287 | 0.5 mL 200 mM stock | 0.1 mM |
| DDM (anagrade) | 510.6 | 0.5 g | 0.05% (w/v) |
Table 2: Protein Purification Buffer B (First Wash Buffer)
| Reagent | MW | Amount per 1 L | Final |
| HEPES | 238.3 | 5.95 g | 25 mM |
| NaCl | 58.44 | 29.2 g | 500 mM |
| Glycerol | 92.09 | 50 mL | 5% (v/v) |
| Imidazole | 68.1 | 5.1 g | 75 mM |
| tris(2-carboxyethyl)phosphine | 287 | 0.5 mL 200 mM stock | 0.1 mM |
| DDM (anagrade) | 510.6 | 0.5 g | 0.05% (w/v) |
Table 3: Protein Purification Buffer C (Second Wash Buffer)
| Reagent | MW | Amount per 1 L | Final |
| HEPES | 238.3 | 7.74 g | 32.5 mM |
| NaCl | 58.44 | 11.686 g | 200 mM |
| Glycerol | 92.09 | 25 mL | 2.5% (v/v) |
| Imidazole | 68.1 | 17.02 g | 250 mM |
| tris(2-carboxyethyl)phosphine | 287 | 0.5 mL 200 mM stock | 0.1 mM |
| DDM (anagrade) | 510.6 | 0.5 g | 0.05% (w/v) |
Table 4: Protein Purification Buffer D (Elution Buffer)
| Reagent | MW | Amount per 1 L | Final |
| HEPES | 238.3 | 9.53 g | 40 mM |
| NaCl | 58.44 | 11.686 g | 200 mM |
| tris(2-carboxyethyl)phosphine | 287 | 0.5 mL 200 mM stock | 0.1 mM |
| DDM (anagrade) | 510.6 | 0.5 g | 0.05% (w/v) |
Table 5: Protein Purification Buffer E (Gel Filtration Buffer)
Discussion
This protocol describes a useful pipeline for expression and purification of mammalian bestrophin ion channels to be used for future in vitro analyses. While the FPLC device is required for size-exclusion chromatography, a syringe pump is sufficient for all steps of affinity chromatography including binding, washing, and eluting. When using a syringe pump to push solutions (in a syringe) through a column, it is essential to underlay the spring side to avoid pushing air bubbles into the column. If the purity of the protein after affinity chromatography is already sufficient for the subsequent experiments, size-exclusion chromatography can be omitted so that a FPLC device may not be required at all.
As it takes about 2 weeks to make the baculoviruses from pBacMam plasmids, to increase efficiency, an initial screen for well-behaved protein homologs can be carried out by harvesting the transiently transfected HEK293 cells (e.g., from a 60 mm dish) and testing how the expressed protein of interest (GFP-tagged) behaves in detergents (e.g., DDM) with fluorescence-detection size-exclusion chromatography (FSEC)18,24. It is most productive to focus on protein constructs that display strong fluorescence in transfected HEK293 cells and good monodispersity and stability in FSEC.
There are several recommendations for increasing the yield of protein purification. First, the conditions for TEV digestion may need to be optimized for each protein. This can be done by testing a range of protein-to-TEV mass ratios (e.g., 10:1, 5:1, 2:1, 1:1, 1:2, 1:5, and 1:10) and incubating times (e.g., 0.5 h, 1 h, 2 h, 5 h, and overnight). Then, it is suggested to run the treated samples in a SDS-PAGE gel to check the cleavage efficiency. In addition, it is important not to let any centrifugal supernatant or eluent leak out of the connections on the FPLC or syringes. Similarly, it is important to avoid bubbles when pipetting the concentrated protein.
Various subsequent in vitro analysis procedures that utilize the proteins from this protocol include lipid bilayer, X-ray crystallography, cryo-electron microscopy, spectroscopy, and high throughput screening8,25,26.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This project was funded by NIH grants EY025290, GM127652, and University of Rochester start-up funding.
References
- Hartzell HC, Qu Z, Yu K, Xiao Q, Chien LT. Molecular physiology of bestrophins: multifunctional membrane proteins linked to best disease and other retinopathies. Physiological Review. 2008;88(2):639–672. doi: 10.1152/physrev.00022.2007. [DOI] [PubMed] [Google Scholar]
- Marmorstein AD, et al. Bestrophin, the product of the Best vitelliform macular dystrophy gene (VMD2), localizes to the basolateral plasma membrane of the retinal pigment epithelium. Proceedings of the National Academy of Sciences of the USA. 2000;97(23):12758–12763. doi: 10.1073/pnas.220402097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt A, et al. Mutations in a novel gene, VMD2, encoding a protein of unknown properties cause juvenile-onset vitelliform macular dystrophy (Best's disease) Human Molecular Genetics. 1998;7(9):1517–1525. doi: 10.1093/hmg/7.9.1517. [DOI] [PubMed] [Google Scholar]
- Petrukhin K, et al. Identification of the gene responsible for Best macular dystrophy. Nature Genetics. 1998;19(3):241–247. doi: 10.1038/915. [DOI] [PubMed] [Google Scholar]
- Li Y, et al. Patient-specific mutations impair BESTROPHIN1's essential role in mediating Ca2+-dependent Cl- currents in human RPE. eLife. 2017;6 doi: 10.7554/eLife.29914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunenari T, et al. Structure-function analysis of the bestrophin family of anion channels. Journal of Biological Chemistry. 2003;278(42):41114–41125. doi: 10.1074/jbc.M306150200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane Dickson V, Pedi L, Long SB. Structure and insights into the function of a Ca(2+)-activated Cl(-) channel. Nature. 2014;516(7530):213–218. doi: 10.1038/nature13913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, et al. Structure and selectivity in bestrophin ion channels. Science. 2014;346(6207):355–359. doi: 10.1126/science.1259723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AA, et al. Bestrophin 1 and retinal disease. Progress in Retinal and Eye Research. 2017. [DOI] [PMC free article] [PubMed]
- Allikmets R, et al. Evaluation of the Best disease gene in patients with age-related macular degeneration and other maculopathies. Human Genetics. 1999;104(6):449–453. doi: 10.1007/s004390050986. [DOI] [PubMed] [Google Scholar]
- Burgess R, et al. Biallelic mutation of BEST1 causes a distinct retinopathy in humans. American Journal of Human Genetics. 2008;82(1):19–31. doi: 10.1016/j.ajhg.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson AE, et al. Missense mutations in a retinal pigment epithelium protein, bestrophin-1, cause retinitis pigmentosa. American Journal of Human Genetics. 2009;85(5):581–592. doi: 10.1016/j.ajhg.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer F, et al. Mutations in the VMD2 gene are associated with juvenile-onset vitelliform macular dystrophy (Best disease) and adult vitelliform macular dystrophy but not age-related macular degeneration. European Journal of Human Genetics. 2000;8(4):286–292. doi: 10.1038/sj.ejhg.5200447. [DOI] [PubMed] [Google Scholar]
- Yardley J, et al. Mutations of VMD2 splicing regulators cause nanophthalmos and autosomal dominant vitreoretinochoroidopathy (ADVIRC) Investigative Ophthalmology & Visual Science. 2004;45(10):3683–3689. doi: 10.1167/iovs.04-0550. [DOI] [PubMed] [Google Scholar]
- Yang T, Justus S, Li Y, Tsang SH. BEST1: the Best Target for Gene and Cell Therapies. Molecular Therapy. 2015;23(12):1805–1809. doi: 10.1038/mt.2015.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce FM, Bucher NL. Baculovirus-mediated gene transfer into mammalian cells. Proceedings of the National Academy of Sciences of the USA. 1996;93(6):2348–2352. doi: 10.1073/pnas.93.6.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kost TA, Condreay JP, Jarvis DL. Baculovirus as versatile vectors for protein expression in insect and mammalian cells. Nature Biotechnology. 2005;23(5):567–575. doi: 10.1038/nbt1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goehring A, et al. Screening and large-scale expression of membrane proteins in mammalian cells for structural studies. Nature Protocols. 2014;9(11):2574–2585. doi: 10.1038/nprot.2014.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, He LL, Chen M, Fang K, Colecraft HM. Bio-inspired voltage-dependent calcium channel blockers. Nature Communications. 2013;4:2540. doi: 10.1038/ncomms3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Hendrickson WA, Colecraft HM. Preassociated apocalmodulin mediates Ca2+-dependent sensitization of activation and inactivation of TMEM16A/16B Ca2+-gated Cl- channels. Proceedings of the National Academy of Sciences of the USA. 2014;111(51):18213–18218. doi: 10.1073/pnas.1420984111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Puckerin A, Colecraft HM. Distinct RGK GTPases differentially use alpha1- and auxiliary beta-binding-dependent mechanisms to inhibit CaV1.2/CaV2.2 channels. Public Library of Science One. 2012;7(5):e37079. doi: 10.1371/journal.pone.0037079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Suhail Y, Dalton S, Kernan T. Genetically encoded molecules for inducibly inactivating CaV channels. Nature Chemical Biology. 2007;3(12):795–804. doi: 10.1038/nchembio.2007.42. [DOI] [PubMed] [Google Scholar]
- Yang T, Xu X, Kernan T, Wu V. Rem, a member of the RGK GTPases, inhibits recombinant CaV1.2 channels using multiple mechanisms that require distinct conformations of the GTPase. Journal of Physiology. 2010;588(Pt 10):1665–1681. doi: 10.1113/jphysiol.2010.187203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawate T, Gouaux E. Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins. Structure. 2006;14(4):673–681. doi: 10.1016/j.str.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Schmidt C, Urlaub H. Combining cryo-electron microscopy (cryo-EM) and cross-linking mass spectrometry (CX-MS) for structural elucidation of large protein assemblies. Currents Opinions in Structural Biology. 2017;46:157–168. doi: 10.1016/j.sbi.2017.10.005. [DOI] [PubMed] [Google Scholar]
- Sun W, Zheng W, Simeonov A. Drug discovery and development for rare genetic disorders. American Journal of Medical Genetics Part A. 2017;173(9):2307–2322. doi: 10.1002/ajmg.a.38326. [DOI] [PMC free article] [PubMed] [Google Scholar]