

Abstract
Conventional next-generation sequencing techniques (NGS) have allowed for immense genomic characterization for over a decade. Specifically, NGS has been used to analyze the spectrum of clonal mutations in malignancy. Though far more efficient than traditional Sanger methods, NGS struggles with identifying rare clonal and subclonal mutations due to its high error rate of ~0.5–2.0%. Thus, standard NGS has a limit of detection for mutations that are >0.02 variant allele fraction (VAF). While the clinical significance for mutations this rare in patients without known disease remains unclear, patients treated for leukemia have significantly improved outcomes when residual disease is <0.0001 by flow cytometry. In order to mitigate this artefactual background of NGS, numerous methods have been developed. Here we describe a method for Error-corrected DNA and RNA Sequencing (ECS), which involves tagging individual molecules with both a 16 bp random index for error-correction and an 8 bp patient-specific index for multiplexing. Our method can detect and track clonal mutations at variant allele fractions (VAFs) two orders of magnitude lower than the detection limit of NGS and as rare as 0.0001 VAF.
Keywords: Genetics, Issue 138, Rare event detection, error-corrected sequencing, bioinformatics, genomics, early detection, molecular tagging
Introduction
As we age, exposure to mutagens and stochastic errors during cell division result in the accumulation of somatic aberrations in the genome, and this underlies the fundamental pathogenesis of malignant transformation, neuro-developmental diseases, pediatric disorders and normal aging1,2. Somatic mutations with disease-driving potential are important diagnostic and prognostic biomarkers for early detection and risk management3,4,5. In order to better understand physiologic clonogenesis, which will inform clinical and research decisions, the accurate quantification and characterization of these mutations is of primary importance. Next-generation sequencing (NGS) is currently used to study clonal mutations in heterogeneous DNA samples; however, NGS is limited to identifying mutations at >0.02 variant allele fraction (VAF) — due to the inherent error-rate of 0.5–2.0% of the sequencing platforms6,7,8. As a result, tracking diagnostically and prognostically significant somatic variants at lower VAF cannot be achieved using standard NGS.
Recently, various methods have been developed in order to circumvent the error rate of NGS8,9,10,11. These methods utilize molecular tagging, which enables error correction after sequencing. Each molecule or genomic fragment in the sequencing library is tagged with a random Unique Molecular Identifier (UMI) that is specific to that molecule. The UMIs are constructed by permutations of a string of randomized nucleotides (8–16 N). A second sample-specific barcode is also integrated into the workflow that enables multiplexing multiple samples into the same NGS sequencing run. PCR amplification is performed on the molecularly tagged library, and subsequently the library is sent for sequencing. During library preparation, it is expected that errors will be randomly introduced to the genomic fragment during PCR amplification and sequencing8. To remove random sequencing errors, raw sequencing reads are grouped according to the UMI. Artifacts from sequencing are not expected to be present in all reads with the same UMI at the same genomic position due to the stochastic nature of introduction, whereas a true variant will be faithfully amplified and sequenced in all reads that share the same UMI. The artifacts are bioinformatically removed. Here, we describe three methods of Error-corrected Sequencing (ECS) optimized in the laboratory for DNA to identify single nucleotide variants (SNVs) and small insertion-deletions (Indels), and for RNA to facilitate quantification of gene expression below the NGS error threshold.
The first method describes a way to look for rare somatic event using gene specific primers designed by researchers. Prior to library preparation, researchers should design primers to target the fragments of interest. We used the web-app Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/). Amplicons of 200–250 bp are ideal for polymerase chain reaction (PCR) as these will, once UMIs have been incorporated, generate overlapping paired-end reads with 150 bp paired-end reads. The optimal primer design conditions to be used are: Minimum primer size = 19; Optimum primer size = 25; Maximum primer size = 30; Minimum Tm = 64 °C; Optimum Tm = 70 °C; Maximum Tm = 74 °C; Maximum Tm difference = 5 °C; Minimum GC content = 45; Maximum GC content = 80; Number to return = 20; Maximum 3' end stability = 100.
In Method 2, we describe a method combining the ECS-DNA protocol with Illumina chemistry to survey for clonal SNVs and small Indels as rare as 0.0001 VAF using commercially available gene panels that include hundreds of amplicons. We have used the TruSight Myeloid Sequencing Panel (Illumina) for our experiment and designed an expanded panel to include additional genes of interest for pediatric myeloid diseases. These panels have not offered unique molecular identifiers (UMIs) that would facilitate error correction, so we have added our own adapter strategy to these panels. ECS should work equally well with any of other panels designed to enrich for genes associated with different diseases. Following DNA isolation and subsequent quantification from the tissue or sample of interest, it is recommended to have at least 500 ng of stock DNA per specimen. We routinely make a single sequencing library using 250 ng of DNA in order to capture as much unique genomic fragment as possible for downstream reads de-duplication and VAF calculation. An optional replicate sequencing library can be made with the remaining 250 ng of DNA. We always make two replicate libraries per specimen, and we consider only those events detected independently in both replicates as true positives. We also implemented a genomic position-specific binomial error model to increase the accuracy of variant calling4,13.
Lastly, we describe a method coupling ECS to RNA sequencing for transcript quantification using off-the-shelf QIAseq Targeted RNA panels (Qiagen). The UMIs required for de-duplication and error correction have been incorporated in the kits, and researchers can make libraries following manufacturer's recommendations. Bioinformatically, researchers can follow the pipeline outlined for ECS-DNA, which will be explained in detail in the PROTOCOL section.
Protocol
1. Targeted Error-corrected Sequencing for DNA
- PCR amplification of genomic fragments of interest.
- Use a high-fidelity DNA polymerase to amplify the amplicons (Materials Table, Item 1). Amplify the PCR reaction with the following conditions in a thermal cycler: 30 s at 98 °C; 18–40 cycles of 10 s at 98 °C, 30 s at 66 °C, and 30 s at 72 °C; 2 min at 72 °C; hold at 4 °C.
- Purify the PCR products with paramagnetic beads (Materials Table, Item 2). Add the PCR reaction to the beads in a 1: 1.8 ratio (PCR reaction volume: bead volume) according to the manufacturer’s protocol. Elute with 20 µL of ddH2O.
- Quantify concentration of DNA (Materials Table, Item 3) to determine final concentration of DNA.
- Run an aliquot of DNA on a 2% agarose gel (Materials Table, Item 4) to confirm the size of the amplicons. NOTE: Alternatively, researchers can opt to perform a Bioanalyzer analysis on the PCR products to determine the size of amplified genomic fragments as well as the concentration of the products.
- Sequencing adapter annealing
- Obtain i7 adapters (Materials Table, Item 5). Use them as they are provided for subsequent steps.
- Purchase 16N i5 adapters commercially with the following oligo sequence (Materials Table Item 6): AATGATACGGCGACCACCGAGATCTACAC(N1:25252525)(N1)(N1)(N1)(N1)(N1)(N1)(N1)(N1)(N1)(N1)(N1)(N1)(N1)(N1)(N1)ACACTCTTTCCCTACACGACGCTCTTCCGATCT NOTE: The 16N i5 adapters replace the standard i5 adapters and they are adapters with a string of 16 random-nucleotide to facilitate ECS.
- Make 16N i5 adapter working solution: 40 µL of 100 µM 16N i5 adapter stock, 10 µL of TE buffer, and 10 µL of 500 µM NaCl solution.
- Aliquot 7.5 µL of the i5 working solution prepared in Step 1.2.3 into separate PCR wells.
- Add 5 µL of sample-specific i7 adapter into corresponding wells.
- Incubate at 95 °C for 5 min then cool by 1 °C every 30 s to 4 °C in a thermal cycler.
- Hold at 4 °C.
- End-repair & dA-tailing of libraries NOTE: In parallel with adapter annealing, one can perform end repair and dA-tailing on the PCR amplicons from Step 1.1. Following completion of these steps, ligation of annealed adapters from Step 1.2 onto the end repaired and dA-tailed PCR amplicons is performed. Following adapter ligation, the ECS library construction is complete.
- Begin with at most 1 µg of starting DNA (minimum ~200 ng)
- Perform end-repair and dA-tail on amplicons (Materials Table, Item 7).
- Add 3.0 µL of End Prep Enzyme Mix and 6.5 µL of End Repair Buffer.
- Incubate the mix for 30 min at 20 °C, then for 30 min at 65 °C and hold at 4 °C.
- Perform ligation on the annealed adapters (Materials Table, Item 8).
- Add 2.5 µL of the annealed adapters from Step 2, 15 µL of Blunt/TA Ligase Mastermix, and 1 µL of Ligation enhancer.
- Incubate the mix for 15 min at 20 °C, then for 15 min at 37 °C.
- Clean up libraries with magnetic beads (Materials Table Item 2): Add the PCR reaction to beads in a modified 1: 0.75 ratio (PCR reaction volume: magnetic bead volume):
- Pipette 62.6 µL of magnetic bead solution into the 83.5 µL of PCR products from Step 1.2.7.
- Transfer the mixture to a 1.5 mL low binding tube.
- Mix thoroughly by pipetting up and down at least 10 times.
- Leave the mixture to stand at room temperature for 5 minutes.
- Place the tube onto a magnetic holder. Incubate for 2 minutes at room temperature or until supernatant is clear.
- Remove supernatant.
- Wash the beads with 200 µL of 70% ethanol.
- Incubate for 30 s. Remove ethanol.
- Repeat ethanol wash step once.
- Air-dry the beads.
- Elute with 20 µL of ddH2O. NOTE: This modification in PCR reaction to magnetic bead ratio will preferentially remove DNA fragments that are smaller than 200 bp.
- Quantification by droplet digital PCR NOTE: Precise mutation quantification requires strict observance of the number of molecules of each library that are loaded onto the sequencer. To achieve this, quantifying the number of molecules for individual libraries per unit volume is performed using the QX200 droplet digital PCR (ddPCR) platform — quantitative PCR is an alternative option. Following ddPCR analysis, the readout will specify the number of molecules per µL per library.
- Dilute ECS libraries 1:1,000 by incrementally diluting by a factor of 10 in PCR strip-tubes.
- Prepare the following mastermix for ddPCR in 1.5 mL tube: 10 µL of PCR Mix (Materials Table, Item 9), 0.2 µL of P5 Primer, 0.2 µL of P7 Primer, 5 µL of ECS cleaned-up product from Step 1.4.1., and 4.5 µL of ddH2O.
- Aliquot 20 µL of the mastermix into each sample well making sure there are multiples of 8.
- Aliquot 70 µL of droplet generation oil (Materials Table, Item 10) into each oil well. Cover the cassette with a rubber gasket.
- Make droplets using the droplet generator (Materials Table, Item 11).
- Using a multichannel pipette, load the droplets generated in Step 1.4.4 into a PCR plate ensuring that the pipetting of the sample is done slowly over a span of 5 seconds to avoid shearing the DNA.
- Amplify the signal in the droplets for 40 cycles in a thermal cycler using the following conditions: 5 min at 95 °C; 40 cycles of 30 s at 95 °C, 1 min at 63 °C; 5 min at 4 °C, 5 min at 90 °C; and then hold at 4 °C.
- Prepare ddPCR template droplet reader machine (Materials Table, Item 11). Ensure specification for parameters for Absolute Quantification and using the QX200 ddPCR Eva Green Supermix.
- Once ddPCR analysis is complete, make sure to set the same divisive threshold across all samples.
- Using the concentration readout from the QX200 Droplet Reader, aliquot the appropriate volume to introduce the desired number of molecules into subsequent step.
- PCR amplification of the libraries for sequencing
- Prepare the following mastermix for the desired number of molecules from Step 1.4.9: 25 µL of Q5 Mastermix (Materials Table, Item 1), 2.5 µL of P5 Primer (10 µM), 2.5 µL of P7 Primer (10 µM), X µL of DNA, 20-X µL of ddH2O.
- Amplify the libraries from Step 1.5.1 in a thermal cycler using the following conditions: 30 s at 98 °C; 20 cycles of 10 s at 98 °C, 30 s at 63 °C, 30 s at 72 °C; 2 min at 72 °C; and then hold at 4 °C.
- Clean up libraries with magnetic beads (Materials Table, Item 2): Add the PCR reaction to magnetic beads in a modified 1: 0.75 ratio (PCR reaction volume: magnetic bead volume).
- Pipette 37.5 µL of magnetic bead solution into the 50 µL PCR products from Step 1.5.2.
- Transfer the mixture to a 1.5 mL low binding tube.
- Mix thoroughly by pipetting up and down at least 10 times.
- Leave the mixture to stand at room temperature for 5 min.
- Place the tube onto a magnetic holder. Incubate for 2 minutes at room temperature or until supernatant is clear.
- Remove supernatant.
- Wash the beads with 200 µL of 70% ethanol.
- Incubate for 30 s. Remove ethanol.
- Repeat ethanol wash step once.
- Air-dry the beads.
- Elute with 20 µL of ddH2O.
- Run an aliquot of DNA on a 2% agarose gel to confirm the size of the amplicons.
- Quantify concentration of DNA (Materials Table, Item 3) to determine concentration of the separate ECS libraries.
- Pool the libraries in equimolar amounts. NOTE: For example, researchers can pool eight libraries in an equimolar group4 with 4 million starting molecules for sequencing using a sequencing platform which outputs up to 400 million reads. Conservatively, it is recommended to use an average of ten raw reads for error-correction per molecules. This would take up 360 million reads (4 million molecules * 8 libraries * 10 reads for error correction). With 4 million unique molecules per library, researchers can expect to get a theoretical mean consensus read coverage of 7042x per amplicon (4 million/568 amplicons from the gene panel).
- Quantify concentration of DNA (Materials Table, Item 3) to determine concentration of pooled ECS library.
- Submit the pooled ECS library at roughly 4 nM.
- Provide the following sequencing settings to Illumina sequencing platforms (MiSeq, HiSeq or NextSeq): 2x144 paired-end reads, 8 cycles Index 1 and 16 cycles Index 2.
2. Gene Panels with Error-corrected Sequencing of DNA
- Hybridization of oligos from gene panels NOTE: In this step, one will construct sequencing libraries using a modified Illumina TruSight or TruSeq protocol to incorporate the UMIs (Materials Table, Item 17).
- Hybridize oligos onto genomic fragment following manufacturer’s protocol. Use 250 ng of DNA (or any desired amount of starting material).
- Remove unbound oligos following manufacturer’s protocol.
- Perform extension-ligation following manufacturer’s protocol. NOTE: Modifications to the manufacturer’s protocol begin below.
- Incorporation of i5 and i7 Adapters via PCR
- Prepare the PCR mastermix by pipetting the following reagents into a tube of appropriate volume size: 37.5 µL of Q5 Mastermix (Materials Table, Item 1), 6 µL of 10 µM 16N i5 adapters (detailed in Method 1, Step 1.2.2), 6 µL of i7 adapters (Use different i7 adapters for separate samples for multiplexing), and 22 µL of extension-ligation solution with beads from Step 2.1.3. NOTE: The Q5 Mastermix replaces the polymerase mastermix provided by Illumina. The Q5 polymerase amplifies the genomic fragment with higher fidelity and fewer introduced errors.
- Run PCR program on a thermal cycler using the following parameters: 30 s at 98 °C, 4–6 cycles of 10 s at 98 °C, 30 s at 66 °C, 30 s at 72 °C; 2 min at 72 °C, and then hold at 4 °C. NOTE: The number of cycles depends on the panel size. From our experience, a 4-cycle PCR is sufficient if the gene panel has about 1,500 different pairs of gene specific oligos, whereas a panel with 500–600 pairs of oligos requires 6 cycles of PCR.
- Clean up PCR reactions with magnetic beads (Materials Table, Item 2): Add the PCR reaction to magnetic beads in a modified 1 PCR reaction: 0.75 magnetic bead ratio:
- Pipette 56.25 µL of magnetic bead solution into the 75 µL of PCR products from Step 2.2.2.
- Transfer the mixture to a 1.5 mL low binding tube.
- Mix thoroughly by pipetting up and down at least 10 times.
- Leave the mixture to stand at room temperature for 5 min.
- Place the tube onto a magnetic holder. Incubate for 2 min at room temperature or until supernatant is clear.
- Remove supernatant.
- Wash the beads with 200 µL of 70% ethanol.
- Incubate for 30 s. Remove ethanol.
- Repeat ethanol wash step once.
- Air-dry the beads.
- Elute with 20 µL of ddH2O.
- Quantify libraries using QX200 ddPCR platform.
- Follow Step 1.4 in Method 1. NOTE: 4 million molecules were normalized per sample library4 in the representative result (Figure 2) in order to obtain a theoretical mean of 7,042 uniquely indexed molecules (4 million divided by 568 gene-specific oligos).
- Amplify and normalize libraries for sequencing.
- Amplify the desired number of molecules using the following mastermix for the final PCR totaling 50 µL: 25 µL of Q5 Mastermix, 2 µL of P5 Primer (1 µM), 2 µL of P7 Primer (1 µM), and 21 µL of DNA molecules.
- Run PCR program on a thermal cycler using the following parameter: 30 s at 98 °C; 16 cycles of 10 s at 98 °C, 30 s at 66 °C, 30 s at 72 °C; 2 min at 72 °C; and then hold at 4 °C.
- Clean up sequencing libraries using magnetic beads (Materials Table, Item 2): Add the PCR reaction to magnetic beads in a modified 1 PCR reaction: 0.75 magnetic bead ratio:
- Pipette 37.5 µL of magnetic bead solution into the 50 µL PCR products from Step 2.4.2.
- Transfer the mixture to a 1.5 mL low binding tube.
- Mix thoroughly by pipetting up and down at least 10 times.
- Leave the mixture to stand at room temperature for 5 min.
- Place the tube onto a magnetic holder. Incubate for 2 min at room temperature or until supernatant is clear.
- Remove supernatant.
- Wash the beads with 200 µL of 70% ethanol.
- Incubate for 30 s. Remove ethanol.
- Repeat ethanol wash step once.
- Air-dry the beads.
- Elute with 20 µL of ddH2O.
- Run an aliquot of eluted DNA (~3 µL) on a 2% agarose gel to confirm the size of the amplicons.
- Quantify concentration of DNA (Materials Table, Item 3) to determine concentration of the separate ECS libraries.
- Pool the libraries in equimolar amounts. Refer to Method 1 Step 1.5.6. and also Discussion for more details on pooling.
- Submit the pooled ECS library at roughly 4 nM.
- Provide the following sequencing settings to Illumina sequencing platforms (MiSeq, HiSeq or NextSeq): 2x144 paired-end reads, 8 cycles Index 1 and 16 cycles Index 2.
- ECS Bioinformatic Processing and Analysis
- Obtain the sample-demultiplexed reads from the sequencer or perform demultiplexing of raw sequence reads into different samples using i7 adapter sequences bioinformatically with a custom script.
- Trim off the first 30 nucleotides of each demultiplexed read to remove oligo sequences from the gene panel.
- Align reads that share the same UMIs to one another to form read families. NOTE: Researchers can use UMI-aware software such as MAGERI13 to extract read families. No hamming distance was allowed within the UMI sequence in this experiment to increase the specificity of the method.
- Perform de-duplication and error-correction using the following recommended parameters.
- Use ≥5 read pairs in the same read family. A minimum of three read pairs is recommended.
- Compare nucleotide at every position across all reads in the same read family, and generate a consensus nucleotide if there is at least 90% concordance among the reads for the particular nucleotide. Call an N if there is less than 90% agreement for the nucleotide position.
- Discard consensus reads that have >10% of the total number of consensus nucleotides being called as N.
- Align all retained consensus reads locally to either hg19 or hg38 human reference genome using researcher’s preferred aligner(s) such as Bowtie2 and BWA.
- Process aligned reads with Mpileup using parameters –BQ0 –d 10,000,000,000,000 to remove coverage thresholds to ensure a proper pileup output regardless of VAF.
- Filter out positions with less than 1000x consensus read coverage. NOTE: The researcher determines the minimum coverage for each nucleotide position arbitrarily, it is recommended to have at least 500x consensus read coverage for downstream analysis.
- Use binomial distribution to call single nucleotide variants (SNPs) in retained data from Step 2.5.7 with the following parameters. The binomial statistic will be based on a genomic position-specific error model. Each genomic position is modeled independently after summing out the error rates of all samples for that particular position. Following the example: Probability of nucleotide profile at a given genomic position, p ∑ Variant RF2 ∑ Total RFs = 26/255505 = 0.000101759 Binomial probability of 24 variant RFs out of 35911 total RFs, P(X ≥ x) in sample K = 1 - binomial(24, 35911, 0.000101759) = 2.26485E-13 NOTE: For each genomic position queried, there would be three possible mutational changes (i.e.,A>T, A>C, A>G), and each of which would be represented as background artifact. Somatic events that are significantly different from the background after Bonferroni correction are retained. In the example shown in Table 1, the number of tests performed was 11, hence a Bonferroni corrected p-value ≤0.00454545 (0.05/11) was required to call an event as statistically significant.
- Somatic events are required to be present in both replicates from the same specimen; otherwise, regard them as false positives.
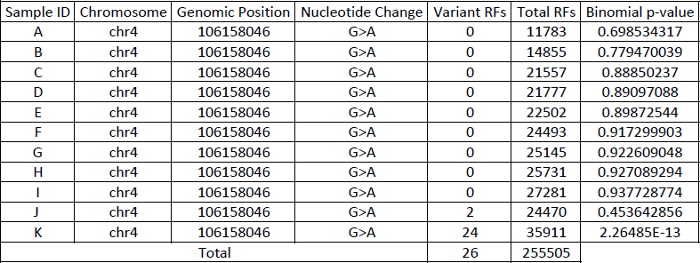 Table 1: Example demonstrating the way to construct a position-specific binomial error model.
Table 1: Example demonstrating the way to construct a position-specific binomial error model.
3. Error-corrected Sequencing of RNA
- In addition to assessing for mutations at the DNA level, integrate ECS with various targeted RNA sequencing panels to detect rare or low abundance transcript at the RNA level. By combining ECS with the off-the-shelf Qiagen RNA sequencing panels, we demonstrated digital quantification of gene expression for transcripts with as few as ten copies without a need for normalization against a housekeeping gene. The UMIs required for error-correction have been integrated into the panel.
- Perform total RNA extraction (Materials Table, Item 20).
- Carry out ECS-RNA library preparation according to manufacturer’s protocol (Materials Table, Item 19).
- Perform bioinformatics pipeline according to Step 2.5.1–2.5.6. of Method 2 outlined in the previous section. After Step 2.5.6, the number of aligned consensus reads per gene represents the expression level of the gene without the need for gene length normalization.
Representative Results
With Targeted Error-Corrected Sequencing for DNA, we have performed a proof of principle experiment diluting mutant patient DNA in commercial genomic DNA. The patient had a mutation in GATA1 (chrX:48650264, C>G) with original VAF of 0.19. We demonstrate in Figure 1 that ECS is quantitative to a level of 1:10,000 for single nucleotide variant.
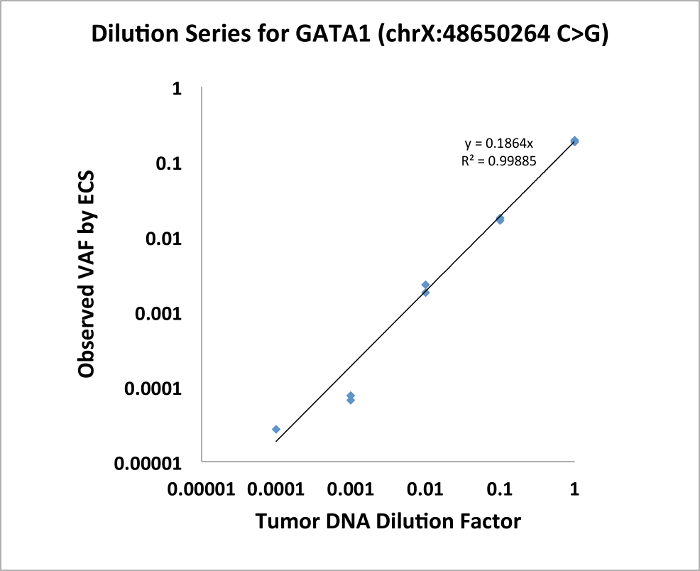
Figure 1: Dilution series of GATA1 SNV demonstrating that ECS is quantitative to the level of 1:10,000. Please click here to view a larger version of this figure.
We also show that the ECS-DNA reliably detects rare clonal mutations in genes recurrently in adult acute myeloid leukemia (AML) in healthy elderly individuals4. We obtained buffy coat samples from 20 healthy individuals in the Nurse's Health Study banked roughly ~10 years apart. We applied the ECS-DNA panel protocol on these samples. For this experiment, we adapted the Illumina TruSight Myeloid Sequencing Panel that consists of 568 amplicons (more information on gene list on https://www.illumina.com/products/by-type/clinical-research-products/trusight-myeloid.html) and sequenced 80 libraries from 20 individuals (2 collections at different time points, 2 replicates per individual per time point) using Illumina NextSeq platform, which generated an average of 47.7 million paired-end reads and an average of 3.4 million error-corrected consensus sequences per library4. The mean nucleotide coverage per library was roughly 6,000x (3.4 millions divided by 568). For each sample, we constructed a position-specific error profile using sequenced libraries that are not from the same sample. We found 109 clonal somatic mutations that were present in both replicates of at least one collection time point. These mutations have VAF ranging from 0.0003–0.1451. We selected 21 mutations with known COSMIC representations, and validated all 21 mutations in one or two collection time point(s) using ddPCR (n = 34, Figure 2, adapted from Young et al. 20164).
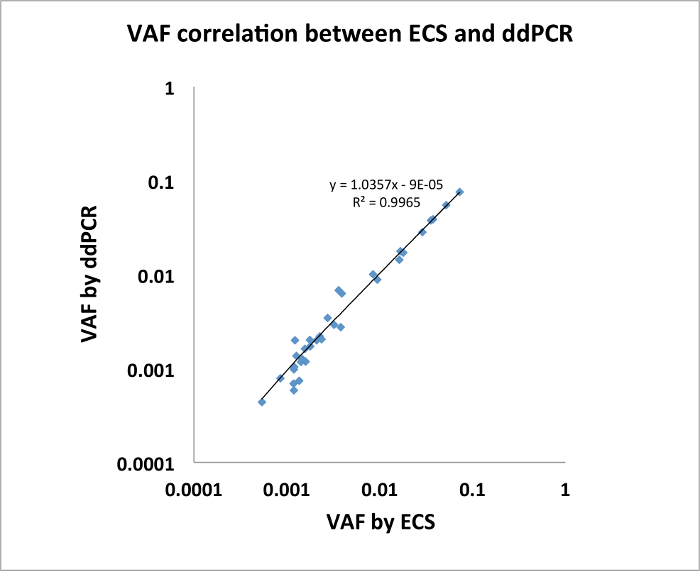
Figure 2: Mutations identified by ECS were verified via ddPCR with highly concordant VAFs. (n=34, modified from Young et al. 20164). Please click here to view a larger version of this figure.
With respect to error-corrected expression level using ECS-RNA protocol, we customized a gene panel using QIAseq chemistry that consists of 416 genes known to be associated with various cancers (adapted from QIAseq Human Cancer Transcriptome panel), and we amplified the most commonly expressed exon of a given gene (Gene list in Supplementary Material 1). We sequenced the libraries using Illumina MiSeq platform in paired-end format that gave an average of 8.3 million reads per library, and we managed to capture an average of 0.417 million error-corrected consensus sequences. We showed that the expression level of low abundance transcript (<1,000 transcript count in 50 ng of total RNA) is highly reproducible between replicates (data point n = 300, Figure 3). Validation by ddPCR (six selected genes of varying degree of expression) demonstrated that the expression level of genes had been correctly captured by the ECS protocol without the need for normalization.
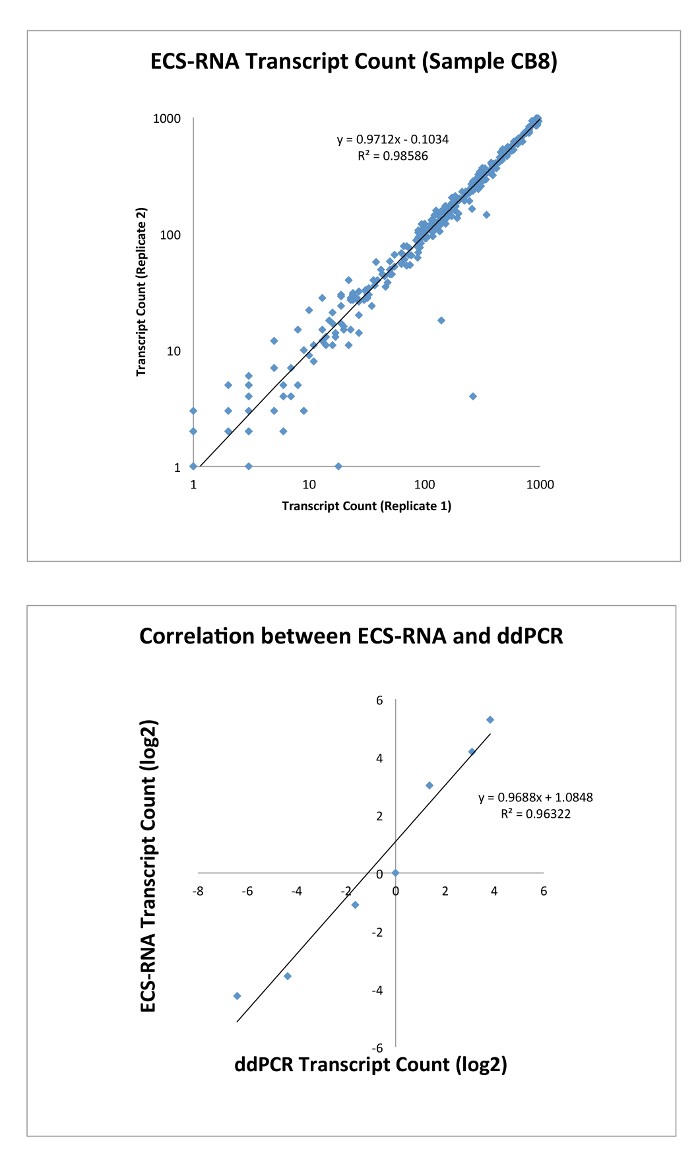
Figure 3: Top, correlation of transcript counts by ECS-RNA between replicates of the same sample (n = 300). Bottom, transcript counts identified by ECS were verified by ddPCR (n = 6). Please click here to view a larger version of this figure.
Discussion
Here, we demonstrate a suite of error-corrected sequencing protocols that can be easily implemented to study mutations with low VAFs in different diseases. The most important factor is the incorporation of UMIs with each molecule before sequencing as they enable error-correction of the raw reads. The methods described here allow researchers to incorporate customized UMIs to both commercially available gene panels and self-designed gene-specific oligos.
Standard NGS protocol precludes the detection of mutations with VAF below 2% due to the sequencing error rate, and this limits the application of NGS in studies where the detection of rare variants is crucial. By circumventing the standard NGS error rate, ECS enables sensitive detection of these raw variants. For instance, detection of pathogenic mutations when these mutations first arise (therefore having low VAF) is imperative to inform early intervention of the disease14,15. In leukemia research, the detection of minimal residual disease (residual leukemic cells post-treatment) informs risk stratification and could be used to inform treatment options in a manner that binary flow cytometric assessments cannot. In addition, ECS is applicable to detect circulating tumor nucleic acid and to evaluate metastatic potential in solid tumor patients by assessing for the presence/absence as well as the variant burden of certain mutations that are characteristics of the primary tumor16.
As demonstrated in Table 1, the power of using binomial distribution-based position-specific error model to call variants depends largely on the number of sequenced libraries as well as the depth of sequencing used to build the error model. The robustness of the error model increases with higher number of samples and more sequencing depth. It is recommended to use at least 10 sequenced samples with an average of error-corrected read coverage of 3000x per sample to build an error profile for each sample. The position-specific approach is similar to MAGERI, but instead of using an aggregate error rate for all six different substitution types (A>C/T>G, A>G/T>C, A>T/T>A, C>A/G>T, C>G/G>C, C>T/G>A)13, we model each substitution independently at every position. For instance, an error rate of C>T at a given genomic position is different from another position. Our approach also takes into account a sequencing batch effect, as the base substitution rate observed in one sequencing run might be different from another run. Hence it is important to model each position for all substitution types especially when samples from different sequencing runs are pooled to build the model.
An important consideration when designing an ECS experiment is the desired detection threshold. The beauty of NGS studies is that they can be easily scaled in terms of genes/targets of interest, detection threshold (dictated by depth of sequencing), and number of individuals queried. For example, if the researchers are interested to find rare mutations in two amplicons with a detection threshold of 0.0001, they can pool maximally 75 samples in a single sequencing run using MiSeq V2 chemistry which outputs up to 15 million reads (2 amplicons * 10,000 molecules * 10 reads for error-correction * 75 samples = 15 million sequencing reads). Researchers can vary the number of molecules going into sequencing or the number of pooled samples in a single sequencing run to adjust the detection threshold. In our studies, we aimed to find mutations with a detection threshold of 0.0001 VAF (1:10,000) using the Illumina gene panel. We routinely use 250 ng of starting DNA to ensure that sufficient molecules are captured in order to achieve the aforementioned detection threshold. Researchers can opt to start with lower amount of DNA (50 ng is recommended) if the desired detection limit is >0.001 VAF.
As the UMIs are appended onto the i5 indexes, sequencing settings have to be amended accordingly. For example, we used 16 N UMIs, and the sequencing settings were 2x144 paired end reads, 8 cycles of Index 1 and 16 cycles of index 2 as opposed to the usual 8 cycles of Index 2. The increase in Index 2 cycle is compensated by a decrease in the total number of cycles allocated to the reads. If researchers opt to use 12N UMIs10,17, the settings should be changed to 12 cycles of Index 2.
This UMI-based sequencing method is optimized to correct for sequencing errors. It remains suboptimal in dealing with PCR jackpotting, which is an issue for all amplification-based method. We performed rounds of post-sequencing and post-bioinformatics validation using ddPCR, and we hardly detect any false positives due to PCR jackpotting. Nonetheless, it is recommended that researchers conduct the experiments using high fidelity polymerase to ensure low amplification errors.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank the participants in the Children's Oncology Group AAML1531 study and the Nurses' Health Study for their contributions in the form of patient samples. This work was funded by the National Institutes of Health (UM1 CA186107, RO1 CA49449 and RO1 CA149445), the Children's Discovery Institute of Washington University and St. Louis Children's Hospital (MC-II-2015-461), and Eli Seth Matthews Leukemia Foundation.
References
- Hoang ML, et al. Genome-wide quantification of rare somatic mutations in normal tissues using massively parallel sequencing. Proceedings of the National Academy of Sciences USA. 2016;113:9846–9851. doi: 10.1073/pnas.1607794113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Roak BJ, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young AL, et al. Quantifying ultra-rare pre-leukemic clones via targeted error-corrected sequencing. Leukemia. 2015;29(7):1608–1611. doi: 10.1038/leu.2015.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young AL, Challen GA, Birmann BM, Druley TE. Clonal hematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. NatureCommunications. 2016;7:12484. doi: 10.1038/ncomms12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JP, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. New England Journal of Medicine. 2012;366:1079–1089. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shendure J, Ji H. Next-generation DNA sequencing. Nature Biotechnology. 2008;26(10):1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- Kohlmann A, et al. Monitoring of residual disease by next-generation deep-sequencing of RUNX1 mutations can identify acute myeloid leukemia patients with resistant disease. Leukemia. 2014;28:129–137. doi: 10.1038/leu.2013.239. [DOI] [PubMed] [Google Scholar]
- Luthra R, et al. Next-generation sequencing-based multigene mutational screening for acute myeloid leukemia using MiSeq: applicability for diagnostics and disease monitoring. Haematologica. 2014;99:465–473. doi: 10.3324/haematol.2013.093765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proceedings of the National Academy of Sciences USA. 2011;108(23):9530–9535. doi: 10.1073/pnas.1105422108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt M, et al. Detection of ultra-rare mutations by next-generation sequencing. Proceedings of the National Academy of Sciences USA. 2012;109(36):14508–14513. doi: 10.1073/pnas.1208715109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden JA, et al. pRESTO: a toolkit for processing high-throughput sequencing raw reads of lymphocyte receptor repertoires. Bioinformatics. 2014;30(13):1930–1932. doi: 10.1093/bioinformatics/btu138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. NatureBiotechnology. 2016;34:547–555. doi: 10.1038/nbt.3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shugay M, et al. MAGERI: Computational pipeline for molecular-barcoded targeted resequencing. PLOSComputationalBiology. 2017;13(5):e1005480. doi: 10.1371/journal.pcbi.1005480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong TN, et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 2014;518:552–555. doi: 10.1038/nature13968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krimmel JD, et al. Ultra-deep sequencing detects ovarian cancer cells in peritoneal fluid and reveals somatic TP53 mutations in noncancerous tissues. Proceedings of the National Academy of Sciences USA. 2016;113(21):6005–6010. doi: 10.1073/pnas.1601311113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phallen J, et al. Direct detection of early-stage cancers using circulating tumor DNA. ScienceTranslationalMedicine. 2017;9:eaan2415. doi: 10.1126/scitranslmed.aan2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egorov ES, et al. Quantitative profiling of immune repertoires for minor lymphocyte counts using unique molecular identifiers. The Journal of Immunology. 2015;194(12):6155–6163. doi: 10.4049/jimmunol.1500215. [DOI] [PubMed] [Google Scholar]