

Abstract
With the rising prevalence of neurodegenerative diseases, it is increasingly important to understand the underlying pathophysiology that leads to neuronal dysfunction and loss. Fluorescence-based imaging tools and technologies enable unprecedented analysis of subcellular neurobiological processes, yet there is still a need for unbiased, reproducible, and accessible approaches for extracting quantifiable data from imaging studies. We have developed a simple and adaptable workflow to extract quantitative data from fluorescence-based imaging studies using Drosophila models of neurodegeneration. Specifically, we describe an easy-to-follow, semi-automated approach using Fiji/ImageJ to analyze two cellular processes: first, we quantify protein aggregate content and profile in the Drosophila optic lobe using fluorescent-tagged mutant huntingtin proteins; and second, we assess autophagy-lysosome flux in the Drosophila visual system with ratiometric-based quantification of a tandem fluorescent reporter of autophagy. Importantly, the protocol outlined here includes a semi-automated segmentation step to ensure all fluorescent structures are analyzed to minimize selection bias and to increase resolution of subtle comparisons. This approach can be extended for the analysis of other cell biological structures and processes implicated in neurodegeneration, such as proteinaceous puncta (stress granules and synaptic complexes), as well as membrane-bound compartments (mitochondria and membrane trafficking vesicles). This method provides a standardized, yet adaptable reference point for image analysis and quantification, and could facilitate reliability and reproducibility across the field, and ultimately enhance mechanistic understanding of neurodegeneration.
Keywords: Neuroscience, Issue 138, aggregate, autophagy, ImageJ, segmentation, ratiometric, neurodegeneration, Drosophila, brain
Introduction
Neurodegenerative diseases affect millions of people each year and the incidence is increasing with an aging population1. While each neurodegenerative disease has a unique etiology, aggregation of misfolded proteins and breakdown of the proteostasis network are common pathological hallmarks of many of these diseases. Elucidating how disruption of these fundamental and interrelated processes goes awry to contribute to neuronal dysfunction and cell death is critical for understanding neurodegenerative diseases as well as guiding therapeutic interventions. Fluorescence-based imaging allows for investigation of these complex and dynamic processes in neurons and has greatly contributed to our understanding of neuronal cell biology. Analysis of fluorescently tagged proteins is challenging, particularly when experiments are carried out in vivo, due to highly compact tissues, diverse cell types, and morphological heterogeneity. Manually assisted quantification is affordable and straightforward, but is often time-consuming and subject to human bias. Therefore, there is a need for unbiased, reproducible, and accessible approaches for extracting quantifiable data from imaging studies.
We have outlined a simple and adaptable workflow using Fiji/ImageJ, a powerful and freely accessible image processing software2,3, to extract quantitative data from fluorescence imaging studies in experimental models of neurodegeneration using Drosophila. By following this protocol to quantify protein aggregation and autophagic flux — two cell biological features that are highly relevant to neurodegenerative disease pathology — we demonstrated the sensitivity and reproducibility of this approach. Analysis of fluorescently tagged mutant huntingtin (Htt) proteins in the Drosophila optic lobe revealed the number, size, and intensity of protein aggregates. We visualized a tandem fluorescent reporter of autophagic flux within the Drosophila visual system, which displays different emission signals depending on the compartmental environment4. Ratiometric-based analysis of the tandem reporter allowed for a quantitative and comprehensive view of autophagy-lysosome flux from autophagosome formation, maturation, and transport to degradation in the lysosome, and additionally highlighted vulnerable compartments disrupted in neurodegenerative conditions. Importantly, in both analyses we implemented semi-automated thresholding and segmentation steps in our protocol to minimize unconscious bias, increase sampling power, and provide a standard to facilitate comparisons between similar studies. The straightforward workflow is intended to make powerful Fiji/ImageJ plugins (developed by computer scientists based on mathematic algorithms) more accessible to neurobiologists and the life sciences community at large.
Protocol
1. Considerations and Preparations for Designing the Image Analysis Experiment
Predetermine suitable anatomical, cellular, or subcellular markers to serve as landmarks for standardizing a region of interest (ROI) across different samples, for example 4',6-diamidino-2-phenylindole (DAPI), membrane markers, localized fluorescent protein, etc.
Select a fluorescent marker that can delimit discrete puncta beyond background levels for the structure of interest. NOTE: This protocol is optimized for proteinaceous structures (e.g., misfolded protein aggregates) and membrane-bound organelles (e.g., autophagy reporter). To visualize protein aggregation, an RFP-tagged N-terminal fragment of human Htt protein with a pathogenic polyQ tract of 138 repeats (RFP-hHttQ138) was used5. The mutant Htt protein forms cytoplasmic aggregates when expressed under neuronal specific drivers such as elav-GAL45,6. To visualize autophagosome-lysosome flux, actin-GAL4 was used to ubiquitously drive expression of the tandem mCherry-GFP-Atg8a reporter. mCherry- and GFP-positive puncta indicate autophagosomes, and the flux is monitored as GFP fluorescence is quenched in the acidic environment of the lysosome, where acid-insensitive mCherry remains until the protein is degraded7. For ratiometric analysis, a single channel that is a consistent marker of the structure can be used for segmentation, or if appropriate a projection of two or more channels can be used to delineate structures, though not explicitly described in this protocol. In this example, mCherry is used for segmentation of Atg8-positive particles, as it is acid-insensitive and will remain in the compartment throughout flux through the pathway.
Download and install the open source image analysis platform ImageJ or Fiji — the preferred distribution of ImageJ with bundled plugins2,3.
Install the plugin for h-maxima interactive watershed. NOTE: This protocol uses Fiji; additional plugins may be required for ImageJ. The plug-in developed by Benoit Lombardot for SCF-MPI-CBG is available online (https://imagej.net/Interactive_Watershed).
Navigate to "Help | Update", select "Manage update sites" from the "ImageJ Updater", choose the SCF-MPI-CBG update site from the list, and then close and restart Fiji.
2. Brain Dissection and Immunofluorescence Staining
- Make silicone elastomer-lined dissection dishes.
- Pour silicone elastomer components into a beaker as indicated by the manufacturer and stir well until thoroughly mixed.
- Use a pipette to fill 5 cm tissue culture dishes one-third to half full (about 10 mL of elastomer mixture). Allow any bubbles to ascend to the surface and gently remove with tissue paper. Cover the dishes and place them on a level surface to cure for 48–72 h at room temperature (RT).
- Dissect Drosophila brain, and visual system if needed.
- To keep the lamina intact during dissection, slide two forceps under the retina and gently tear through the middle of the eye to remove the retina. Pull away any retinal tissue attached to the lamina with forceps held parallel to the lamina surface. NOTE: Residual pigment will wash off in the following steps and usually does not interfere with antibody staining and imaging.
- Dilute 37% formaldehyde in phosphate-buffered saline (PBS) to make a final concentration of 3.7% fresh before use in a 500 µL microcentrifuge tube. Transfer the dissected brain to microcentrifuge tube with fix solution and incubate for 15 min at RT with gentle rocking. NOTE: Formaldehyde has a natural tendency to be oxidized, producing formic acid. Therefore, it is suggested to aliquot the 37% formaldehyde for storage and dilute to 3.7% freshly before each use. Over-fixation or under-fixation will affect the integrity of the tissue and fluorescence10.
- Wash 3 times with PBTx (PBS with 0.4% Triton X-100) for 15 min each.
- Perform immunohistochemistry and mount specimens.
- If antibody staining is needed, remove PBTx, add primary antibody diluted in PBTx with 5% normal goat serum, and incubate on an aliquot mixer overnight at 4 °C.
- Remove primary antibody and wash 3 times with PBTx for 15 min each. Add secondary antibody diluted in PBTx with 5% normal goat serum and incubate for 1 h at RT or overnight at 4 °C. Wash 3 times with PBTx for 15 min each.
- If DAPI staining is needed, remove PBTx, add DAPI solution (stock solution: 5 mg/mL, dilute to a working concentration of 15 µg/mL in PBTx), and incubate for 10 min at RT. Wash 3 times with PBTx for 15 min each.
- To clear the tissue, place a drop of mounting medium onto the center of a microscopy slide with a spacer of one layer of clear tape on both sides. Transfer the brain onto the drop of mounting medium, and wait for 1–2 min until the brain becomes transparent. Carefully remove the mounting medium with a pipette.
- To mount, apply a drop of fresh mounting medium onto the brains on the slide. Carefully overlay a cover glass avoiding bubbles. Seal the edges with rubber cement, and air-dry at RT for 20 min. Proceed to image acquisition for best results.
3. Image Acquisition
Optimize microscope acquisition parameters, including spatial resolution, objective, zoom, scanning speed, and step size, to maximize resolution of the structures of interest to facilitate semi-automated segmentation during image analysis. NOTE: It may be useful to capture a sample image and test semi-automated segmentation (in Step 5 of this protocol) before imaging all experimental samples. In this way the user can adjust imaging settings so that analytical tools can readily discern the biological structure of interest.
- Adjust image detector controls to capture the highest signal-to-noise ratio and highest dynamic range of the specimen.
- Use a LUT (look up table) that indicates pixel saturation (exposure too high) or undersaturation (offset too low) while adjusting settings (Figure 3B, red indicates pixel saturation by a Hi/Low LUT).
- Verify gain and offset settings are appropriate for all experimental groups, as experimental manipulations likely alter the structure of interest. NOTE: It is imperative that the same settings are used for all groups to make quantitative comparisons.
Image through the tissue to capture all data. NOTE: It is recommended to capture all of the available data during one imaging session and to define a more precise area during ROI selection in Step 4 if needed.
Save the image as the file type supported by the imaging software in order to preserve metadata such as acquisition parameters and spatial calibration. Save the image with a file name including experimental date, genetic background, and fluorescent reporters, antibodies, or dyes corresponding to channels scanned such as [Experimental group name]_[Specimen name]_[Experiment date]_Ch1.[Fluorophore-target/Dye]_Ch2.[Fluorophore-target/Dye], etc.
4. Fiji/ImageJ Image Import and ROI Selection
Open an image in Fiji. Use the "Bio-Formats Import Options" dialog box, choose "View stack with: Hyperstack" and set "Color mode: Grayscale". NOTE: It is recommended to open the microscope file type, as metadata such as spatial calibration can be read by Fiji.
- Identify an area from a single z-plane or a projection based on the marker channel(s) that can be used as a standardized ROI across specimens (Figure 1, ROI selection).
- Use the "C" scrollbar to view the channel(s) used to capture the marker(s) designated in step 1.1.
- Use the "Z" scrollbar to move through the focal planes. Choose a single slice or create a projection of multiple slices by clicking "Image | Stacks | Z project" and set "Start slice" and "Stop slice" to encompass the ROI. NOTE: Setting the "Projection type" to "Max Intensity" in the "Z project" dialog box is often best suited to emphasize structures for segmentation in Section 5, though this may not be appropriate for all applications. If another type of projection is required for analysis, take care to save and document this file as such for feature extraction in Section 6 and/or 7.
- Generate a new image containing the channel(s) and focal plane(s) of interest to simplify the following steps in the image analysis protocol. Select "Image | Duplicate", set the desired "Channels" (c) and "Slices" (z), and change the file name to reflect the selection (e.g., [Experimental group name]_[Specimen name]_[Experiment date]_[Channel(s)]_[z-plane(s)]).
Use one of the "selection tools" on the Fiji toolbar to manually select a standardized ROI based on the selection criteria determined in step 4.2.
Add the ROI to the ROI manager by clicking "Analyze | Tools | ROI Manager" and click "Add" on the ROI Manager menu. Save the ROI from the ROI Manager menu by clicking "More | Save", and then name the file to reflect the ROI (e.g., [Experimental group name]_[Specimen name]_[Experiment date]_[Channel(s)]_[z-plane(s)]_[ROI description]). NOTE: This can be reopened in Fiji for later analyses or can be applied to other channels or images as in Section 5.3.
5. Preprocessing and Segmentation with h-maxima Watershedding
- Perform preprocessing on the channel used to capture the structures of interest.
- If the image file consists of multiple channels, separate the channels by clicking "Image | Color | Split channels" to isolate the channel of interest.
- Apply a filter such as a median filter to reduce noise or gaussian blur for smoothing by clicking "Process | Filters | Filter type" (e.g., "Median Filter" or "Gaussian Blur").
- Sample different filters and filter parameters on images from each experimental group. Proceed through step 6.2 with a representative example from each group to check segmentation performance. Select the filter and settings that facilitate segmentation of the structures of interest.
- Record the filter and settings. Apply these parameters across experimental groups.
- Save a copy of the image as a tiff file including the applied filter for reference. Use a detailed name such as [Experimental group name]_[Specimen name]_[Experiment date]_[Channel]_[z-plane(s)]_[Filter with parameters].
- Perform segmentation of structures of interest with the interactive h-maxima watershed tool on the preprocessed image generated and saved in step 5.1.
- With the preprocessed image active in Fiji, initiate the h-maxima interactive watershed tool by clicking "SCF | Labeling | Interactive H_Watershed". NOTE: This plugin first calculates the watershed and then allows the user to explore the effects of local maxima and threshold options on segmentation through an instantly updated output image.
- From the control panel, adjust seed dynamics (h), intensity threshold (T), and peak flooding (%) to optimize segmentation. NOTE: Always refer to the raw image before preprocessing from Section 4.2 to manually inspect performance of segmentation of the structures of interest.
- When satisfied with segmentation results, select "Export regions mask" and choose "Export". This will generate a binary image of the watershed results (Figure 1, Segmentation).
- Record the segmentation parameters; these parameters need to be applied across experimental groups for quantitative comparisons. Save the binary results mask if desired for reference with segmentation parameters detailed in the name such as [Experimental group name]_[Specimen name]_[Experiment date]_[Channels]_[z-plane(s)]_[Filter with parameters]_[H_Watershed parameters].
- Extract the structures of interest from the watershed results.
- Open the saved ROI from Step 4.4, in the ROI manager. With the binary regions mask from Step 5.2.4 active, select the ROI from the ROI manager to apply to the image to limit feature extraction to the standardized ROI.
- With the ROI active on the binary watershed mask, choose "Analyze | Analyze particles" with "Add to Manager" selected in the dialog box to extract features. NOTE: This will add a particle identifier for each structure delineated during segmentation to the ROI Manager. Options are available on this menu to exclude features based on size or circularity if needed to refine structures included in the analysis.
- Save the particles by clicking "More | Save" from the ROI manager menu. Ensure that identifiers are deselected and all particles will all be saved in a zip file. Use a detailed name such as [Experimental group name]_[Specimen name]_[Experiment date]_[Channels]_[z-plane(s)]_[Filter with parameters]_[H_Watershed parameters]_[structures].
6. Quantity, Area, and Intensity Quantification and Analysis
Open the raw image generated and saved in step 4.2. Scroll to the channel used for capturing the structures of interest. NOTE: It is critical to take intensity measurements from the raw image (before preprocessing) as applying filters in step 5.1 changes the pixel values.
Open the particles obtained from feature extraction in step 5.3 and select "Show All" from the ROI Manager. NOTE: This action will overlay an outline of each particle on the "ROI Manager" onto the image and should match the edges of the structures of interest (Figure 1, Feature extraction).
Set the desired measurements by clicking "Analyze | Set measurements". NOTE: The area, intensity, and density of particles can be measured by choosing "area" and "integrated density". When integrated density is selected, Fiji will produce two values: ‘Integrated density’ calculated as the product of the area and mean gray value and ‘Raw integrated density’ measured as the sum of pixel values. Density of the fluorescent protein in the particles is calculated as the sum of pixel values divided by the area. The number of particles generated in step 5.3 indicates the quantity of particles within the tissue ROI defined in step 4.4.
From the ROI Manager menu choose "Measure". The results are expressed in calibrated units as long as the measurements are taken from files derived in Fiji from the imaging software. Copy the results from the results window and paste into spreadsheet software for compilation and further calculations.
7. Ratiometric Quantification and Data Analysis
Follow steps 6.1 and 6.2 to open particle identifiers on the first raw channel of interest.
Set the desired measurements by clicking "Analyze | Set measurements". NOTE: The average intensity per particle can be measured by choosing "Mean Gray Value".
From the ROI Manager select "Measure". Copy data from results window and paste into spreadsheet software.
Move the "C" scrollbar to the second raw channel of interest, GFP in this example (Figure 4A), and repeat steps 7.2. and 7.3. NOTE: The particles saved in step 5.3 will default to the channel and slice from which it was generated. Take care to ensure particles are applied to the correct channel of interest.
Calculate the ratio of the intensity from one channel to the intensity of the second for each particle in the spreadsheet software.
- Use a scatter plot to visualize and quantify ratiometric data from fluorophores that exhibit individual changes reflective of underlying biological processes.
- Generate a scatter plot in the graphing software. NOTE: Each data point represents a structure of interest where the intensity of the first fluorophore of interest is the "x value" and the intensity of the second fluorophore is the "y value" measured from each particle.
- Set gating thresholds for each fluorophore to define the quadrants.
- Use line profile to visualize fluorophore intensity across a structure of interest.
- Select an ROI generated in step 5.3 and use the straight-line tool on the toolbar to draw a straight line through the ROI. Add the line ROI to the ROI Manager.
- For each channel of interest, with the line ROI active, select "Analyze | Plot profile". NOTE: The plot profile function will generate a histogram plot and a table of the values of the intensity along the line. Copy results measured from each channel and paste into spreadsheet software to generate a plot showing both channels along the line.
Representative Results
Quantification of the number, area, and intensity of fluorescently tagged mutant Htt aggregates in the Drosophila optic lobe
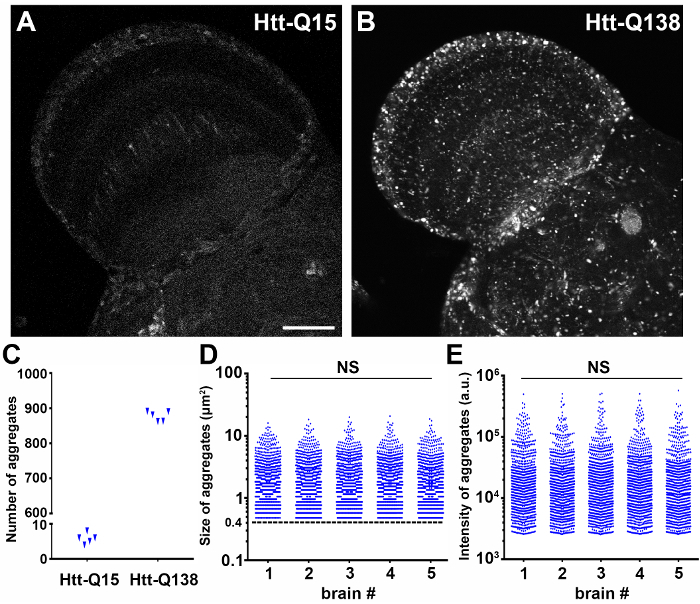
To investigate misfolded protein aggregation in the central nervous system of a Drosophila model of Huntington's disease, RFP-tagged mutant human Htt with a non-pathological (UAS-RFP-hHttQ15) or pathological expansion (UAS-RFP-hHttQ138) and membrane GFP (UAS-mCD8::GFP)11 were co-expressed under a pan-neuronal driver elav-GAL4. Brains of female flies 2 days after eclosion (DAE) were dissected, fixed, and stained with DAPI according to section 2 of the protocol. Confocal laser scanning microscopy was performed focusing on the optic lobe with a 40X oil immersion objective lens, 1.30 numerical aperture (NA), using a scan speed of 8.0 µs per pixel and spatial resolution of 1024 by 1024 pixels (12 bits per pixel). The optimal step size determined by imaging software was 1.08 µm per slice for the objective with the following combination of excitation/emission wavelengths: Channel 1) 488/510 for GFP, Channel 2) 543/581 for RFP, and Channel 3) 405/461 for DAPI. Image detector settings applied to all groups to capture images suitable for comparisons, were based on an RFP-Htt-Q138 specimen, which accumulates high intensity RFP-Htt aggregates, rather than a non-pathogenic RFP-Htt-Q15 control specimen, which maintains low levels of diffuse RFP-Htt (Figure 2). If image acquisition settings were set to maximize the dynamic range of an RFP-Htt-Q15 specimen, then the RFP-Htt-Q138 aggregates would be saturated. As an example, a micrograph of protein aggregation in the Drosophila optic lobe was saved as HttQ138_Opticlobe.01_01012018_Ch1.RFP-Htt_Ch2.GFP_Ch3.DAPI.
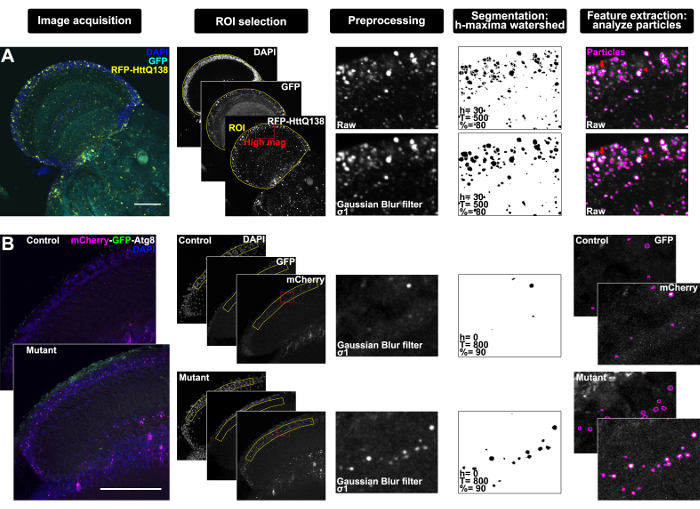
The entire optic lobe was imaged during microscopy and consisted of about 15 z-slices, but after imaging it was determined that analyzing a single z-plane was best to resolve individual structures that would otherwise appear overlapping in a projection due to the high density within the tissue. To determine a standardized ROI, the C2 and C3 cell bodies located between the medulla and lobula plate of the optic lobe12 were used as an anatomical landmark. In Fiji, the image was thus duplicated to reflect the analysis requirements and was saved as a tiff file named HttQ138_Opticlobe.01_01012018_Ch1.GFP_Ch2.RFP-Htt_Ch3.DAPI_z8, where z8 reflects the eighth slice where C2 and C3 neuronal cell bodies were most prominent by DAPI staining (Figure 1A, Image acquisition). The polygon selection tool was used to manually select an ROI around the optic lobe using DAPI staining and neuronal GFP signal as a guide (Figure 1A, ROI selection, where yellow indicates the ROI and red designates the high magnification image shown for clarity in the following panels of the workflow). Next, a Gaussian Blur filter with σ value set to 1 was applied to the RFP-Htt channel to smooth the image and facilitate segmentation (Figure 1A, Preprocessing), and the performance of downstream segmentation and feature extraction with and without this preprocessing step is illustrated in Figure 1A. Interactive H-Watershed was performed on images of the RFP-Htt channel with the following parameters: seed dynamics, 30; intensity threshold, 500; peak flooding (in%), 80; with 'allow splitting' and 'export regions mask' selected (Figure 1A, Segmentation). The resulting binary regions mask was saved as HttQ138_Opticlobe. 01_01012018_Ch2.RFP-Htt_z8_GuassianBlur-sigma1_HWatershed-h30_T500_80percent and used to generate particles with the analyze particles tool with a minimum size exclusion of 0.4 µm2 to define protein aggregates13,14. The resulting particles were saved from the ROI manager as HttQ138_Opticlobe.01_01012018_Ch1.RFP-Htt_z8_GuassianBlur-sigma1_HWatershed-h30_T500_80percent_aggregates and were applied to the raw RFP-Htt channel (before preprocessing) to extract quantitative imaging data (Figure 1A, Feature extraction).
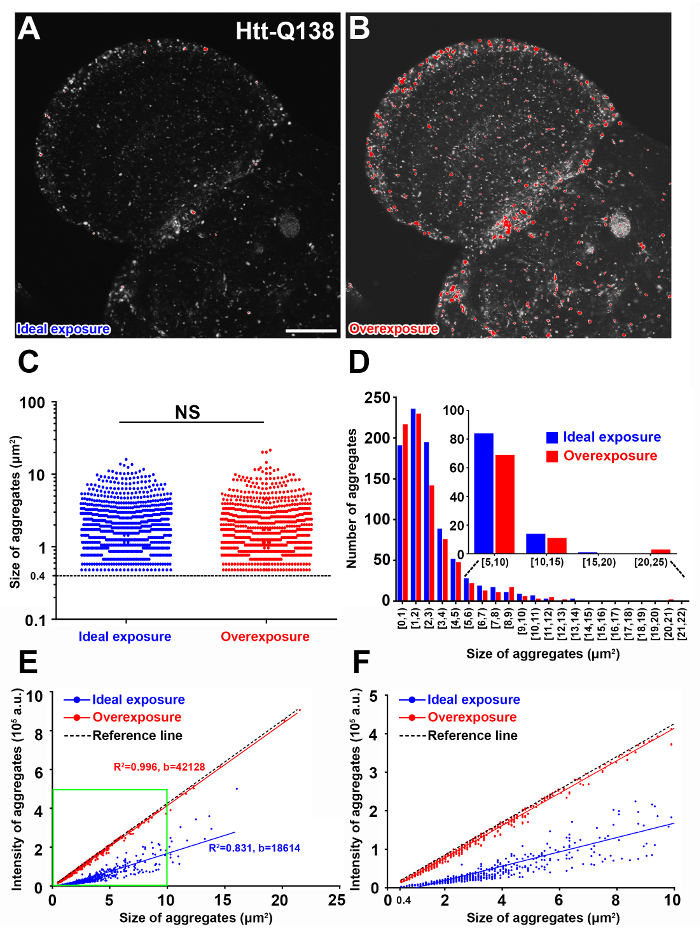
Quantification of the number of semi-automatically segmented RFP-Htt aggregates from standardized focal planes through the optic lobe of Drosophila revealed diffuse expression of the non-pathogenic Htt-Q15 expansion and accumulation of protein aggregates by the disease-causing Htt-Q138 expansion (Figure 2A-C). The sum of all pixel values in each particle given by 'Raw integrated density' was used for comparing the RFP-Htt content of each aggregate. Size and intensity profiles of all aggregates from a standardized focal plane of five different optic lobes expressing Htt-Q138 illustrate the robustness and reproducibility of this workflow (Figure 2D, E). Notably, when aggregates were overexposed during image acquisition (Figure 3A, B), quantification of size and number of aggregates are comparable to aggregates captured with ideal exposure (Figure 3C, D), however the intensity of the overexposed aggregates becomes a function of size as saturated pixels exhibit the maximum intensity value (Figure 3E, F). Therefore, setting up appropriate scanning exposure parameters is critical to extract the full range of quantitative molecular composition of protein aggregates.
Ratiometric-based quantification of autophagy flux in the Drosophila visual lamina using a tandem fluorescent Atg8a reporter
To assess autophagy-lysosome flux in the Drosophila visual system, mCherry-GFP-Atg8a reporter was ubiquitously expressed in control or Drosophila spermine synthase (dSms) homozygous mutant (dSmse/e) flies with actin-GAL4 driver. dSmse/e flies exhibit neurodegeneration that is associated with impaired autophagic flux due to lysosomal dysfunction15. Brains with attached lamina of female flies at 5 DAE were dissected, fixed, and stained with DAPI. The lamina was imaged by laser scanning confocal microscopy with a 100X oil immersion objective lens, 1.35 numerical aperture (NA), at a sampling speed of 8.0 µs per pixel, and 12 bits per pixel optical resolution, and step size of 1.2 µm.
This analysis was performed on a max z-projection of seven z-slices through the lamina to increase sampling power of the sparse Atg8-positive structures (Figure 1B, Image acquisition). The standardized ROI was manually drawn with the polygon selection tool around the cell body layer revealed by DAPI and based on the well-characterized anatomy and cellular organization of the tissue (Figure 1B, ROI selection, where yellow indicates the ROI and red designates the high magnification image shown for clarity in the following panels of the workflow)16. The tandem reporter relies on the stability properties of GFP and mCherry moieties to mark progression through the autophagy-lysosome pathway, such that GFP is quenched by the acidity of the lysosome where mCherry remains until it is degraded. Therefore, the longer-lived mCherry fluorophore was used for segmentation in this protocol. A Gaussian Blur filter with σ value 1 was applied to the mCherry channel (Figure 1B, Preprocessing), and Interactive H-Watershed was performed with the following parameters: seed dynamics, 0; intensity threshold, 800; peak flooding (in%), 90; with 'allow splitting' and 'export regions mask' selected (Figure 1B, Segmentation). The resulting binary regions mask was used to generate particles with the analyze particles tool. The resulting particles were applied to both the raw mCherry and raw GFP channels (before preprocessing) to extract quantitative ratiometric data from autophagy-lysosome compartments (Figure 1B, Feature extraction).
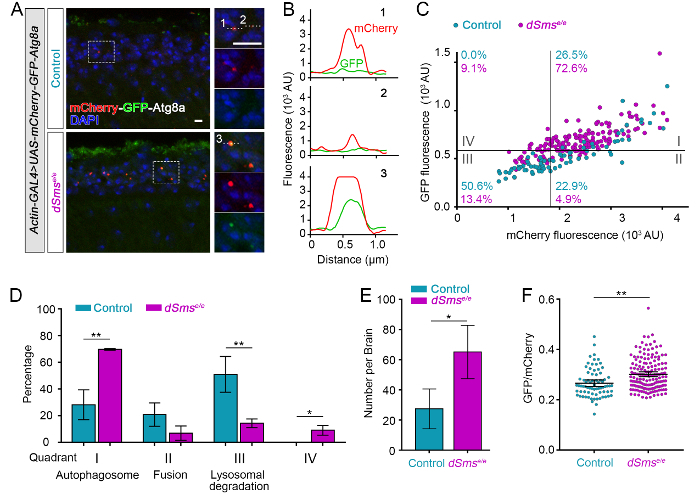
Visual inspection of mCherry-GFP-Atg8-positive structures indicated enrichment in the cell body region of the lamina, consistent with previous reports of retrograde transport of autophagic vesicles to the cell body in neurons17. Plot profile was used to visualize mCherry and GFP intensity across several Atg8-positive structures (see line ROIs in Figure 4A and resulting intensity profiles along the line in Figure 4B), revealing differences in the reporter where high intensity of both fluorophores indicates autophagosomes (Figure 4B3), high mCherry and low GFP indicates fusion with the lysosome as GFP is quenched in this acidic compartment (Figure 4B1), and finally low mCherry reflects degradation within the lysosome (Figure 4B2). A scatter plot generated from the mean fluorophore intensity of each semi-automatically segmented mCherry-Atg8-positive particle illustrates flux through the autophagy-lysosome pathway that was then separated into discrete steps by quadrant analysis (Figure 4C). The plot was divided into four quadrants using gating thresholds of GFP = 570, mCherry = 1,800. The thresholds were set based on the control group using the following principals: (1) the design of the tandem fluorescent reporter and the properties of the two fluorophores, so that theoretically GFP should not fluoresce where there is no mCherry signal (quadrant IV), and (2) the distribution of Atg8-positive particles reflects the distribution of autophagy-lysosome pathway-related compartments in wild-type neurons where basal autophagy is highly efficient18,19. In the laminas of control flies, around 25% of the Atg8-positive particles were binned as autophagosomes (quadrant I), and half of the puncta are processed beyond autophagosome-lysosome fusion (quadrant II and III); dSmse/e laminas had significantly increased autophagosomes or dysfunctional lysosomes (quadrant I) and decreased autolysosomes (quadrant II and III), suggestive of autophagosome accumulation through the defects in autophagosome-lysosome fusion and/or lysosomal degradation (Figure 4C, D). Quantitative data also supported obvious accumulation of Atg8-positive structures in lamina from dSmse/e flies (Figure 4E). Ratiometric analysis of mean mCherry and GFP signal highlighted a difference between control and dSmse/e group (Figure 4F) but could not detail the overall influence on the pathway nor the specific steps revealed by the quadrant analysis.
Figure 1: Workflow through bias-minimized, semi-automated segmentation of subcellular structures in the Drosophila brain using Fiji with examples. (A) Representative confocal micrographs from single focal plane through the optic lobe of a Drosophila adult brain expressing pan neuronal RFP-tagged mutant Htt and membrane-targeted GFP (elav-GAL4 > UAS-mCD8:GFP, UAS-RFP-hHttQ138) with DAPI staining (Image acquisition). DAPI and GFP were used to select an ROI (yellow) around the optic lobe; red box indicates region magnified through workflow for clarity (ROI selection). Preprocessing panel shows high magnification of raw image of the RFP-HttQ138 channel (top) and after application of a Gaussian Blur filter, σ1 (bottom). Segmentation by interactive h-maxima watershed using the indicated parameter settings for seed dynamics determined by h-maxima (h), global watershed stop criteria determined by intensity threshold (T), and regional stop criteria determined by peak flooding (%) performed on the RFP-HttQ138 raw (top) and Gaussian blurred (bottom) images, and watershed results from regions mask exported from watershed plugin and inverted for clarity. Particles generated by analyze particles (magenta) overlaid on the RFP-HttQ138 raw images (Feature extraction). Arrows indicate aggregates that are over-segmented without preprocessing and arrowheads indicate structures that fail to separate after filtering. (B) Representative confocal micrographs from maximum z-projection of 7 focal planes through the horizontal section of lamina of a control and mutant (dSmse/e) Drosophila adult expressing ubiquitous GFP-mCherry-Atg8a (actin-GAL4 > UAS-GFP-mCherry-Atg8) with DAPI staining (Image acquisition). DAPI was used to select an ROI (yellow) around the cell body layer of the lamina; red box indicates region magnified through workflow for clarity (ROI selection). Preprocessing panel shows high magnification of mCherry channel with Gaussian Blur filter, σ1 applied to both control and mutant lamina. Segmentation by interactive h-maxima watershed performed on the filtered mCherry images with the indicated parameter settings, and watershed results shown as binary regions mask inverted for clarity. Particles generated by analyze particles (magenta) overlaid on the raw mCherry and GFP images (Feature extraction). Scale bars = 50 µm. Please click here to view a larger version of this figure.
Figure 2: Quantification of the size and intensity of mutant RFP-Htt aggregates is robust and reproducible. (A-B) Drosophila brains with RFP-hHttQ15 (A) or RFP-hHttQ138 expression (B) under a pan-neuronal driver elav-GAL4 were dissected and fixed and the optic lobes were imaged by laser scanning confocal microscopy. Scale bar = 50 µm. (C) The average number of aggregates in each group quantified from a manually selected ROI around the optic lobe and focused on a standardized z-slice (n = 5). (D-E) The size (D) and total intensity (E) of RFP-hHttQ138 aggregates quantified from standardized ROIs in the optic lobes of 5 different animals. Aggregate data is plotted in log scale. Aggregates were defined as objects greater than 0.4 µm2 (dashed line in D). Statistical analysis, one-way ANOVA, NS: not significant; a.u.: arbitrary unit Please click here to view a larger version of this figure.
Figure 3: Image overexposure leads to inaccurate intensity measurements. (A-B) Representative confocal micrograph of the same optic lobe with ideal exposure (A) and overexposure (B) pseudocolored with the HiLo LUT in Fiji. Red in B indicates overexposed aggregates. Scale bar = 50 µm. (C) Quantification of aggregate size after implementing h-maxima watershed parameters to generate similar segmentation results. Statistical analysis, independent samples t-test, NS: not significant. (D) Frequency distribution of the aggregate size in ideal and overexposed images. Aggregates with a size from 5 to 25 µm2 were further binned in 4 categorical groups. (E -F) Correlation analysis of the intensity of the aggregates as a function of aggregate size (E). A reference line is drawn (black) when all the pixels have a maximum intensity value of 4095. Green box indicates aggregates below 10 µm2 area shown in F. b: regression coefficient, R2: coefficient of determination; a.u.: arbitrary unit Please click here to view a larger version of this figure.
Figure 4: Quantification of a tandem fluorescent mCherry-GFP-Atg8a genetic reporter reveals distinct features of autophagic flux in Drosophila. (A) Representative confocal micrographs of Drosophila lamina from control and dSmse/e mutant flies ubiquitously expressing mCherry-GFP-Atg8a (actin-GAL4 > mCherry-GFP-Atg8a) and stained for DAPI. Dashed boxed areas of the lamina cell body layer are shown in high magnification (right). Numbers indicate representative Atg8a-positive structures plotted in B and quantified in C. (B) Histogram plot of the fluorescence intensity in arbitrary units (AU) along line ROIs indicated in high magnification images in A. (C) mCherry-GFP-Atg8 structures plotted by mean GFP intensity as a function of mean mCherry intensity measured from control and dSmse/e lamina (particles measured from 3 laminae from each group). The plot was divided into four quadrants using gating thresholds of GFP = 570 and mCherry = 1800 with the percentage of structures in each quadrant indicated by genotype.(D) Quantification of the mean percentages of mCherry-GFP-Atg8 categorized into each quadrant in C (n = 3 lamina).(E) Quantification of the numbers of mCherry-GFP-Atg8 puncta per lamina (Mean ± S.D., n = 3 lamina). (F) Ratiometric analysis of mCherry-GFP-Atg8 structures observed in control and mutant fly lamina (Mean with 95% CI, n = 3 lamina). Student t-test. *P <0.05, **P <0.01. Sscale bars = 2 µm. Please click here to view a larger version of this figure.
Discussion
The protocol outlined here can be used to robustly and reproducibly quantitate cell biological processes visualized by fluorescence-based imaging. Biological context and technical limitations need to be carefully considered to guide the experimental design. Fluorescent markers of subcellular structures of interest, whether immunohistochemical, dye-based, or genetically expressed, need to be distinguishable above background by morphology and intensity. The UAS/GAL4 system is widely used to drive targeted gene expression in Drosophila. The examples presented here used transgenic lines carrying GAL4 drivers to activate transcription of fluorescently tagged proteins under the control of the UAS enhancer to investigate protein aggregation and autophagy in the nervous system. If simultaneous expression of an experimental UAS transgene is required along with a fluorescent reporter transgene, then the control group should also include co-overexpression of an inconsequential transgene to control for GAL4 dosage, for example UAS-GFP or UAS-lacZ. Standardized sample preparation, as outlined here for Drosophila central nervous system and visual system, from dissection, fixation, antibody staining, through mounting, is critical for optimal imaging and quantification.
Good imaging practices that maximize signal-to-noise ratio and dynamic range are necessary for semi-automated analysis and are paramount for accurate comparisons between experimental groups. Image acquisition parameters should be set so that the reporter is not oversaturated so that the full range of protein intensity can be extracted and quantified (Figure 3). For quantitative comparisons, imaging should be performed with the same acquisition parameters and preferably on the same day.
ROI selection, image processing, feature segmentation, and feature extraction using the open-sourced platform ImageJ/Fiji are application-dependent; while initial parameters must be defined by the user, their application across specimens will minimize bias while maximizing quantitative data output. It is critical to carefully document workflow and recommended to save an image as a tiff file after each step with file name detailing applied processing. This can be especially useful when working out protocol parameters for the first time and will serve as a reference point to ensure consistency between experimental groups at each step in image processing and analysis. ROI selection criteria using markers or anatomical landmarks should be defined from a control group, but care should be taken to ensure that experimental manipulations do not alter the markers or landmarks in a way that would prevent reliable indication of a standardize ROI. A single slice or a z-projection of multiple slices can be selected to best address the objectives of the analysis, where a single focal plane can be useful to improve separation of neighboring structures, and a z-projection can be useful for analyzing structures localized throughout the tissue volume. The type of z-projection used for segmentation and for image analysis should be carefully and independently determined. A maximum intensity projection will produce a sharp image, since structures are brightest in the planes where they are in focus, and are usually well suited for segmentation. Image analysis may be acceptable on a maximum projection, as in the example of autophagy-lysosome analysis; Atg8-positive structures are smaller than the step size used for sampling, thus are most accurately represented by the maximum intensity from the sole focal plane from which they were captured. However, for analysis of structures larger than the step size, a sum-projection that adds all the pixels with the same xy coordinates may more accurately represent the intensity information of each structure. While this protocol is designed for analyzing a single slice or a z-projection of multiple slices in 2D, the workflow could be readily adapted to analyze 3D structures in a z-series using several of the same tools. 3D analysis would be desirable for a small sample size of irregularly shaped structures, where a z-slice that obliquely intersects the structure would skew the analysis.
Preprocessing with a filter, such as those suggested in this protocol, can reduce noise while preserving edges to aid detection of structures of interest during segmentation; background noise and fluorescent marker inhomogeneity without sufficient smoothing will result in over-segmentation, while settings that result in too much blurring will remove detail and prevent accurate edge detection. Segmentation by watershed considers the image as a topographic surface with a water source at local maxima — high intensity pixels in a fluorescent image — that floods out until it meets neighboring flooded areas where a dam, or segment boundary, is built20. The h-maxima watershed uses a user-defined threshold (h-maxima dynamics in SCF-MPI-CBG plugin) to distinguish meaningful local maxima to avoid over-segmentation of structures that do not have a unique local maximum21. The interactive h-maxima watershed plugin implemented in this protocol allows the user to adjust the h-maxima dynamics (h), global watershed stop criteria determined by intensity threshold (T), and regional stop criteria determined by peak flooding (%), while instantly viewing watershed results on an output image to qualitatively evaluate segmentation performance. Segmentation results will closely agree with most structure boundaries, though both over-segmentation and incomplete separation (Figure 1A, red arrows in Feature extraction) may occur where fluorescent labeling is inhomogeneous or the tissue is densely populated. The efficiency of the semi-automated segmentation and analysis steps can be leveraged to increase sampling power and mitigate segmentation pitfalls.
After segmentation and feature extraction, it is straightforward to extract morphological information such as area, dimensions, and circularity. Furthermore, if images are collected appropriately to avoid overexposure, molecular information can also be inferred from this image analysis by intensity measurements. Once measures of neurodegenerative disease pathology are extracted and quantified by this approach, these measures can be used to address a number of important questions. Specifically, the protocol has demonstrated the potential to use image analysis and quantification of protein aggregates to identify factors that influence protein aggregation and correlate distribution, number, size, and protein density of the aggregates with neuronal dysfunction and organismal health. Ratiometric-based quantification can be used to inform aggregate interaction with subcellular components or processes during disease progression. While biochemical and flow cytometry have been applied to analyze the interaction of aggregates and with other subcellular structures, cell lysis can lead to protein loss, change aggregate properties, and disrupt protein-protein interactions22. mCherry-GFP-Atg8 has been widely used to monitor autophagy through fluorescence microscopy, especially in cultured cells23, but unbiased and quantitative analytical methods are lacking. Furthermore, in vivo analysis using the genetically encoded tandem reporter is challenging and requires highly controlled genetic background and tissue preparation. This protocol outlines verified methodology from tissue preparation to imaging practices critical to quantify autophagy-lysosome compartments. With good genetic practices, the image analysis approaches including Atg-positive particle intensity profiles and quadrant analysis with gating by individual fluorescent signals, can afford powerful detection and comparison of normal and pathological autophagic flux in vivo in diverse disease models.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work is supported by the Sheila and David Fuente Neuropathic Pain Research Program Graduate Fellowship (to J.M.B.), the Lois Pope LIFE Fellows Program (to C.L., Y.Z., and J.M.B.), the Snyder-Robinson Foundation Predoctoral Fellowship (to C.L.), the Dr. John T. Macdonald Foundation (to C.L.), contracts, grants from National Institutes of Health (NIH) HHSN268201300038C, R21GM119018, and R56NS095893 (to R.G.Z.), and by Taishan Scholar Project (Shandong Province, People's Republic of China) (to R.G.Z.).
References
- Erkkinen MG, Kim M-O, Geschwind MD. Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harbor perspectives in biology. 2017. p. a033118. [DOI] [PMC free article] [PubMed]
- Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nature methods. 2012;9(7):676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature methods. 2012;9(7):671. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauvezin C, Ayala C, Braden CR, Kim J, Neufeld TP. Assays to monitor autophagy in Drosophila. Methods. 2014;68(1):134–139. doi: 10.1016/j.ymeth.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss KR, Kimura Y, Lee W-CM, Littleton JT. Huntingtin aggregation kinetics and their pathological role in a Drosophila Huntington's disease model. Genetics. 2012;190(2):581–600. doi: 10.1534/genetics.111.133710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock DT, Ganetzky B. Transcellular spreading of huntingtin aggregates in the Drosophila brain. Proceedings of the National Academy of Sciences. 2015;112(39):E5427–E5433. doi: 10.1073/pnas.1516217112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVorkin L, Gorski SM. Monitoring autophagy in Drosophila using fluorescent reporters in the UAS-GAL4 system. Cold Spring Harbor Protocols. 2014;2014(9) doi: 10.1101/pdb.prot080341. [DOI] [PubMed] [Google Scholar]
- Williamson WR, Hiesinger PR. Preparation of developing and adult Drosophila brains and retinae for live imaging. Journal of visualized experiments: JoVE. 2010. [DOI] [PMC free article] [PubMed]
- Sweeney ST, Hidalgo A, de Belle JS, Keshishian H. Dissection of adult Drosophila brains. Cold Spring Harbor Protocols. 2011. [DOI] [PubMed]
- Fox CH, Johnson FB, Whiting J, Roller PP. Formaldehyde fixation. Journal of Histochemistry & Cytochemistry. 1985;33(8):845–853. doi: 10.1177/33.8.3894502. [DOI] [PubMed] [Google Scholar]
- Lee T, Luo L. Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends in neurosciences. 2001;24(5):251–254. doi: 10.1016/s0166-2236(00)01791-4. [DOI] [PubMed] [Google Scholar]
- Fischbach K-F, Dittrich A. The optic lobe of Drosophila melanogaster. I. A Golgi analysis of wild-type structure. Cell and tissue research. 1989;258(3):441–475. [Google Scholar]
- Ruan K, Zhu Y, Li C, Brazill JM, Zhai RG. Alternative splicing of Drosophila Nmnat functions as a switch to enhance neuroprotection under stress. Nature Communications. 2015;6:10057. doi: 10.1038/ncomms10057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai RG, et al. NAD synthase NMNAT acts as a chaperone to protect against neurodegeneration. Nature. 2008;452(7189):887. doi: 10.1038/nature06721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, et al. Spermine synthase deficiency causes lysosomal dysfunction and oxidative stress in models of Snyder-Robinson syndrome. Nat Commun. 2017;8(1):1257. doi: 10.1038/s41467-017-01289-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokop A, Meinertzhagen IA. Development and structure of synaptic contacts in Drosophila. Semin Cell Dev Biol. 2006;17(1):20–30. doi: 10.1016/j.semcdb.2005.11.010. [DOI] [PubMed] [Google Scholar]
- Maday S, Holzbaur EL. Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev Cell. 2014;30(1):71–85. doi: 10.1016/j.devcel.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Molecular biology of the cell. 2004;15(3):1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Sato Y, Nixon RA. Primary lysosomal dysfunction causes cargo-specific deficits of axonal transport leading to Alzheimer-like neuritic dystrophy. Autophagy. 2011;7(12):1562–1563. doi: 10.4161/auto.7.12.17956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent L, Soille P. Watersheds in digital spaces: an efficient algorithm based on immersion simulations. IEEE Transactions on Pattern Analysis & Machine Intelligence. 1991. pp. 583–598.
- Najman L, Schmitt M. Geodesic saliency of watershed contours and hierarchical segmentation. IEEE Transactions on pattern analysis and machine intelligence. 1996;18(12):1163–1173. [Google Scholar]
- Shiber A, Breuer W, Ravid T. Flow cytometric quantification and characterization of intracellular protein aggregates in yeast. Prion. 2014;8(3):276–284. doi: 10.4161/19336896.2014.968445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]