

Abstract
Phosphoproteomics involves the large-scale study of phosphorylated proteins. Protein phosphorylation is a critical step in many signal transduction pathways and is tightly regulated by kinases and phosphatases. Therefore, characterizing the phosphoproteome may provide insights into identifying novel targets and biomarkers for oncologic therapy. Mass spectrometry provides a way to globally detect and quantify thousands of unique phosphorylation events. However, phosphopeptides are much less abundant than non-phosphopeptides, making biochemical analysis more challenging. To overcome this limitation, methods to enrich phosphopeptides prior to the mass spectrometry analysis are required. We describe a procedure to extract and digest proteins from tissue to yield peptides, followed by an enrichment for phosphotyrosine (pY) and phosphoserine/threonine (pST) peptides using an antibody-based and/or titanium dioxide (TiO2)-based enrichment method. After the sample preparation and mass spectrometry, we subsequently identify and quantify phosphopeptides using liquid chromatography-mass spectrometry and analysis software.
Keywords: Cancer Research, Issue 138, Prostate cancer, mass spectrometry, proteomics, phosphoproteomics, phosphorylation, kinases, cell signaling
Introduction
An estimated 165,000 new cases and approximately 29,000 deaths will occur in 2018 due to prostate cancer, representing the most common cancer and second leading cause of cancer-related death in men in the United States1. Early stages of prostate cancer are treatable with resection or radiation therapy of organ-confined disease, where the ten-year recurrence rate is between 20% and 40% for patients who undergo prostatectomy and between 30% and 50% for patients who receive radiation therapy2. Because prostate cancer relies on androgen signaling for growth, surgical and chemical castration therapies are also employed for high-risk patients. However, relapse occurs when the cancer no longer responds to androgen deprivation therapy as evidenced by biochemical recurrence, where the prostate-specific antigen in serum rises again. At this point in the progression, metastases are often detected as well. This advanced stage, called metastatic castration-resistant prostate cancer, represents the lethal form of the disease where the prognosis is a median survival time of less than two years3. Few treatment options are available in late-stage disease, including second-generation antiandrogens such as enzalutamide and abiraterone, as well as taxane-based chemotherapy like docetaxel. Despite available treatments, the disease often progresses. Therefore, the discovery and development of novel treatment modalities are necessary to improve the care of prostate cancer patients with advanced disease.
Mass spectrometry (MS)-based approaches provide a global analysis of the proteome through the detection of hundreds to thousands of peptide analytes4. In particular, discovery proteomics, also known as data-dependent acquisition (DDA), can yield the identification and quantitation of thousands of peptides4,5. MS-based discovery proteomics can be further delineated into top-down proteomics, where intact proteins are characterized, and bottom-up (also known as shotgun) proteomics, where peptides are analyzed to characterize proteins5. Thus, in shotgun proteomics, a proteolysis step takes place in the sample preparation preceding the MS analysis to cleave proteins into peptides. At the end, a database search is performed to map the peptides back to the proteins for identification. Label-free as well as several isotope-labeling [e.g., stable isotope labeling by amino acids in cell culture (SILAC)] methods can be used to quantitatively compare peptides between samples6,7. While isotope labeling techniques are the gold standard, label-free methods have demonstrated similar quantification accuracies8,9 and have comparable tradeoffs between sensitivity and specificity10. Label-free quantitation provides greater coverage and permits comparisons between many more samples, whereas label-based methods are limited by cost and multiplexing capacities6,7,8.
Furthermore, shotgun MS can be also used to interrogate post-translational modifications (PTMs) such as phosphorylation11. Due to the lower stoichiometric nature of phosphopeptides compared to total peptides, several methods are employed to enrich for phosphopeptides, including antibody-based immunoprecipitation of phosphotyrosine (pY) peptides, titanium dioxide (TiO2), and immobilized metal affinity chromatography (IMAC)5,12. Because protein phosphorylation is a key step in many cell signaling pathways, shotgun phosphoproteomics allows researchers to investigate cell signaling changes in different cancers, including breast13, prostate14, renal15, and ovarian,16,17 to better understand cancer biology and to identify potential new targets for therapy.
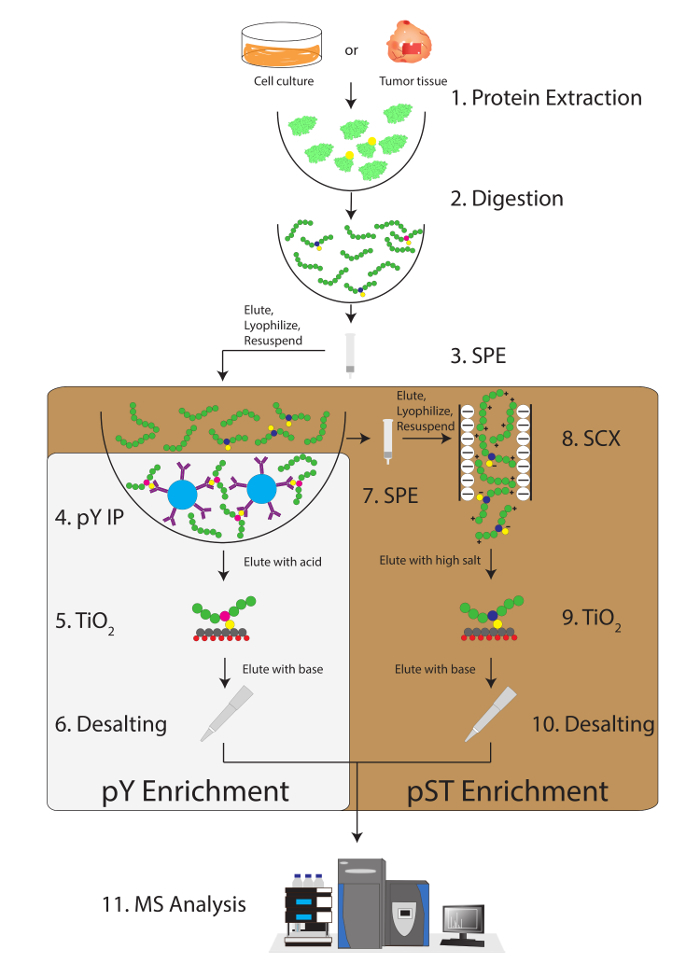
This label-free shotgun phosphoproteomic method was built and refined based on previous work by the Graeber group18,19,20. This protocol begins by describing the extraction and digestion of proteins and phosphoproteins from tissue into peptides. We then detail the enrichment of pY peptides using specific phosphotyrosine antibodies and TiO2. We also describe the enrichment of phosphoserine/threonine (pST) peptides using strong cation exchange (SCX) followed by TiO2. This protocol concludes with the submission of samples to an MS facility and the use of MS analysis software to identify and quantify phosphopeptides and their corresponding phosphoproteins. The application of this protocol can extend beyond prostate cancer into other cancers and fields outside of oncology.
Protocol
Experiments using xenograft tumors were approved by the Rutgers University Institutional Animal Care and Use Committee as set forth under the guidelines of the National Institutes of Health.
1. Protein Extraction
Prepare lysis buffer (Table 1). (The volume depends on the number of samples to be harvested.) For in vitro cell samples, proceed to step 1.2. For tumor tissue, proceed to step 1.3.
- Harvesting cells
- Collect the cells in a 50 mL conical tube and spin them at 700 x g for 5 min at 4 °C. Discard the supernatant and keep the pellet on ice. Repeat this step for all dishes to collect the cells into one pellet. (Typically, about 5 nearly confluent 15-cm dishes of cells are needed for 5 mg of protein, but this may be dependent on the cell line and needs to be determined empirically by each investigator.)
- Wash the pellet with 30 mL of chilled phosphate-buffered saline (PBS) and spin at 700 x g for 5 min at 4 °C before aspirating the PBS. Add 1.5 mL of lysis buffer per 5 mg of protein used to the cell pellet. Pipet up and down a couple of times. Skip to step 1.4.
- Harvesting tissues
- Weigh the tumor and add 2 mL of ice-cold lysis buffer for every 100 mg of tissue in a culture test tube. (Typically, 50 to 150 mg of tissue wet weight is needed.)
Homogenize the lysate using a hand-held or benchtop homogenizer (pulse 2x for 15 s.) Clean the homogenizer before the first sample and between samples by using 10% bleach, 70% ethanol, and deionized water in succession.
To reduce and alkylate, heat the homogenized samples at 95 °C for 5 min. Then cool them on ice for 15 min. On ice, sonicate the lysate 3x (i.e., pulse for 30 s with 60 s pauses between pulses). The sample should not be viscous or clumpy at this point. Heat the lysate at 95 °C for 5 min21.
Centrifuge the lysate in the same sonication tube using a swing bucket rotor at 3,500 x g at 15 °C for 15 min. Collect the supernatant and discard the pellet.
Determine the protein concentration by performing a Bradford assay22. If necessary, dilute the lysate to 5 mg/mL with a lysis buffer. Store it at -20 °C. Note: The experiment can be paused here. Freeze the samples at -80 °C and continue at a later date.
2. Lysate Digestion
Dilute the sample 12-fold by using 100 mM Tris (pH = 8.5) to reduce the amount of guanidinium. Dilute all samples to the same volume to minimize the effects of unequal digestion. Save 12.5 µg of the undigested lysate to confirm it on a Coomassie-stained gel23.
For 5 mg of protein, add 10 µg of Lysyl Endopeptidase (Lys-C) and incubate it at room temperature for 5 - 6 h. Adjust pH to 8.0 by adding 1 M untitrated Tris (pH ~11).
Prepare 1 mg/mL of L-1-tosylamido-2-phenylethyl chloromethyl ketone (TPCK)-treated trypsin in 1 mM HCl (with 20 mM CaCl2). Add the trypsin at a 1:100 trypsin:protein ratio and incubate it at 37 °C for 3 h.
Add the same amount of fresh trypsin as in step 2.3. Incubate it at 37 °C overnight.
Save 12.5 µg of the digested lysate to confirm the complete digestion on a Coomassie-stained gel23.
3. Reverse Phase Extraction
Record the lysate volume. Filter the sample by using a 15 mL 10 kDa cutoff filter. Centrifuge the sample at 3,500 x g using the swing bucket rotor (or 3,500 x g in a fixed angle rotor) at 15 °C until the retentate volume is less than 250 µL (this takes approximately 45 - 60 min). Collect the flow-through and discard the retentate. Note: The experiment can be paused here. Freeze the samples at -80 °C and continue at a later date.
To acidify the sample, add approximately 20 µL of 5% trifluoroacetic acid (TFA) per mL of lysate. Mix them well and measure the sample pH by using pH strips. Adjust pH to 2.5 using 5% TFA.
Connect the shorter end of a C-18 column to a vacuum manifold. Set the vacuum between 17 and 34 kPa (or according to the manufacturer’s instructions). Using glass pipettes, wet the column with 3 mL of 100% acetonitrile (ACN). Do not let the column dry.
Using glass pipettes, equilibrate the column with 6 mL of 0.1% TFA applied as 2x 3 mL. Load the acidified sample into the column. Do not add more than 3 mL at a time. Adjust the vacuum to target about 1 to 2 drops per second.
Using glass pipettes, wash the column with 9 mL of 0.1% TFA applied as 3x 3 mL. Elute the column with 2 mL of 40% ACN, 0.1% TFA. Collect two 2 mL fractions into glass culture tubes. Discard the column.
Cover the eluate tubes with parafilm and punch 3 - 5 holes on the cover using a 20G needle. Freeze the eluate on dry ice for at least 30 min until it is completely solid.
Lyophilize the fractions overnight. On the following day, make sure the samples are completely dry before stopping the lyophilizer. Store the tubes in a 50 mL conical tube with delicate wipes at -80 °C. Note: The experiment can be paused here.
4. Immunoprecipitation and Enrichment of pY Peptides24
Resuspend the lyophilized powder with 0.5 mL of ice-cold immunoprecipitation (IP) binding buffer in each fraction. Pool the fractions by transferring the 0.5 mL resuspension volume from the second fraction to the first fraction and save the pipette tip. Vigorously vortex (instead of pipetting up and down) to make sure the sample is completely dissolved before transferring it to a 3.6 mL screw cap cryotube.
As in step 4.1., rinse the lyophilization tubes with another 0.5 mL of IP binding buffer (Table 1) in each tube. Transfer the solution to the 3.6 mL screw cap tube using the same pipette tip to minimize any sample loss. Repeat the rinse 1x more, making the final resuspension volume 2 mL (for 5 mg of protein). Measure the sample pH to make sure it is approximately 7.4. If it is too acidic, iteratively add 10 µL of 1M Tris (untitrated, pH ~11). If it is too basic, iteratively add 10 µL of dilute HCl (1:25 or 1:100).
- Pre-wash the pY beads (for 5 mg of starting lysate)
- 25 µg of 4G10 antibody and 12.5 µg of 27B10.4 antibody are needed per sample. After using a p200 pipette with a cut tip to transfer the antibodies into separate microcentrifuge tubes, wash the antibodies with 450 µL of ice-cold IP binding buffer 2x. Centrifuge them at 100 x g for 1 min at 4 °C and aspirate out the supernatant.
- Resuspend the beads to a stock concentration of 0.5 mg/mL using IP binding buffer. (Do not vortex the beads.) After aliquoting the necessary slurry (50 µL of 4G10 antibody slurry and 25 µL 27B10.4 antibody slurry per sample) into a single tube, spin down the stock centrifuge tubes at 200 x g for 1 min at 4 °C. Wash the sidewalls with supernatant before returning the beads to storage in the refrigerator.
Add pre-washed pY beads to the resuspended sample solution in the screw cap cryotubes. Incubate them at 4 °C on an end-over-end rotator overnight.
Place the screw cap cryotubes in a 50 mL centrifuge tube lined with a delicate wipe. Spin down the beads at 100 x g for 1 min. Save the supernatant, which will be used to enrich for pST peptides. (The enrichment for pST begins at step 7 and can be performed in parallel to the pY peptide processing).
Resuspend the beads with 300 µL of IP binding buffer. Transfer them to a 2 mL microcentrifuge tube and spin them down at 100 x g for 1 min at 4 °C.
Rinse the incubation tube 3x with 200 µL of IP binding buffer. Transfer the contents to the same Microcentrifuge tube each time. Then spin them down.
Wash the beads in the microcentrifuge tube 3x with 500 µL of IP binding buffer and spin them down at 100 x g for 1 min. Then wash the beads 4x with 450 µL of 25 mM NH4HCO3, pH 7.5, and spin them down at 100 x g for 1 min. Use a fresh 25 mM NH4HCO3 solution from powder every time.
Centrifuge the beads at 1,500 x g for 1 min. Use a gel-loading tip to remove the supernatant completely by dipping the tip of the gel-loading tip slightly below the beads’ surface.
Add 4x the bead volume of 0.1% TFA to the beads (i.e., add 300 µL of 0.1% TFA for 75 µg of pY bead slurry). Mix them well and incubate the mixture in a thermomixer at 1,000 rpm for 15 min at 37 °C.
Transfer the resuspension to a 0.2 µm spin filter. Quickly spin down the elution tube and transfer the residual volume to the same spin filter using a P10 pipet. Spin down the spin filter at 850 x g for 1 min. Transfer the elution to a low protein-binding microcentrifuge tube. Vacuum concentrate the eluate to dryness overnight at 40 °C and with a heat time of 300 min. Note: The experiment can be paused here. Freeze the samples at -80 °C and continue at a later date.
5. Titanium Dioxide Enrichment25 of pY Peptides
Resuspend the dried down phosphopeptides in 200 µL of 50% ACN, 0.1% TFA. Vortex and centrifuge them at 10,000 x g for 30 s. Repeat this 1x to resuspend them well.
- Preparing the TiO2 beads contained in tips that have a capacity for 200 µL samples.
- Gently tap on the small tip-side of the tip to move the material to that end. Rinse the tip by adding 200 µL of 100% ACN, followed by inverting the tip and flicking the small end to move the liquid towards the cap.
- Using a razor blade, cut the small tip of the tip and place it over a low protein-binding tube. (Avoid using polystyrene tubes as the TiO2 will stick to the sides of the tube.) Remove the cap and insert a micropipette to plunge out the remaining ACN. Repeat the wash with 200 µL of 100% ACN. The TiO2 beads are now located in the low protein-binding tube for the following steps.
- Precondition TiO2 with 500 µL of 100% ACN 2x. Pipet it to mix the beads with the solvent. Centrifuge them at 100 x g for 1 min.
- Condition TiO2 with 500 µL of 0.2 M sodium phosphate buffer (pH ~7) 2x. Wash the beads with 300 µL of equilibration buffer 3x. Because TiO2 is very dense, the beads will settle quickly.
Add 400 µL of 50% ACN, 0.1% TFA into the low protein-binding tube, followed by adding 84 µL of lactic acid. Transfer the resuspended phosphopeptides into the low protein-binding tube and incubate them for 1 h at room temperature using an end-over-end rotator.
Centrifuge the beads at 100 x g for 1 min to pellet them. Wash them with 300 µL of equilibration buffer (Table 1) 2x and spin them down at 100 x g for 1 min.
Rinse the beads with 300 µL of rinsing buffer 2x. Transfer them to a 0.2 µm spin filter. Spin them at 1,500 x g for 1 min.
Transfer the filter unit to a clean 1.5 mL low protein-binding tube. Elute the contents 2x with 200 µL of 0.9% NH3 in H2O. Measure the pH with pH strips, which should be between 10 and 11. Vacuum concentrate the eluate to dryness overnight to evaporate the ammonia.
6. Desalting pY Peptides for MS Analyses
Reconstitute the phosphopeptides with 15 µL of 0.1% TFA by vortexing and centrifuging them at 10,000 x g for 30 s to resuspend them. Repeat this 1x to resuspend them well. Do not pipette up and down.
Clean the sample using a C-18 tip with a binding capacity of 5 µg and follow the manufacturer’s protocol.
Completely dry the elution volume by vacuum concentration. This takes 1 - 2 h. Resuspend the dried phosphopeptides in 12.5 µL of mass spectrometry solution (see Table 1) (or as recommended by the researcher’s MS proteomics core facility). Vortex and briefly spin the solution down at 10,000 x g for 30 s. Repeat this 2x to resuspend them well. The samples are ready for submission to a mass spectrometry facility (step 11). Note: The following steps below are related to pST peptide enrichment only.
7. Reverse Phase Extraction of pST Peptides
Measure the peptide concentration of the supernatant acquired from step 4.6 by performing a peptide assay. A sufficient amount for pST mass spectrometry is 2.5 mg.
Adjust pH to 3.5 with 5% TFA.
Connect the shorter end of a C-18 column to a vacuum manifold. Set the vacuum between 17 and 34 kPa (or according to the manufacturer’s instructions). Wet the column with 3 mL of 100% ACN. Do not let the column dry.
Equilibrate the column with 6 mL of 0.1% TFA applied as 2x 3 mL. Load the acidified sample into the column. Do not add more than 3 mL at a time. Adjust the vacuum to target about 1 - 2 drops per second.
Wash the column with 9 mL of 0.1% TFA applied as 3x 3 mL. Elute the column with 2 mL of 40% ACN, 0.1% TFA. Collect two 2 mL fractions into glass culture tubes. Discard the column.
Cover the eluate tubes with parafilm and punch 3 - 5 holes on the cover using a 20G needle. Freeze the eluate on dry ice for at least 30 min until it is completely solid.
Lyophilize the selected fractions overnight. On the following day, make sure the samples are completely dry before stopping the lyophilizer. Store the tubes in a 50 mL conical tube with delicate wipes at -80 °C. Note: The experiment can be paused here.
8. Strong Cation Exchange (SCX) of pST Peptides
Resuspend the lyophilized peptides in 2 mL of Buffer A (Table 1). Pool the fractions for each sample. (The solution will be cloudy.)
Prepare the vacuum manifold. Connect an SCX column to a 3 mL syringe with the plunger removed. Set the vacuum between 17 and 34 kPa (or according to the manufacturer’s instructions).
Condition the SCX column with 4 mL of ACN, followed by 4 mL of Buffer A.
Load the 2 mL of the sample from step 8.1 and collect the eluate immediately. Load 3 mL of A:B (80.9:19.1) buffer and collect the eluate. Pool the eluates of each sample and aliquot them into 2 mL low protein-binding tubes.
Vacuum concentrate all samples until approximately 30% of the volume remains. (This step takes approximately 2 - 4 h.) Pool the aliquots into 1 low protein-binding tube for each sample.
Connect the shorter end of a C-18 column to a vacuum manifold. Set the vacuum between 17 and 34 kPa (or according to the manufacturer’s instructions). Wet the column with 3 mL of 100% ACN 2x. Do not let the column dry.
Equilibrate the column with 3 mL of 0.1% TFA 2x. Load the sample into the column. Do not add more than 3 mL at a time. Adjust the vacuum to target about 1 - 2 drops per second.
Wash the column with 3 mL of 0.1% TFA 2x. Elute the column with 4 mL of 50% ACN, 0.1% TFA.
9. Titanium Dioxide Enrichment of pST Peptides
- Preparing the TiO2 beads contained in tips that have a capacity for 200 µL samples
- Gently tap on the small tip side of the tip to move the beads to that end. Remove the cap and pour the beads into a polypropylene 15 mL conical tube.
- Rinse the tip by adding 200 µL of 100% ACN, inverting the tip a couple times and flicking the small end to move the liquid towards the cap. Using a razor blade, cut the small tip of the tip and place it over the polypropylene 15 mL conical tube. Remove the cap and insert a micropipette to plunge out the remaining ACN. Repeat the wash with 200 µL of 100% ACN. The TiO2 beads are now located in the 15 mL conical tube for the following steps.
- Precondition TiO2 with 500 µL of 100% ACN 2x. Pipet it to mix the beads with the solvent. Centrifuge them at 100 x g for 1 min.
- Condition TiO2 with 500 µL of 0.2 M sodium phosphate buffer (pH ~7) twice. Wash the beads with 300 µL of equilibration buffer 3x.
Transfer the eluted phosphopeptides into the polypropylene 15 mL conical tube. Add 560 µL of lactic acid and incubate it for 1 h at room temperature using an end-over-end rotator.
Centrifuge the mixture at 100 x g for 1 min to pellet the beads. Wash them with 300 µL of equilibration buffer (Table 1) 3x. Spin them down at 100 x g for 1 min.
Rinse the beads with 300 µL of rinsing buffer 2x. Transfer them to a 0.2 µm spin filter. Spin them down at 1,500 x g for 1 min.
Transfer the filter unit to a clean 1.5 mL low protein-binding tube. Elute the contents 2x with 200 µL of 0.9% NH3 in H2O. Let the solution sit on the phosphopeptides for 2 min before eluting them. Measure the pH, which should be between 10 and 11.
Vacuum concentrate the eluate to dryness overnight to evaporate the ammonia.
10. Desalting pST Peptides for MS Analyses
Gently tap on the small tip-side of the tip to move the material to that end. Rinse the tip by adding 200 µL of 100% ACN, followed by inverting the tip and flicking the small end to move the liquid towards the cap.
Using a razor blade, cut the small tip of the tip and place it over a polypropylene 15 mL conical tube. Remove the cap and insert a micropipette to plunge out the remaining ACN. Repeat the wash with 200 µL of 100% ACN. The TiO2 beads are now located in the polypropylene 15 mL conical tube for the following steps.
Clean the sample using a C-18 tip with a binding capacity of 100 µg. (Follow the manufacturer’s instructions.)
Completely dry the elution volume by vacuum concentration. This takes 1 - 2 h.
Resuspend the dried phosphopeptides in 12.5 µL of mass spectrometry solution (or as recommended by the researcher’s MS proteomics core facility). Vortex and centrifuge them at 10,000 x g for 30 s. Repeat 2x to resuspend them well. (Do not pipette up and down.)
11. Mass Spectrometry Analysis
- Submit the samples to the MS proteomics core facility to perform liquid chromatography-tandem MS (LC-MS/MS) using their recommended settings. Example settings are as follows (see Table 2 for the summary):
- Load 5 µL of the samples onto a trap column (2 cm long x 75 µm diameter) and wash them with 0.1% TFA for 5 min with a flow rate of 5 µL/min.
- Bring the trap in line with a nano analytical column (20 cm x 75 µm) with a flow rate of 300 nL/min.
- The segmented linear gradients (a percentage of 0.16% formic acid, 80% ACN in 0.2% formic acid) are different between pY and pST samples:
- For the pY samples, elute them using a gradient of 4 - 15% in 5 min, 15 - 50% in 40 min, and 50 - 90% in 5 min.
- For pST samples, elute them using a gradient of 4 - 15% in 30 min, 15 - 25% in 40 min, 25 - 50% in 44 min, and 50 - 90% in 11 min.
- Acquire MS data in data-dependent acquisition mode with a cyclic series of a full scan with a resolution of 120,000 followed by MS/MS (HCD, relative collision energy of 27%) of the 20 most intense ions and a dynamic exclusion duration of 20 s.
After the MS run completion, import the MS raw files into an MS analysis software program to identify and quantify phosphopeptides. (MaxQuant software8,26,27 was used in this experiment. Unless specified in Table 3, the default settings were used.)
Representative Results
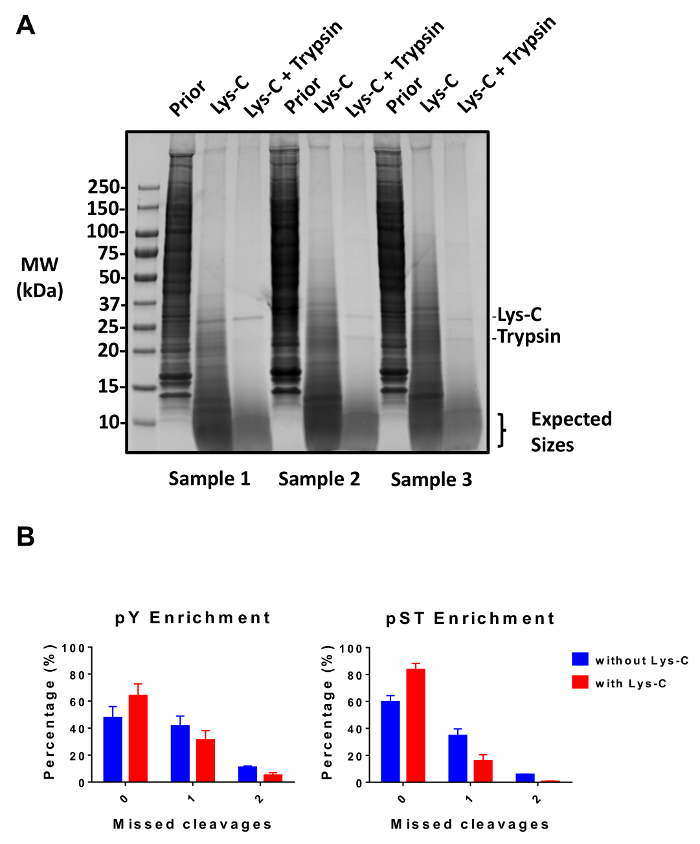
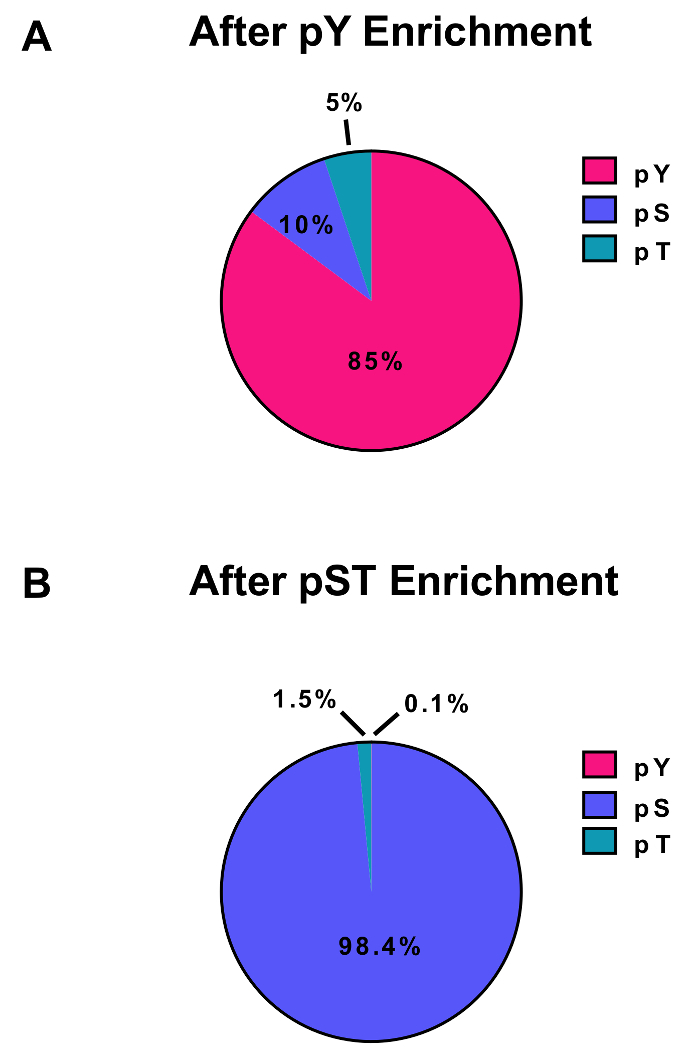
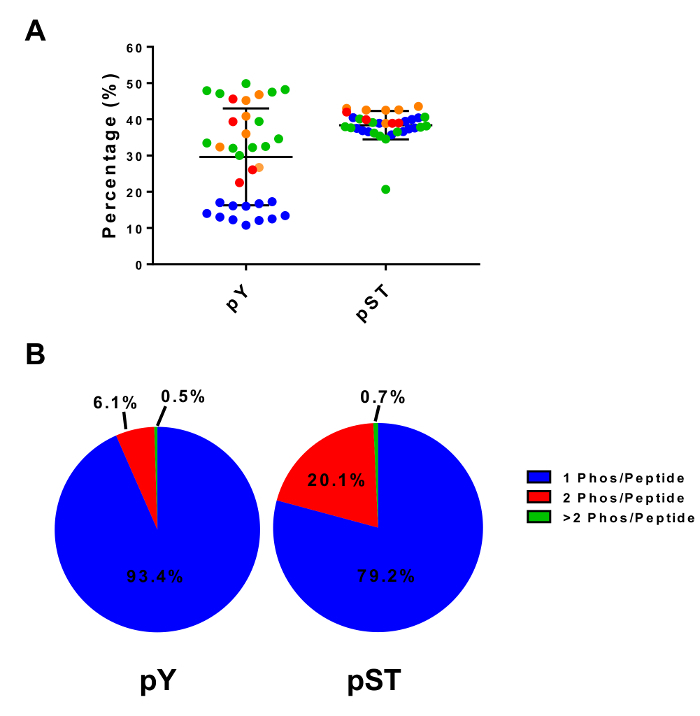
This protocol describes in detail a method for protein extraction and digestion followed by phosphopeptide enrichment and subsequent MS analysis (Figure 1). The compositions of all the buffers and solutions that are used in this protocol are listed in Table 1. The sequential use of Lys-C and trypsin provides an efficient digestion. A Coomassie-stained gel of pre-digested lysate confirms the presence of proteins, while staining of post-digested lysate confirms the complete digestion (Figure 2A). For a complete digestion, no bands should appear above 15 kDa, except the 30 kDa and 23.3 kDa bands for Lys-C and trypsin, respectively. The addition of Lys-C also reduces the number of missed cleavages (Figure 2B). Because pY peptides represent only 2% of the phosphoproteome28, immunoprecipitation of the pY peptides using a pY-specific antibody is the first step of pY peptide enrichment. The resulting supernatant becomes the input for pST peptide enrichment. The pY immunoprecipitation effectively separates pY peptides from pST peptides where on average 85% of the phosphopeptides identified from the pY preparation are pY (Figure 3A) and over 99% of the phosphopeptides identified from the pST preparation are pST (Figure 3B). Titanium dioxide is used to enrich for phosphopeptides in both preparations. The expected percentage of peptides in the MS-ready preparation that are phosphorylated is between 30 - 50% (Figure 4A). The variability in the phosphopeptide enrichment percentage may be greater in the pY preparation as a result of there being many fewer pY peptides than pST peptides. In terms of phosphopeptide species, the majority of the phosphopeptides detected have a single or double phosphoryl group (Figure 4B).
After performing mass spectrometry, the MS raw files are loaded into an MS analysis software. The parameter settings used in the experiment are listed in Table 3 but will vary from software to software and may vary from version to version. The parameters that are not listed were left as default, including an FDR cutoff of 1% for peptide-spectrum matching (PSM) with a minimum Andromeda score of 40 for the identification of modified peptides27. Setting a localization probability cutoff of greater than 0.75 filters out approximately 5% of the pY peptides and 15% and 34% of the pS and pT peptides, respectively (Figure 5A). After applying these filters, the expected number of phosphopeptide identifications at the end of the MS analysis is approximately 300 pY peptides (for 5 mg of the starting protein) and about 7,500 pS peptides and 640 pT peptides (for 2.5 mg of the starting peptide amount) from the respective enrichment preparations (Figure 5B). The number of replicates and the variability of the phosphopeptide signal intensity determines adequate powering for statistical comparisons. In four separate experiments with groups containing either biological duplicates or triplicates, the percent coefficients of variation (%CV) for detected phosphopeptides were calculated. Distributions of lower variability (e.g., pST groups 1 - 5 in Figure 5C) indicate that the sample collection, preparation, and mass spectrometry runs were consistent. On the other hand, distributions of higher variability (e.g., pST group 6 in Figure 5C) indicates noisier data that would require larger fold-changes to detect significant differences in downstream differential analyses.
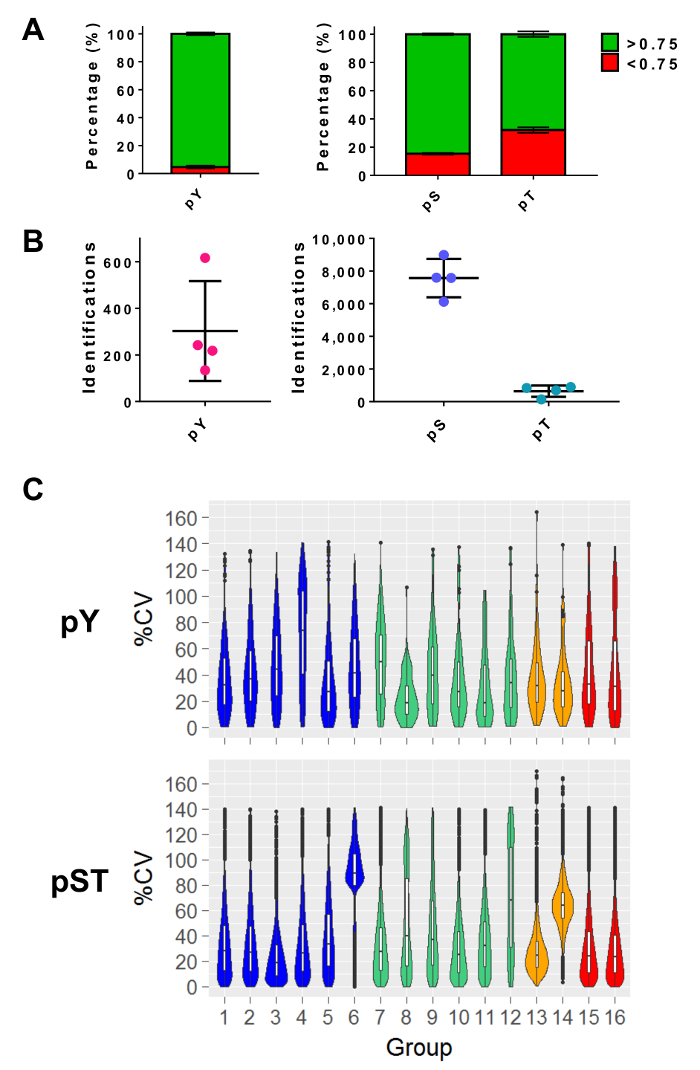
Figure 1: Workflow diagram. Proteins from samples are extracted and digested. Peptides are extracted by solid phase extraction (SPE), and phosphotyrosine (pY) peptides are immunoprecipitated. In parallel, the phosphoserine/threonine (pST) peptides are enriched from the supernatant in the pY immunoprecipitation step. Strong cation exchange (SCX) is performed on the supernatant to remove highly charged peptides to reduce the ion suppression12. Both preparations undergo phosphopeptide enrichment via titanium dioxide (TiO2). After sample cleanup, liquid chromatography-tandem mass spectrometry (LC-MS/MS) is performed to measure the phosphopeptide abundance. The raw data is then loaded into an MS analysis software to identify phosphopeptides. Please click here to view a larger version of this figure.
Figure 2: Evaluation of digestion. (A) Three samples with 12.5 µg of lysate pre-digestion, post-Lys-C digestion, and post-trypsin digestion are shown. A Coomassie gel-stain test shows a clean digestion after sequential use of Lys-C and trypsin. The molecular weight (MW) size markers are in kilodaltons (kDa). (B) A reduction in missed cleavages is observed after Lys-C was added to the protocol. The percentage of phosphopeptides without missed cleavages increased from 48% to 64% and from 60% to 84% on average for pY and pST enrichment preparations, respectively. The graphs summarize the data obtained from two experiments performed without Lys-C and five experiments performed with Lys-C. The error bars are standard deviations representing 38 pY and 38 pST samples from 2 separate experiments (without Lys-C) and 62 pY and 60 pST samples from 5 separate experiments (with Lys-C). Please click here to view a larger version of this figure.
Figure 3: Enrichment of pY and pST phosphopeptides. These panels show the percentages of pSTY phosphopeptides from either (A) the pY or (B) the pST enrichment preparations. The pY enrichment by pY immunoprecipitation and titanium dioxide resulted in 85% phosphopeptides being for pY peptides, while only 0.1% of the phosphopeptides in the pST enrichment are pY. These values were drawn from examining the Phospho (STY)Sites.txt file of one representative experiment after filtering out contaminants, reverse sequences, and phosphopeptides with localization probabilities less than 0.75. Please click here to view a larger version of this figure.
Figure 4: Phosphopeptide enrichment with titanium dioxide. (A) The percentage of detected phosphopeptides (relative to total peptides) from samples in four separate experiments is shown. (B) This panel shows the average composition of mono-, double-, and multi-phosphorylated peptides in four separate experiments. The error bars in panel A are standard deviations. Please click here to view a larger version of this figure.
Figure 5: Expected phosphoresidue identifications. (A) This panel shows the phosphorylation localization probabilities of IDs from pY enrichment (left) and pST enrichment (right). The mean percentage of IDs that meet the > 0.75 probability cutoff is 93%, 75%, and 52% for pY, pS, and pT, respectively. (B) The mean number of IDs with a >0.75 localization probability is 300 for pY, 7,500 for pS, and 640 for pT. (C) This panel shows violin plots of the percent coefficient of variation (%CV) of the phosphopeptides. An evaluation of %CV was only performed if a signal intensity value was detected in each biological replicate or triplicate group. Data was taken from four separate experiments. The error bars in panels A and B are standard deviations from 34 pY and 34 pST samples from 4 separate experiments. Please click here to view a larger version of this figure.
| Buffer | Volume | Composition |
| 6 M guanidinium chloride lysis buffer | 50 mL | 6 M guanidinium chloride, 100 mM tris pH 8.5, 10 mM tris (2-carboxyethyl) phosphine, 40 mM chloroacetamide, 2 mM sodium orthovanadate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 500 mg n-octyl-glycoside, ultra-pure water to volume |
| 100 mM sodium pyrophosphate | 50 mL | 2.23 g sodium pyrophosphate decahydrate, ultra-pure water to volume |
| 1M β-glycerophosphate | 50 mL | 15.31 g β-glycerophosphate, ultra-pure water to volume |
| 5% trifluoroacetic acid | 20 mL | Add 1 mL of 100% trifluoroacetic acid into 19 mL ultra-pure water |
| 0.1% trifluoroacetic acid | 250 mL | Add 5 mL 5% trifluoroacetic acid to 245 mL ultra-pure water |
| pY elution buffer | 250 mL | 0.1% trifluoroacetic acid, 40% acetonitrile, ultra-pure water to volume |
| pST elution buffer | 250 mL | 0.1% trifluoroacetic acid, 50% acetonitrile, ultra-pure water to volume |
| IP binding buffer | 200 mL | 50 mM tris pH 7.4, 50 mM sodium chloride, ultra-pure water to volume |
| 25 mM ammonium bicarbonate, pH 7.5 | 10 mL | Dissolve 19.7 mg into 10 mL sterile ultra-pure water, pH to 7.5 with 1 N hydrochloric acid (~10-15 µL/10 ml solution), make fresh |
| 1M phosphate buffer, pH 7 | 1,000 mL | 423 mL 1 M sodium dihydrogen phosphate, 577 mL 1 M sodium hydrogen phosphate |
| Equilibration buffer | 14 mL | 6.3 mL acetonitrile, 280 µL 5% trifluoroacetic acid, 1740 µL lactic acid, 5.68 mL ultra-pure water |
| Rinsing buffer | 20 mL | 9 mL acetonitrile, 400 µL 5% trifluoroacetic acid, 10.6 mL ultra-pure water |
| Mass spectrometry solution | 10 mL | 500 µL acetonitrile, 200 µL 5% trifluoroacetic acid, 9.3 mL ultra-pure water |
| Buffer A | 250 mL | 5 mM monopotassium phosphate (pH 2.65), 30% acetonitrile, 5 mM potassium chloride,ultra-pure water to volume |
| Buffer B | 250 mL | 5 mM monopotassium phosphate (pH 2.65), 30% acetonitrile, 350 mM potassium chloride, ultra-pure water to volume |
| 0.9% ammonium hydroxide | 10 mL | 300 μL 29.42% ammonium hydroxide, 9.7 mL ultra-pure water |
Table 1: Buffers and solutions. This table shows the compositions of the buffers and solutions used in this protocol.
| LC-MS/MS Settings | ||
| Parameter | pY Setting | pST Setting |
| Sample loading (µL) | 5 | |
| Loading flow rate (µL/min) | 5 | |
| Gradient flow rate (nL/min) | 300 | |
| Linear gradient (percentage 0.16% formic acid, 80% ACN in 0.2% formic acid) | 4 - 15% for 5 min | 4 - 15% for 30 min |
| 15 - 50% for 40 min | 15 - 25% for 40 min | |
| 50 - 90% for 5 min | 25 - 50% for 44 min | |
| 50 - 90% for 11 min | ||
| Full scan resolution | 120,000 | |
| Number of most intense ions selected | 20 | |
| Relative collision energy (%) (HCD) | 27 | |
| Dynamic exclusion (s) | 20 |
Table 2: LC-MS settings. This is an example of LC-MS settings in a typical shotgun phosphoproteomic experiment. The samples were loaded on to a trap column. The trap was brought in-line with an analytical column. These settings were optimized for using the LC-MS system listed in the Table of Materials and Reagents. These settings would need to be adjusted for other LC-MS systems.
| MaxQuant Parameter Settings | ||
| Setting | Action | |
| Group-Specific Parameters | ||
| Type | Type | Select Standard |
| Multiplicity | Set to 1 | |
| Digestion Mode | Enzyme | Select Trypsin/P |
| Max. missed cleavages | Set to 2 | |
| Modifications | Variable modifications | Add Phospho (STY) |
| Label-free quantification | Label-free quantification | Select LFQ |
| LFQ min. ratio count | Set to 1 | |
| Fast LFQ | Check off | |
| Miscellaneous | Re-quantify | Check off |
| Global Parameters | ||
| Sequences | FASTA files | Select fasta file downloaded from UniProt |
| Fixed modifications | Add Carbamidomethyl (C) | |
| Adv. Identification | Match between runs | Check off |
| Match time window | Set to 5 min | |
| Alignment time window | Set to 20 min | |
| Match unidentified features | Check off | |
| Protein quantification | Min. ratio count | Set to 1 |
| Folder locations | Modify accordingly |
Table 3: MS analysis software settings. In MaxQuant, the group-specific and global parameters in this table were selected or adjusted. All other parameters remained at default. These experiments were conducted using version 1.5.3.30. The parameters may vary from version to version and from software to software.
Discussion
Before utilizing this protocol to enrich for phosphopeptides, a careful consideration of the experimental design is critical. Using biological replicates is a more cost-effective use of mass spectrometry resources than technical replicates. The number of replicates that are necessary will depend in part on the variability of the data. A recent study demonstrated that, while increasing the number of replicates beyond three only marginally increases the number of identifications, the number of significant identifications between groups increases with more replicates10.
Due to the lower abundance of phosphoproteins in the cell, sufficient starting protein amounts are necessary to obtain a global phosphoproteome from prostate cancer samples in discovery mode. In these experiments, 5 mg of protein was used. Approximately five nearly confluent 15-cm dishes of cells provide enough protein as input into this protocol, although this will be cell line-dependent. As for tumor tissue, the expected yield of protein is about 6 - 8% of tissue weight. In the in vitro setting, a positive control sample to consider is the addition of 1 mM vanadate for 30 min before harvesting the cells. Vanadate, a competitive protein phosphotyrosyl phosphatase inhibitor, will preserve the tyrosine phosphorylation, thus increasing the number of pY peptide identifications29.
Clean digestion is a key step to maximize phosphopeptide identification. In addition to the Coomassie stain test, the percent of missed cleavages in the data can be used to evaluate digestion efficiency (Figure 2). Quality-control software is available that analyzes missed cleavages and other metrics to assess MS data quality30. While trypsin is the most common, alternative proteases are available5 to address coverage gaps in the proteome where optimal tryptic peptides cannot be generated31. The settings of the MS analysis software would then need to be modified accordingly to adjust for changes in proteases.
The protocol employs immunoprecipitation (for pY enrichment) as well as titanium dioxide (TiO2) to enrich for phosphopeptides. Alternative approaches to enrich for peptides include immobilized metal affinity chromatography (IMAC), other metal oxides for metal oxide affinity chromatography (MOAC) such as aluminum hydroxide, and polymer-based metal ion affinity capture (PolyMAC)5,12. Previous studies have shown that different enrichment methods enrich for different populations of phosphopeptides32. For instance, IMAC enriches more multi-phosphorylated peptides while MOAC preferably enriches for mono-phosphorylated peptides33. The Representative Results of this protocol reflect this observation (Figure 4B). A recent publication demonstrated that combining IMAC and MOAC using a hybrid material could potentially provide greater coverage of phosphopeptide species34. Thus, this protocol could be modified to utilize other enrichment methods in parallel to allow for even more comprehensive phosphoproteomic analyses.
The MaxQuant26 software suite is used to analyze the MS data in this protocol, but commercial applications35 are also available for phosphopeptide identification and quantification. For phosphopeptide identification, a localization probability cutoff is applied. This filter is performed to select for phosphopeptides with a high confidence (i.e., greater than 0.75) in phosphoresidue identification10,28. In other words, the summed probability of all other residues that could potentially contain the phospho-group is less than 0.25. This cutoff could be raised to increase the stringency of the phosphopeptide selection. In regard to the number of identifications, the expected number of pY peptides is in the hundreds, while the expected number of pST peptides is in the high thousands. These values reflect previously observed phosphoproteome distribution where about 2%, 12%, and 86% of the phosphosites are pY, pT, and pS, respectively28.
If the pY and pST enrichment steps are performed in parallel, the sample preparation steps in the protocol can be completed in six days. By pairing with the powerful tool of MS, phosphopeptide enrichment protocols such as this provide a global approach for scientists to collect data to analyze the phosphoproteome in their respective research fields.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank members of the Drake lab for providing advice and input on the manuscript. We also thank the members of the Biological Mass Spectrometry Facility of Robert Wood Johnson Medical School and Rutgers, The State University of New Jersey, for providing advice and performing mass spectrometry on our samples. Larry C. Cheng is supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number T32 GM008339. Thomas G. Graeber is supported by the NCI/NIH (SPORE in Prostate Cancer P50 CA092131; P01 CA168585) and an American Cancer Society Research Scholar Award (RSG-12-257-01-TBE). Justin M. Drake is supported by the Department of Defense Prostate Cancer Research Program W81XWH-15-1-0236, Prostate Cancer Foundation Young Investigator Award, the New Jersey Health Foundation, and a Precision Medicine Initiative Pilot Award from the Rutgers Cancer Institute of New Jersey.
References
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA: A Cancer Journal for Clinicians. 2018;68(1):7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- Paller CJ, Antonarakis ES. Management of biochemically recurrent prostate cancer after local therapy: evolving standards of care and new directions. Clinical Advances in Hematology & Oncology. 2013;11(1):14–23. [PMC free article] [PubMed] [Google Scholar]
- Lowrance WT, Roth BJ, Kirkby E, Murad MH, Cookson MS. Castration-resistant prostate cancer: AUA guideline amendment 2015. The Journal of Urology. 2016;195(5):1444–1452. doi: 10.1016/j.juro.2015.10.086. [DOI] [PubMed] [Google Scholar]
- Domon B, Aebersold R. Options and considerations when selecting a quantitative proteomics strategy. Nature Biotechnology. 2010;28(7):710–721. doi: 10.1038/nbt.1661. [DOI] [PubMed] [Google Scholar]
- Zhang YY, Fonslow BR, Shan B, Baek MC, Yates JR. Protein analysis by shotgun/bottom-up proteomics. Chemical Reviews. 2013;113(4):2343–2394. doi: 10.1021/cr3003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantscheff M, Schirle M, Sweetman G, Rick J, Kuster B. Quantitative mass spectrometry in proteomics: a critical review. Analytical and Bioanalytical Chemistry. 2007;389(4):1017–1031. doi: 10.1007/s00216-007-1486-6. [DOI] [PubMed] [Google Scholar]
- Bantscheff M, Lemeer S, Savitski MM, Kuster B. Quantitative mass spectrometry in proteomics: critical review update from 2007 to the present. Analytical and Bioanalytical Chemistry. 2012;404(4):939–965. doi: 10.1007/s00216-012-6203-4. [DOI] [PubMed] [Google Scholar]
- Cox J, et al. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Molecular & Cellular Proteomics. 2014;13(9):2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubbi L, et al. Global phosphoproteomics reveals crosstalk between Bcr-Abl and negative feedback mechanisms controlling Src signaling. Science Signaling. 2011;4(166):ra18. doi: 10.1126/scisignal.2001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogrebe A, et al. Benchmarking common quantification strategies for large-scale phosphoproteomics. Nature Communications. 2018;9(1):1045. doi: 10.1038/s41467-018-03309-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush J, et al. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nature Biotechnology. 2005;23(1):94–101. doi: 10.1038/nbt1046. [DOI] [PubMed] [Google Scholar]
- Fila J, Honys D. Enrichment techniques employed in phosphoproteomics. Amino Acids. 2012;43(3):1025–1047. doi: 10.1007/s00726-011-1111-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P, et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534(7605):55–62. doi: 10.1038/nature18003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JM, et al. Phosphoproteome integration reveals patient-specific networks in prostate cancer. Cell. 2016;166(4):1041–1054. doi: 10.1016/j.cell.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue HW, et al. Metabolic reprogramming ensures cancer cell survival despite oncogenic signaling blockade. Genes & Development. 2017;31(20):2067–2084. doi: 10.1101/gad.305292.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francavilla C, et al. Phosphoproteomics of primary cells reveals druggable kinase signatures in ovarian cancer. Cell Reports. 2017;18(13):3242–3256. doi: 10.1016/j.celrep.2017.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, et al. Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell. 2016;166(3):755–765. doi: 10.1016/j.cell.2016.05.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaggs BJ, et al. Phosphorylation of the ATP-binding loop directs oncogenicity of drug-resistant BCR-ABL mutants. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(51):19466–19471. doi: 10.1073/pnas.0609239103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimman A, et al. Activation of aortic endothelial cells by oxidized phospholipids: a phosphoproteomic analysis. Journal of Proteome Research. 2010;9(6):2812–2824. doi: 10.1021/pr901194x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimman A, Berliner JA, Graeber TG. Phosphoproteomic analysis of aortic endothelial cells activated by oxidized phospholipids. Methods in Molecular Biology. 2013. pp. 53–69. [DOI] [PubMed]
- Humphrey SJ, Azimifar SB, Mann M. High-throughput phosphoproteomics reveals in vivo insulin signaling dynamics. Nature Biotechnology. 2015;33(9):990–995. doi: 10.1038/nbt.3327. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Meyer TS, Lamberts BL. Use of coomassie brilliant blue R250 for the electrophoresis of microgram quantities of parotid saliva proteins on acrylamide-gel strips. Biochimica el Biophysica Acta. 1965;107(1):144–145. doi: 10.1016/0304-4165(65)90403-4. [DOI] [PubMed] [Google Scholar]
- Bergstrom Lind S, et al. Immunoaffinity enrichments followed by mass spectrometric detection for studying global protein tyrosine phosphorylation. Journal of Proteome Research. 2008;7(7):2897–2910. doi: 10.1021/pr8000546. [DOI] [PubMed] [Google Scholar]
- Pinkse MW, Uitto PM, Hilhorst MJ, Ooms B, Heck AJ. Selective isolation at the femtomole level of phosphopeptides from proteolytic digests using 2D-NanoLC-ESI-MS/MS and titanium oxide precolumns. Analytical Chemistry. 2004;76(14):3935–3943. doi: 10.1021/ac0498617. [DOI] [PubMed] [Google Scholar]
- Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature Biotechnology. 2008;26(12):1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Cox J, et al. Andromeda: a peptide search engine integrated into the MaxQuant environment. Journal of Proteome Research. 2011;10(4):1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- Olsen JV, et al. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127(3):635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Huyer G, et al. Mechanism of inhibition of protein-tyrosine phosphatases by vanadate and pervanadate. The Journal of Biological Chemistry. 1997;272(2):843–851. doi: 10.1074/jbc.272.2.843. [DOI] [PubMed] [Google Scholar]
- Bielow C, Mastrobuoni G, Kempa S. Proteomics quality control: quality control software for MaxQuant results. Journal of Proteome Research. 2016;15(3):777–787. doi: 10.1021/acs.jproteome.5b00780. [DOI] [PubMed] [Google Scholar]
- Swaney DL, Wenger CD, Coon JJ. Value of using multiple proteases for large-scale mass spectrometry-based proteomics. Journal of Proteome Research. 2010;9(3):1323–1329. doi: 10.1021/pr900863u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodenmiller B, Mueller LN, Mueller M, Domon B, Aebersold R. Reproducible isolation of distinct, overlapping segments of the phosphoproteome. Nature Methods. 2007;4(3):231–237. doi: 10.1038/nmeth1005. [DOI] [PubMed] [Google Scholar]
- Leitner A, Sturm M, Lindner W. Tools for analyzing the phosphoproteome and other phosphorylated biomolecules: a review. Analytica Chimica Acta. 2011;703(1):19–30. doi: 10.1016/j.aca.2011.07.012. [DOI] [PubMed] [Google Scholar]
- Yang DS, et al. Design and synthesis of an immobilized metal affinity chromatography and metal oxide affinity chromatography hybrid material for improved phosphopeptide enrichment. Journal of Chromatography A. 2017;1505:56–62. doi: 10.1016/j.chroma.2017.05.025. [DOI] [PubMed] [Google Scholar]
- Al Shweiki MR, et al. Assessment of label-free quantification in discovery proteomics and impact of technological factors and natural variability of protein abundance. Journal of Proteome Research. 2017;16(4):1410–1424. doi: 10.1021/acs.jproteome.6b00645. [DOI] [PubMed] [Google Scholar]