

Abstract
White adipose tissue (WAT) plays a crucial role in regulating weight and everyday health. Still, there are significant limitations to available primary culture models, all of which have failed to faithfully recapitulate the adipose microenvironment or extend WAT viability beyond two weeks. The lack of a reliable primary culture model severely impedes research in WAT metabolism and drug development. To this end we have utilized NIH's standards of a microphysiologic system to develop a novel platform for WAT primary culture called 'SWAT' (sandwiched white adipose tissue). We overcome the natural buoyancy of adipocytes by sandwiching minced WAT clusters between sheets of adipose-derived stromal cells. In this construct, WAT samples are viable over eight weeks in culture. SWAT maintains the intact ECM, cell-to-cell contacts, and physical pressures of in vivo WAT conditions; additionally, SWAT maintains a robust transcriptional profile, sensitivity to exogenous chemical signaling, and whole tissue function. SWAT represents a simple, reproducible, and effective method of primary adipose culture. Potentially, it is a broadly applicable platform for research in WAT physiology, pathophysiology, metabolism, and pharmaceutical development.
Keywords: Behavior, Issue 138, Adipose tissue, adipocytes, obesity, microphysiological systems, organs on chips, drug screening, drug development, environmental toxicity, primary culture, diabetes, metabolism
Introduction
Adipose tissue is the primary organ of obesity, which carries direct annual medical costs between $147 billion and $210 billion in the U.S.1. The accumulation of adipose tissue also contributes to other leading causes of death such as heart disease, type II diabetes, and certain types of cancer2. In vitro culture models are essential for metabolic studies and drug development, but current research models of adipose tissue have major deficiencies. Adipocytes are fragile, buoyant, and terminally differentiated cells that will not adhere to cell culture plastics, and therefore cannot be cultured using conventional cell culture methods. Since the 1970s, several methods have been used in attempts to overcome these barriers, including the use of glass coverslips, ceiling culture, suspension culture, and extracellular matrices3,4,5,6,7. However, these methods have been marked by cell death and dedifferentiation, and they are typically used for no more than a two-week study period. Moreover, these models do not attempt recapitulate the native adipose microenvironment as they do not maintain the intact ECM, the interactions between adipocytes and stromal support cells, nor the contractile forces cells exert on each other in in vivo WAT.
In the absence of a gold-standard primary adipose culture method, adipose research has relied primarily on differentiated pre-adipocytes (diffAds). DiffAds are multilocular, adherent, and metabolically active. By contrast, primary white adipocytes are unilocular, nonadherent, and demonstrate relatively low metabolism. The failure of current adipose culture models to recapitulate the physiology of healthy mature adipose tissue is likely a major factor in the absence of FDA-approved medications that directly target adipocytes. In fact, the lack of physiologic in vitro organ models is a major problem across most organs and disease.
In its position paper announcing the creation of its Microphysiological Systems (MPS) program, the National Institutes of Health (NIH) reported that the 2013 success rate across all human pharmaceutical clinical trials was only 18% for phase II and 50% for phase III clinical trials8.The MPS program is designed to directly address the inability of in vitro monoculture to model human physiology. The NIH defines MPSs as culture systems comprised of human primary or stem cells in multicellular 3D constructs that recapitulate organ functioning. Unlike reductionist models of homogeneous, immortalized cell cultures, MPSs should accurately model cell-cell, drug-cell, drug-drug, and organ-drug interactions9. Unlike short-term primary culture methods, NIH standards dictate MPS sustainability over 4 weeks in culture8. Further details of the MPS program can be found at the NIH's RFAs (#RFA-TR-18-001)10.
We have developed a simple, novel, adaptable, and inexpensive adipose MPS termed "sandwiched white adipose tissue" (SWAT)11. We overcome the natural buoyancy of adipocytes by "sandwiching" minced primary adipose tissue between sheets of adipose-derived stromal cells (ADSCs) (Figure 1). The resulting 3D construct recapitulates the cell-cell contact and the native adipose microenvironment by surrounding mature adipocytes with a natural adipocyte support cell population. SWAT has been validated by demonstrating 8-week viability, response to exogenous signaling, adipokine secretion, and engraftment into an animal model.
Protocol
All tasks were performed in adherence to protocols #8759 and #9189, as approved by the IRB Office of LSUHSC-NO. All animal work was performed in adherence to protocol #3285 approved by the IACUC Office at LSUHSC-NO.
1. Seeding of Sandwiching Cell Sheets
NOTE: See Figure 1.
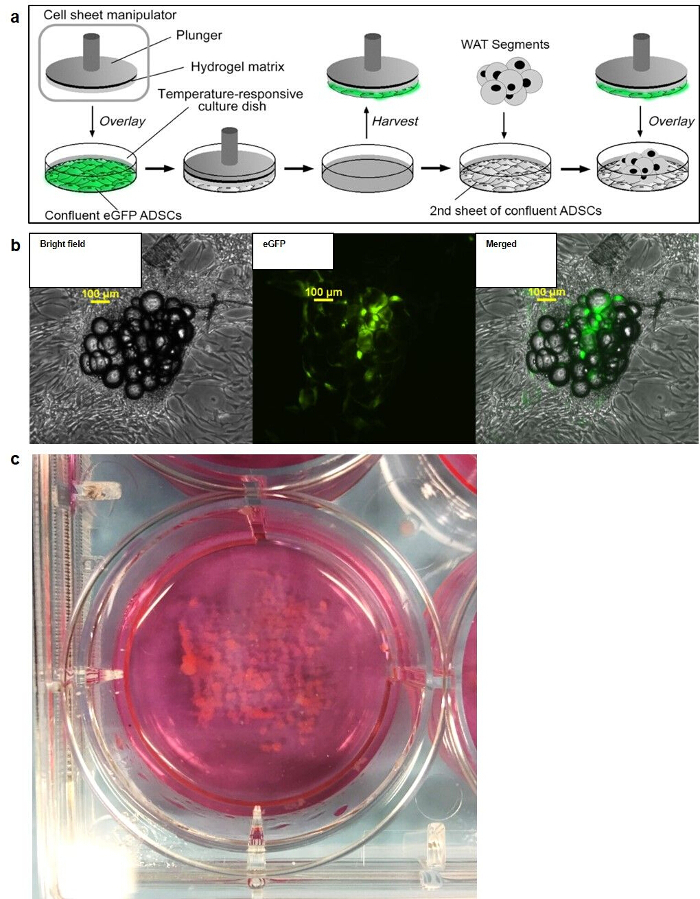
Seed ADSCs at approximately 80% confluency in tissue culture plates (6 cm or 6-well plates). For each well of SWAT desired, seed 1 conventional tissue culture well and 1 well of corresponding size on poly(N-isopropylacrylamide (pNIPAAm)-coated tissue culture plastic plate. NOTE: Accordingly, a 6-well plate of SWAT will require seeding cells on one 6-well standard tissue culture plate (base layer) and one 6-well pNIPAAm-coated tissue culture plate (upper layer). pNIPAAm-coated plates can be bought commercially or produced in-lab12,13,14.
Maintain ADSCs at 37 °C and 5% CO2 in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% FBS and 1x Penicillin/Streptomycin. Change media every 2 days.
Allow cells to coalesce until they become 100% confluent and take on a striated pattern (approximately 6–8 days). NOTE: These cells will need to function as a single intact cell sheet in order to stabilize the buoyant adipose tissue. Insufficiently confluent cells will fragment upon seeding with WAT.
2. Preparation of SWAT Supplies
Prepare several 10 mL aliquots of 1x Hank's Balanced Salt Solution (HBSS); prepare enough for the desired number of plates with volume to spare.
- Prepare the plunger apparatus for SWAT seeding. Construct the plunger using simple acrylic plastics, comprised of a stem attached to a round disk. Ensure that it fits within the circumference of the tissue culture wells (diameter: <6 cm for 6 cm dish, <3.5 cm for 6-well plate; approximate mass: 6.7 g for a 6 cm dish, 5.1 g for a 6-well plate).
- Prepare extra plungers to ensure that the procedure will run smoothly in case there is a problem with any individual plunger.
- Wrap lab tape around the rim of the plunger disk, at least 2x, to prevent leakage of gelatin solution.
- Spray a biosafety cabinet (BSC) with 70% EtOH. Spray down a 15 mL conical tube rack inside the BSC with empty, uncapped 15 mL conical tubes; tubes will help secure the placement of the gelatin plungers.
- Spray each tape-wrapped plunger thoroughly with 70% EtOH and place into a 15mL conical tube on the rack. Spray metal washers (approximate mass 6.3 g) and place them on the rack along with a pair of hooked forceps; the washers will add weight to the plungers during SWAT seeding.
- Close the BSC sash and turn on the UV light to facilitate drying and sterilization. NOTE: Ideally, conduct this step 24 h prior to SWAT seeding. Alternatively, conduct the process on the day of tissue collection/SWAT seeding; however, in this case, allow 15–45 min for UV drying/sterilization.
3. Preparation of Gelatin Plungers — Application to Upper Cell Sheets
Heat a water bath to 75 °C.
- Prepare the gelatin solution by adding 0.75 g of gelatin powder to 10 mL stock of 1x HBSS. Under a fume hood add 100 µL of 1 M NaOH to balance the pH of the solution.
- Add the 10 mL stocks to the water bath and shake vigorously every 5 min until the powder dissolves into solution. Aim to dissolve the powdered gelatin soon after adding to the heated water bath.
- Turn on the BSC blower, turn off the UV light, and raise the sash. Spray the BSC surface and prepare the filter supplies (5 mL luer-lock syringes and 0.2 µm syringe filters). Prepare multiple filters to efficiently strain sufficient volumes of gelatin to apply to plungers.
When the gelatin solution reaches a homogenous consistency, filter the solution and apply it to the plungers. Load the syringe with gelatin. Apply the gelatin to the plastic plungers through the syringe filter (~2.5 mL for a 6-well plate, ~4.5 mL for a 6 cm dish) and allow to solidify (~20 min).
Once the gelatin is solid, unwrap the tape from the edge of the plungers. With hooked forceps, remove the gelatin from the outer edge of the plunger (i.e., the raised edges of the meniscus). Ensure that the remaining gelatin in the center of the plunger is completely level to maximize contact with ADSC sheet.
Once excess gelatin is removed, gently apply the gelatin plungers to the pNIPAAm-coated ADSC plates. Use the metal washers to weigh down the plunger. Do not shear cell sheets while applying the plungers.
- Leave the plungers on the cell sheets for 1.5 h at room temperature. Incubate plates/plungers in an ice water bath for 1.5 h to complete dissociation of the cell sheet from the pNIPAAm-coated plate surface.
- Exercise caution while incubating in the ice bath; do not allow the ice bath to contaminate the cell media during incubation.
- After completing incubation, clean the bottom of the pNIPAAm-coated plates to remove non-sterile water.
4. White Adipose Tissue Processing
When collecting human adipose tissue from the operating room, keep all samples in a sterile container that is on ice until SWAT is to be seeded. Add sterile maintenance media, such as phosphate-buffered saline (PBS), to adipose tissue container for tissue stability.
Add cold cell culture medium to 1.5 mL microcentrifuge tubes for each well/dish to be seeded (100 µL for a 6-well plate, 200 µL for a 6 cm dish).
- Mince the adipose tissue.
- For solid adipose tissue segments, do as follows.
- Wash large segments of adipose tissue 3x in sterile PBS and remove as much PBS as possible.
- Coarsely mince tissue with forceps and sterile razor and remove as much vasculature and fascia as possible (see Discussion for more detail).
- Finely mince fat with razor until minced tissue takes on thick, liquid consistency. NOTE: Ideally tissue will appear homogenous with no visible individual segments of WAT although this is not always possible.
- Process lipoaspirate as follows.
- Under the BSC, tape sterile gauze over the top of a beaker, and place this beaker in a larger beaker to collect any excess liquid.
- Using a 25 mL serological pipette, draw as much lipoaspirate as needed and apply it to the surface of the sterile gauze. Apply PBS directly to this surface to wash lipoaspirated fat and to remove excess blood and lipid residue.
- Use forceps to recover drained tissue, transfer it to a sterile mincing surface, and mince the lipoaspirate.
Use a sterile razor to cut off the distal end of p1000 pipette tips to transfer minced tissue; this will minimize shear stress that can lead to adipocyte lysis. Once a proper tissue consistency is reached transfer the desired volume of minced tissue to each 1.5 mL tube (300–400 µL for 6-well plate, 500–600 µL for 6 cm dish). Mix minced WAT and media briefly in the tubes.
- Take the base ADSC plates and decant/aspirate the media. Replace the media with the WAT/media mixture from each 1.5 mL tube.
- Gently remove the gelatin plungers from the pNIPAAm-coated plates and apply them to the WAT mixture on the base ADSC plates. Examine the monolayer of the pNIPAAm-coated plates under a microscope to confirm cellular detachment.
Set a heat block to approximately 37–40 °C under the BSC. With the plungers still in place, move the plates to the heat block's surface. Add 2–3 mL of warmed culture media to incubate the cells and facilitate gelatin melting.
After ~30 min, gently remove the plungers from the plate surface. Replace the lids of the base tissue culture plates and move to a cell culture incubator. Once the gelatin has completely liquefied at 37 °C, aspirate and replace cell culture media.
Maintain SWAT at 37 °C and 5% CO2 in phenol red-free M199 medium with 7 µM insulin, 30 µM dexamethasone, 1x Penicillin/Streptomycin. Maintain in approximately 2 mL media for 6-well plates and 3 mL for 6 cm dishes. Change media every 2 days.
5. SWAT Harvest
Prepare collagenase (0.5 mg/mL collagenase, 500 nM adenosine, in PBS) aliquots in 15 mL conical tubes (approximate volume of 10 mL). Freeze the tubes and store them at -20 °C.
Aspirate any culture medium from cells, wash 1x with PBS, and then aspirate PBS. Prep the collagenase aliquots by thawing them in a 37 °C water bath. NOTE: Ideally, the collagenase solution will reach 37 °C; immediately thereafter, add tissue.
- Add all tissue from the SWAT plate to the individual aliquots. Harvest SWAT using a sterile cell scrapper and transfer it to the collagenase aliquots with a cut-off p1000 pipette tip. Add tissue directly to the 15 mL conical tubes containing collagenase solution.
- Alternatively, incubate the tissue/collagenase mixture in a 50 mL conical tube; the increased surface area will further facilitate enzymatic digestion.
Place sample tubes in an incubated orbital shaker at a 45° angle. Incubate at 200 rpm, at 37 °C for 30–60 min.
Place a 250 µm mesh filter into a new 15 mL conical tube for collection. Pour the digested adipocyte solution through the filter. NOTE: This will allow all cells to pass through while filtering out fibrous tissue.
Allow flow-through to sit for 5 min at room temperature to allow for phase separation. NOTE: The adipocytes will float to the top of the solution while the adipose stromal cells (ASCs) will settle into the lower phases. Centrifugation for 5 min at 500 x g can also maximize cell separation or harvest surrounding ADSCs in the pellet.
- Using a cut-off p1000 pipette tip, transfer the adipocytes (the floating layer at top of collagenase solution) to a 1.5 mL microcentrifuge collection tube. Taking ~250 µL at a time, pipette slowly along the edge of the tube to collect the adipocytes.
- Rotate the tube slowly while collecting the adipocytes to maximize the recovery of cells adhering to the inside. Keep drawing more cells until the 1.5 mL microcentrifuge tube is full.
Remove excess liquid from the isolated adipocytes using a syringe attached to a needle (~21 G). Submerge the needle under the floating adipocyte layer. Agitate the needle briefly to dislodge any cells adhering to the needle shaft, and then wait for the dislodged adipocytes to float to the top.
Slowly draw the excess liquid, being careful to avoid unintended removal of adipocytes. Use microcentrifuge tube graduations to consistently isolate sample volumes (e.g., .0.1 mL for each sample).
Use the isolated cells for DNA/RNA extraction, glucose uptake assay, lipolysis assay, etc.
Representative Results
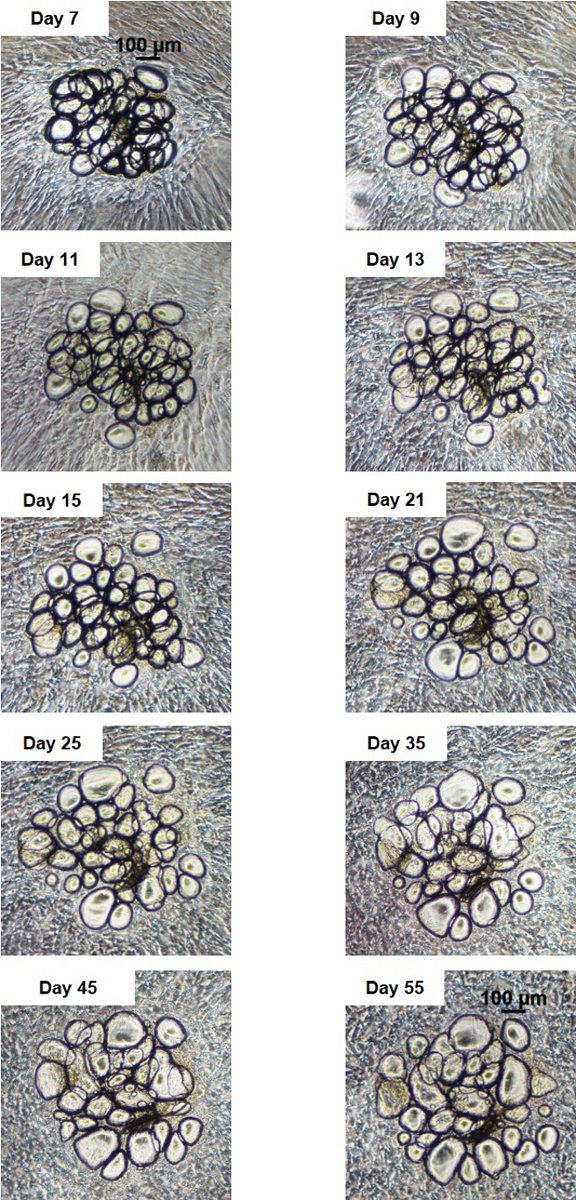
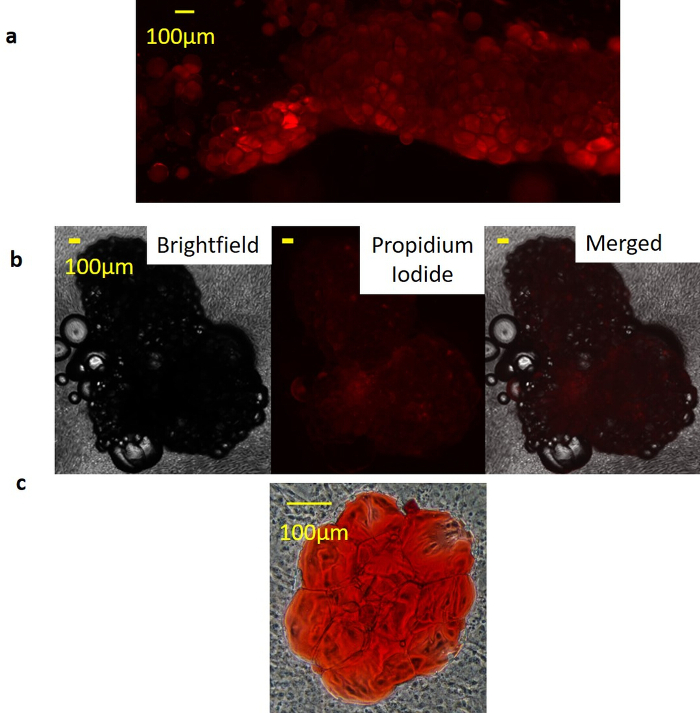
Viability of SWAT was initially assessed by serial brightfield imaging of individual WAT clusters (n = 12) over approximately 7.6 weeks. Clusters remained secured in place on the monolayer throughout this time. Slight morphological changes were observed with individual adipocytes warping slightly or shifting positions. However, adipocytes neither become multilocular over time, indicating a lack of dedifferentiation, nor did they exhibit any visible signs of cell death such as cellular blebbing, lysis, or fragmentation (Figure 2). Adipocyte identity of clusters was confirmed on two-week-old SWAT with lipid stain Nile Red (NR). Clear NR staining of unilocular adipocyte clusters indicated that these were indeed adipocytes with intact membranes, rather than artifact, cysts, or dead adipocytes. The absence of NR staining in the surrounding ADSC sheets indicates that these cells are not accumulating lipid and therefore not differentiating towards an adipogenic lineage themselves (Figure 3a). Propidium iodide staining was performed on 53 day-old SWAT. Exclusion of the stain within adipocyte clusters at advanced timepoints demonstrates a lack of cell death (Figure 3b). Oil-Red-O (ORO) stain was performed on 51 day-old SWAT with the lipid stain localizing within the fixed adipocyte clusters, indicating that SWAT adipocytes maintain viability with intact adipocytes even at advanced timepoints (Figure 3c).
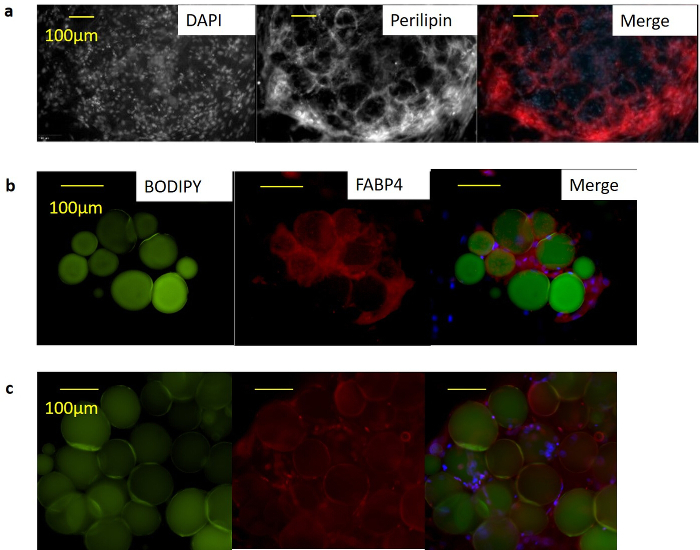
Immunocytochemistry (ICC) was performed to demonstrate expected protein expression and localization of mature adipocyte markers within SWAT. Intact SWAT plates were stained with antibodies against perilipin (ab3526, 1:200 dilution), PPARγ2 (sc-166731, 1:100 dilution), and FABP4 (ab92501, 1:1,000 dilution). Confocal images of 12 day-old SWAT demonstrated expected localization of perilipin around lipid droplet surface (Figure 4a). Fluorescence microscopy of 3 day-old SWAT with the lipid counter stain BODIPY demonstrated FABP4 localization within the cytoplasm (Figure 4b), and overlapping signal from PPARG and DAPI staining demonstrated PPARG localization to the nucleus (Figure 4c).
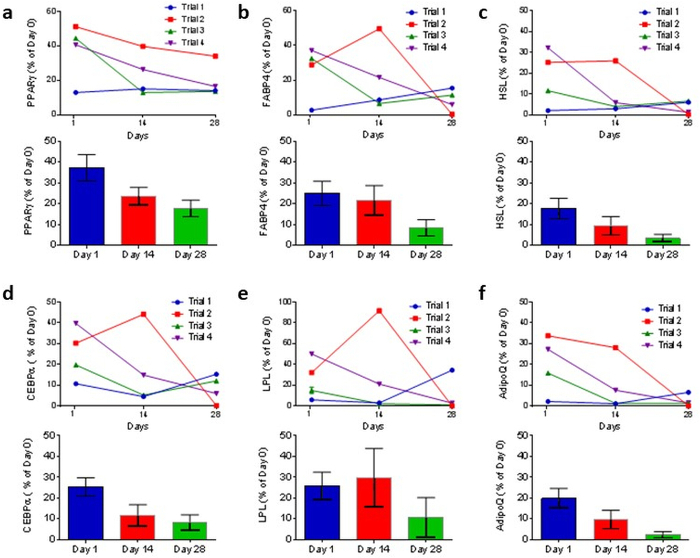
SWAT transcriptional profile was evaluated in tissue seeded from multiple subjects (n = 4 donors). Upon collection, a portion of adipose tissue was used for a baseline RNA extraction (d0), while the rest was used to seed SWAT plates. These plates were then harvested at 24 h, 14 days, and 28 days in SWAT culture. One-step quantitative reverse transcription polymerase chain reaction (RT-qPCR) was performed to determine transcription levels. Six key adipocyte genes were examined: PPARG, FABP4, LPL, CEBPA, HSL, and ADIPOQ (Figure 5). ACTB was used as an internal control and transcriptional levels were expressed as a percent of subject-matched baseline (d0) expression. All genes demonstrated robust transcription in SWAT, although levels did trend downwards over time. We believe that observed transcriptional profiles could be improved with further media optimization as we elaborate in the discussion section.
Basal endocrine function was evaluated on the same cultures used for the transcriptional profile. Supernatant was collected from SWAT plates and leptin levels were measured utilizing enzyme-linked immunosorbent assay (ELISA) (ab100581). SWAT displayed basal leptin secretion at days 1, 14, and 28 (Figure 6a). ADSC sheets were maintained in parallel with SWAT plates as a control (i.e., ADSC sheets without sandwiched WAT). Secreted leptin could not be detected in these controls (data not shown).
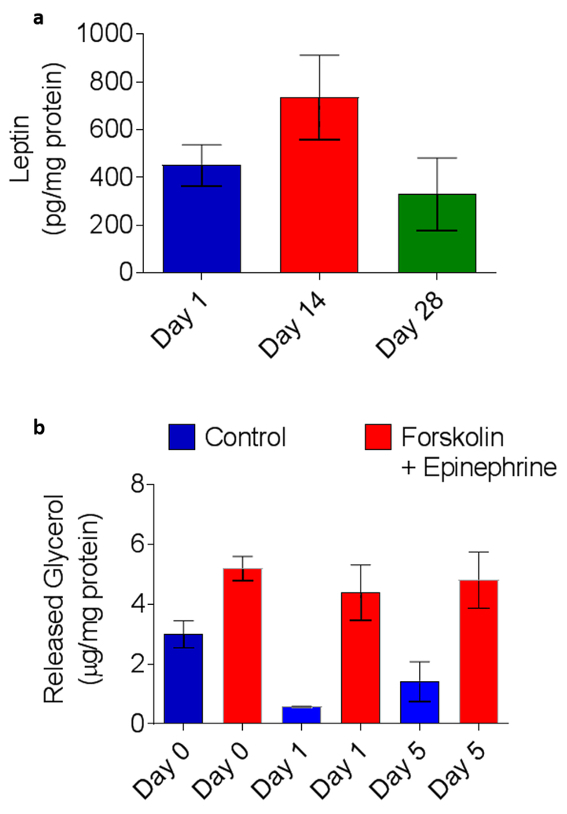
To determine whether SWAT could maintain capacity for an inducible response over time, lipolysis was examined (n = 4). To evaluate inducible lipolytic capacity, a portion of freshly collected WAT (d0) was minced and adipocytes were enzymatically isolated (similar to SWAT harvest in Protocol Section 5), while the rest was used to seed SWAT plates. Isolated adipocytes were incubated for 3 h at 37 °C in 300 µL of 2% fatty-acid-free bovine serum albumin in HBSS with either 100 µM forskolin, 1 µM epinephrine, or control buffer. Glycerol levels in collected media was measured with Free Glycerol Reagent. SWAT was then harvested at days 1 and 5 and lipolytic capacity were evaluated in the same manner. Chemical stimulation increased glycerol release significantly at all three timepoints (Figure 6b): glycerol release of treated adipocytes was increased in d0 WAT by 82 ± 46% (p = 0.01), 1-day SWAT by 577 ± 387% (p = 0.02), and five-day SWAT by 348 ± 343% (p = 0.03). Mean glycerol levels in stimulated adipocytes were very similar across all three timepoints: primary WAT 5.2 ± 0.8, 1-day SWAT 4.4 ± 1.9 and 5-day SWAT 4.8 ± 1.8 µg glycerol/mg total protein. Mean glycerol levels in control (i.e., basal lipolysis) were relatively high in freshly collected WAT (d0) as compared to SWAT-harvested cells. This may be due to the increased stress to WAT tissue at d0 during surgical excision, transport to lab, and mincing. However, these experiments demonstrate no diminishment in lipolytic capacity in SWAT at either 1 or 5 days as compared to freshly collected WAT.
SWAT was demonstrated to be capable of transplantation and recovery in a mouse model (n = 4) as a demonstration of complex, whole tissue function. It is also a possible application of the platform should a researcher wish to utilize SWAT to manipulate WAT in vitro before transplanting tissue into an animal model. SWAT was cultured for 10 days and harvested utilizing the standard protocol; isolated adipocytes from SWAT plates were then loaded into a 1 mL syringe (without a needle). An incision was made under the skin on the dorsal side of an immunocompromised eGFP-expressing mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl Tg(CAG-EGFP)1Osb/SzJ). This created a small dorsal subcutaneous pocket into which the SWAT-cultured adipocytes were injected with the syringe, and the pocket was then sutured closed. After 10 days post-transplantation, mice were sacrificed. Transplants were clearly visible and readily recovered (Figure 7a). Both the recovered transplant and a mouse contralateral fat depot were fixed, paraffin embedded, and sectioned. Immunohistochemistry was performed on the sectioned slides with primary antibodies against GFP (A10262, 1:100 dilution), and human PLIN (sc-390169, 1:100 dilution). The selected anti-PLIN antibody was experimentally validated to stain human PLIN but not mouse. Adipocytes from the recovered transplant were approximately 50–120 µm in size, which is the expected range for human adipocytes, and were completely negative for GFP (Figure 7b). However, approximately 31% of the adipocytes were positive for human PLIN, indicating successful engraftment into the host. The adipocytes in the mouse fat depots were sized 20–40 µm, which is consistent with mouse adipocytes, while staining completely positive for GFP and completely negative for human PLIN (Figure 7c). ADSC sheets (with no sandwiched WAT) were scraped from cell culture dishes and transplanted into mice as control. However, these did not yield any recoverable tissue (data not shown). This indicated that the recovered adipocytes were from the mature cultured SWAT, and not the result of differentiated human ADSCs.
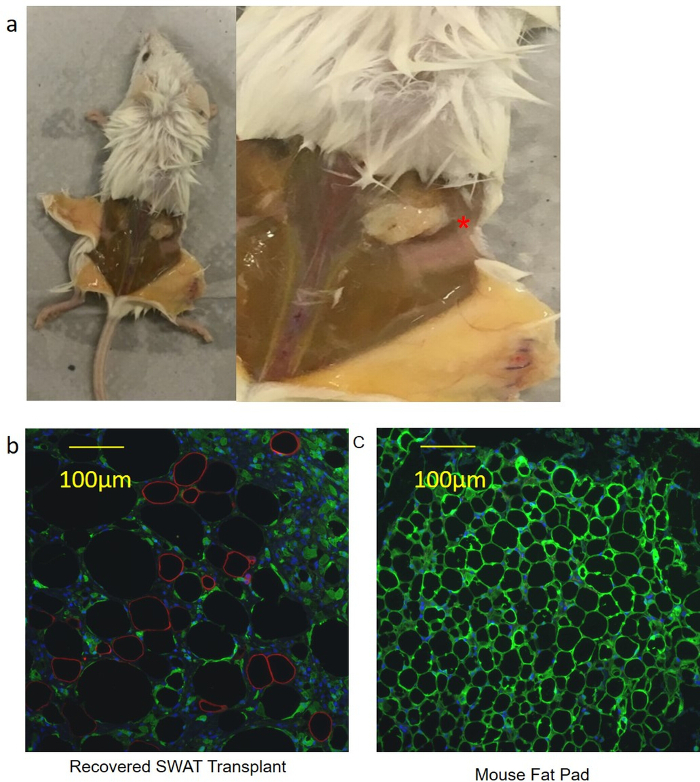
Figure 1: The SWAT method. (a) Schematic of SWAT showing the transfer of a sheet of eGFP-labeled ASCs from a pNIPAAm-coated tissue culture dish onto a second, unlabeled sheet of ASCs grown on a standard tissue culture dish. Minced, primary human WAT is sandwiched between the 2 ASCs sheets. (b) Fluorescence microscopy demonstrating Brightfield, GFP, and merged channels of a SWAT created by the technique described in (a). Scale bar = 100 µm. (c) Digital photograph of a representative, 5 day-old SWAT cultured in a standard 6-well plate. The large volume of WAT is stably secured to the bottom of the well. Figure is modified from Lau et al.11 Please click here to view a larger version of this figure.
Figure 2: SWAT remains viable at 8 weeks in culture. Serial brightfield imaging of a single SWAT cluster over 55 days demonstrates no morphologic changes consistent with cell death. Scale bars = 100 µm. Figure is modified from Lau et al.11 Please click here to view a larger version of this figure.
Figure 3: SWAT remains viable at greater than 50 days. (a) Nile Red staining demonstrates staining restriction to SWAT cultured for 15 days, indicating live viable adipocytes. Surrounding stromal cells did not stain. Figure are at 40X magnification, and Scale bar = 100 µm. (b) Propidium iodide (PI) staining of SWAT cultured for 53 days reveals complete PI exclusion, indicating no adipocyte death; Scale bar = 100 µm. (c) Oil-Red-O staining of a 51 day-old SWAT demonstrates restriction of neutral lipids to the adipocytes, indicating long-term stability of adipocyte cell membranes. Figure is modified from Lau et al.11 Please click here to view a larger version of this figure.
Figure 4: Immunocytochemistry demonstrates that SWAT stains positively for mature adipocyte cell markers with expected localization. (a) Confocal microscopy of 2 week-old SWAT revealed human PLIN+ unilocular cells with localization to lipid droplet surface. Scale bar = 100 µm. (b) Fluorescent microscopy of 3 day-old SWAT revealed human FABP4+ cells with localization to the cytoplasm and (c) PPARγ+ cells with localization to cell nucleus. Scale bar = 100 µm. Figure is modified from Lau et al.11 Please click here to view a larger version of this figure.
Figure 5: SWAT maintains key adipocyte identity gene expression. SWAT clusters were collagenase digested after 1, 14, and 28 days in culture and gene expression determined by RT-qPCR. RT-qPCR demonstrates that 1-day-old and 14-day-old SWAT preserve high levels of (a) PPARG, (b) FABP, (c) HSL, (d) CbEBPA, (e) LPL and (f) ADIPOQ. ACTB was used as reference gene. Error bars = standard error. Figure is modified from Lau et al.11. Please click here to view a larger version of this figure.
Figure 6: SWAT basally secretes adipokines and responds physiologically to catecholamine stimulation. (a) ELISA quantification of 24 h basal leptin secretion shows no significant change after 28 days in culture. (b) Adrenergic stimulation with epinephrine and forskolin generates a significant glycolytic response in freshly harvested primary WAT (1.7x) and subject-matched SWAT at 1 and 5 days in culture. Day 1 SWAT responded to catecholamine stimulus with a 6.3-fold increase over basal glycolysis while Day 5 SWAT demonstrated a 3.3-fold increase over basal glycolysis. Stimulated glycolysis was not significantly different in SWAT compared to matched fresh WAT (p = 0.53, n = 4). Error bars = standard error. Figure is modified Lau et al.11. Please click here to view a larger version of this figure.
Figure 7: SWAT can be reimplanted in vivo, demonstrating preserved whole tissue function. (a) SWAT was readily visualized 10 days after transplantation into an immunocompromised mouse (asterisk). Had SWAT not successfully implanted, by 10 days the necrotic SWAT would have been liquefied. (b) Sections of recovered SWAT demonstrated large, unilocular adipocytes (diameters 50 to 120 µm) that were eGFP-. 31.3% of the adipocytes were human PLIN+. The human PLIN+ adipocytes tended to be smaller, which would fit with clinical observations of fat grafting in humans where smaller adipocytes are more likely to survive the transfer. Scale bar = 100 µm, 200X magnification. (c) Contralateral fat pads taken from the same mice demonstrated much smaller adipocytes (20–40 µm) and were human PLIN-. SCale bar = 100 µm, 200X magnification. Figure is modified from Lau et al.11. Please click here to view a larger version of this figure.
Discussion
This protocol details the use of ADSCs to sandwich human white adipose tissue; human ADSC cell lines can be isolated via well-established protocols15. However, the system can be adapted for individualized research requirements (such as using 3T3L-1 cells to sandwich mouse WAT). This process involves handling primary human tissue. Standard safety precautions should be employed; handle human tissues as BSL-2 pathogens (e.g., HIV, HepC). Only handle tissue directly under a BSC. Wear all appropriate PPE as dictated by institutional safety standards — double gloves are highly recommended. Properly disinfect all supplies and wastes with 10% bleach solution or 70% ethanol solution before re-use or disposal.
The first critical step in SWAT culture is procuring and maintaining pNIPAAm-coated tissue culture plates. Such plates are available commercially, or they can be produced through established methods12,13,14. These plates are temperature-responsive cell culture surfaces. Above the critical temperature (~32 °C), the surface is hydrophobic and cells will adhere, similar to other treated plastics for tissue culture. However, below this critical temperature, the surface becomes hydrophilic and the cells will dissociate from the plastic surface16. As a result, care should be taken to establish: 1) how quickly a given cell type will dissociate from the monolayer before reaching full confluency (i.e., during normal media changes) and 2) the necessary time for lowering the temperature with the gelatin plunger applied to the pNIPPAm-coated plate, to ensure that the entire cell sheet dissociates. Media changes should be performed with media warmed to 37 °C; if cells are prone to quick dissociation, perform media changes on a heat block set to 37 °C under the BSC.
The second critical step is processing the adipose tissue to prepare for SWAT seeding. We performed our characterization by mincing WAT with sterilized forceps and sterilized razors. However, human fascia cannot be cut easily with a razor. Maximizing the adipocyte-to-fascia ratio is important to retain as much adipose tissue within SWAT as possible over a long-term period. Large pieces of intact fascia will prevent cell-cell contact between the ADSCs and adipocytes, which is essential to keeping SWAT together. Excessive amounts of fascia can also have a "pulling on a thread" effect. Whereas a single WAT cluster can dissociate from a plate without affecting the neighboring clusters, multiple clusters connected by fascia can pull enough clusters off a monolayer to render a SWAT well unusable. Liposuction, which involves the mechanized mincing of WAT, can circumvent this issue as fascia is finely minced during surgery. However, during liposuction, patients are routinely treated with high doses of epinephrine; the possible consequences of this treatment on the tissue (and subsequent experiments) must be accounted for during a study.
The final critical step in SWAT culture is selecting an experimental culture media. We performed our initial characterization experiments utilizing a standard cell culture formulation (10% FBS, 1x Penicillin/Streptomycin in DMEM). However, based on available literature regarding maximizing adipocyte transcription, we used a specialized formulation (7 µM insulin, 30 µM dexamethasone, 1x Penicillin/Streptomycin in M199 media)17. This formulation improved the early levels of transcription, but levels decreased over time. We believe this is because in the body, adipocytes are exposed to a wide variety of chemical signals, often cycling between fed and starved states over the course of a day. Thus, it should be possible to improve the observed transcriptional profile over time by further optimizing the supplements to our cell culture media.
SWAT harvest (Protocol, Section 5) isolates adipocytes from the culture, reduces the sample volume, and removes interference from the surrounding ADSC sheets. This can precede adipocyte homogenization (which is necessary for protein or nucleic acid isolation), or functional assays of intact adipocytes (such as glycolysis or glucose uptake assays). Alternatively, the intact SWAT plate can be harvested for immunocytochemical staining, which allows the ADSC sheets to secure the WAT clusters to the monolayer throughout the staining process. In this case, aspirate the culture media and fix the entire culture via standard protocols. However, we recommend adding a glass coverslip before imaging to flatten the 3D adipocyte clusters and improve the resulting image quality.
We believe SWAT has major potential as a drug screening platform. The technique is simple, reproducible, and utilizes conventional tissue culture plates. Therefore, drug screening experiments are executable by adding a pharmacologically active compound to the culture medium. Adipocytes can then be harvested to examine the effect of the compound on gene expression, epigenetic profile, insulin sensitivity, etc. The addition of environmental toxins to culture medium can similarly be used to examine their effects on adipocytes. Because SWAT allows the researcher to track individual adipocyte clusters over time, it is uniquely suited to evaluate a treatment that makes a visible phenotypic change to adipocytes. A prominent example would be human adipocyte "browning," wherein a white, lipid-storing adipocyte becomes "browned" or similar to a thermogenic, multilocular brown adipocyte18. This is a phenomenon of potentially major clinical relevance that has been demonstrated in mouse models, but never in a human. Co-cultured variations on SWAT also hold tremendous research potential. SWAT is a natural co-culture system that utilizes standard cell culture wells. Therefore, one can maintain the adipocytes among other clinically relevant cell types by changing the sandwiching cells or by utilizing tissue culture membrane inserts. For example, hepatocytes could function as the upper sandwiching cell sheet to screen a drug that is filtered through the liver before reaching the adipose tissue. Likewise, cancer cells could be grown on a membrane insert over SWAT to examine how adipocytes contribute to cancer pathology.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors would like to acknowledge the institutional support provided by LSU Health Sciences Center, which funded the project.
References
- Cawley J, Meyerhoefer C. The medical costs of obesity: An instrumental variables approach. Journal of Health Economics. 2012;31(1):219–230. doi: 10.1016/j.jhealeco.2011.10.003. [DOI] [PubMed] [Google Scholar]
- Pi-Sunyer X. The Medical Risks of Obesity. Postgraduate Medicine. 2009;121(6):21–33. doi: 10.3810/pgm.2009.11.2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith U. Morphologic studies of human subcutaneous adipose tissue in vitro. Anatomical Record. 1971;169(1):97–104. doi: 10.1002/ar.1091690109. [DOI] [PubMed] [Google Scholar]
- Sugihara H, Yonemitsu N, Miyabara S, Yun K. Primary cultures of unilocular fat cells: characteristics of growth in vitro and changes in differentiation properties. Differentiation. 1986;31(1):42–49. doi: 10.1111/j.1432-0436.1986.tb00381.x. [DOI] [PubMed] [Google Scholar]
- Sugihara H, Yonemitsu N, Toda S, Miyabara S, Funatsumaru S, Matsumoto T. Unilocular fat cells in three-dimensional collagen gel matrix culture. The Journal of Lipid Research. 1988;29:691–697. [PubMed] [Google Scholar]
- Hazen SA, Rowe WA, Lynch CJ. Monolayer cell culture of freshly isolated adipocytes using extracellular basement membrane components. The Journal of Lipid Research. 1995;36:868–875. [PubMed] [Google Scholar]
- Fried SK, Kral JG. Sex differences in regional distribution of fat cell size and lipoprotein lipase activity in morbidly obese patients. International Journal of Obesity. 1987;11(2):129–140. [PubMed] [Google Scholar]
- Sutherland ML, Fabre KM, Tagle DA. The National Institutes of Health. Microphysiological Systems Program focuses on a critical challenge in the drug discovery pipeline. Stem Cell Research and Therapy. 2013;4 doi: 10.1186/scrt361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikswo JP. The relevance and potential roles of microphysiological systems in biology and medicine. Experimental Biology and Medicine. 2014;239(9):1061–1072. doi: 10.1177/1535370214542068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Institutes of Health. NIH-CASIS Coordinated Microphysiological Systems Program for Translational Research in Space (#RFA-TR-18-001) 2017. Available from: https://grants.nih.gov/grants/guide/rfa-files/RFA-TR-18-001.html.
- Lau FH, Vogel K, Luckett JP, Hunt M, Meyer A, Rogers CL, Tessler O, Dupin CL, St Hilaire H, Islam KN, Frazier T, Gimble JM, Scahill S. Sandwiched White Adipose Tissue: A Microphysiological System of Primary Human Adipose Tissue. Tissue Engineering Part C: Methods. 2018;24(3) doi: 10.1089/ten.TEC.2017.0339. [DOI] [PubMed] [Google Scholar]
- Ebara M, Hoffman JM, Stayton PS, Hoffman AS. Surface modification of microfluidic channels by UV-mediated graft polymerization of non-fouling and 'smart' polymers. Radiation Physics and Chemistry. 2007;76:1409–1413. [Google Scholar]
- Lin JB, Isenberg BC, Shen Y, Schorsch K, Sazonova OV, Wong JY. Thermo-responsive poly(N-isopropylacrylamide) grafted onto microtextured poly(dimethylsiloxane) for aligned cell sheet engineering. Colloids and Surfaces B: Biointerfaces. 2012;99:108–115. doi: 10.1016/j.colsurfb.2011.10.040. [DOI] [PubMed] [Google Scholar]
- Turner WS, Sandhu N, McCloskey KE. Tissue Engineering: Construction of a Multicellular 3D Scaffold for the Delivery of Layered Cell Sheets. Journal of Visualized Experiments. 2014. p. e51044. [DOI] [PMC free article] [PubMed]
- Bunnell B, Flaat M, Gagliardi C, Patel B, Ripoll C. Adipose-derived stem cells: Isolation, expansion and differentiation. Methods. 2008;45(2):115–120. doi: 10.1016/j.ymeth.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama Y, Kikuchi A, Yamato M, Okano T. Ultrathin poly(N-isopropylacrylamide) grafted layer on polystyrene surfaces for cell adhesion/detachment control. Langmuir. 2004;20(13):5506–5511. doi: 10.1021/la036139f. [DOI] [PubMed] [Google Scholar]
- Fried SK, Russell CD, Grauso NL, Brolin RE. Lipoprotein lipase regulation by insulin and glucocorticoid in subcutaneous and omental adipose tissues of obese women and men. Journal of Clinical Investigation. 1993;92(5):2191–2198. doi: 10.1172/JCI116821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keipert S, Jastroch M. Brite/beige fat and UCP1 - is it thermogenesis? Biochimica et Biophysica Acta. 2014;1837(7):1075–1082. doi: 10.1016/j.bbabio.2014.02.008. [DOI] [PubMed] [Google Scholar]