

Abstract
The efficacy of CAR-T cell therapy against poorly responding tumors has been enhanced by administering the cells in combination with immune checkpoint blockade inhibitors. Alternatively, the CAR construct has been engineered to co-express factors that boost CAR-T cell function in the tumor microenvironment. Here we modified CAR-T cells to secrete PD-1-blocking single-chain variable fragments (scFv). These scFv-secreting CAR-T cells work in both a paracrine and autocrine manner to improve the anti-tumor activity of CAR-T cells and bystander tumor-specific T cells in clinically relevant syngeneic and xenogeneic mouse models of PD-L1+ hematologic and solid tumors. Efficacy was similar or better to that achieved by combination therapy with CAR-T cells and a checkpoint inhibitor. This approach could improve safety as the secreted scFv remained localized to the tumor, protecting CAR-T cells from PD-1 inhibition, which could potentially avoid toxicities associated with systemic checkpoint inhibition.
T cells can be directed to target tumor cells through expression of a chimeric antigen receptor (CAR). CARs are synthetic receptors consisting of an extracellular antigen recognition domain, which are most commonly a single chain variable fragment (scFv) but can also take the form of any antigen-binding peptide. This binding domain is then linked, with or without a hinge domain, to intracellular T cell activation and costimulation domains. Although CAR-T cell therapy has shown remarkable results in patients with B-cell acute lymphoblastic leukemia (B-ALL) 1 , its efficacy in treating other hematological and solid tumors has been less impressive 1.
These modest responses may relate to the tumor microenvironment (TME). When infused into patients, CAR-T cells often encounter an inhibitory TME with cells and inhibitory ligands that can bind to inhibitory receptors on T cells and hinder T cell anti-tumor responses. For instance, in ovarian cancer, immunosuppressive M2-polaraized tumor associated macrophages (TAM) 2 and regulatory T cells (Treg) 3,4 have been found to populate the TME, and presence of these cells correlate with reduced tumor-infiltrating lymphocytes 5 and poor outcomes in patients 2,3. Both TAM and Treg suppress infiltrating T-cells via contact and cytokine-medicated mechanisms 5,6. Furthermore, upon activation, T-cells secrete IFN-γ, an effector cytokine, which has been shown to dynamically upregulate programmed death ligand-1 (PD-L1) expression on OC cells in both clinical 7 and preclinical models 8. PD-L1 bindings to the inhibitory receptor programmed death 1 (PD-1) on T cells and suppresses T cell function 9. Interruption of PD-1/PD-L1 ligation via CRISPR-mediated deletion of PD-L1 on OC cells significantly improved the efficacy of adoptively transferred second-generation CAR-T cells in preclinical models 8. Taken together, these factors may contribute to the lack of clinical efficacy of CAR-T cells for this solid tumor malignancy 10.
Checkpoint blockade therapy, which uses antibodies to disrupt the interaction between inhibitory receptors on T cells –particularly CTLA-4 and PD-1- and their suppressive ligands on tumors cells, has shown clinical responses in patients with a range of solid tumors 11,12,13 and hematological malignancies 14. Correlates for efficacy of checkpoint blockade therapy include T cell activation markers, tumor cell expression of PD-L1, a pre-existing CD8+ T cell infiltrate in the tumor 15,16 and tumor mutational burden 15,17,18,19,20. Together, these studies suggest that tumor-specific T cells are an integral mechanism of action of checkpoint blockade and that re-engagement of pre-existing tumor-specific T cells is critical to the success of this therapeutic modality.
We previously described a strategy for “armored” CAR-T cell, which are CAR-T cells that are co-modified to express immunomodulatory ligands such as CD40L 21 or to secrete cytokines such as IL-12 22,23,24,25,8 or IL-18 26 to enhance CAR-T cell function in the tumor microenvironment. Therefore, rather than combining CAR-T cells with existing systemic checkpoint blockade antibody treatment, as studied previously in preclinical models 27,8,28, we aimed to use our armored CAR-T cell platform to create a single therapy in which CAR-T cells secrete an immune checkpoint blockade single-chain variable fragment (scFv). Given that CAR-T cells traffic to the tumor, the PD-1-blocking scFv would be delivered locally to the site of disease, thereby minimizing the toxicities associated with immune checkpoint blockade. We demonstrate that CAR-T cells that secrete a PD-1-blocking scFv enhance the survival of PD-L1+ tumor-bearing mice in syngeneic and xenogeneic mouse models through both autocrine and paracrine mechanisms. This strategy has the potential to enhance the efficacy of CAR-T cell therapy in cancers with an immune-suppressive TME.
Results
Mouse CAR-T cells can be co-modified to secrete an anti-mouse PD-1-blocking scFv.
To test our approach in an immunocompetent syngeneic mouse model, retroviral second-generation CAR constructs were generated containing binding domains recognizing CD19 or the retained portion of MUC16 (MUC16ecto) 29 and mouse CD28 and zeta T cell signaling domain. These conventional CARs are labeled 19m28mZ or 4H11m28mZ, respectively. Armored mouse CAR constructs, labeled 19m28mZ/RMP1–14 or 4H11m28mZ/RMP1–14, utilized the same backbone, binding domain, and mouse signaling domain as the second-generation mouse CAR and were additionally co-modified to include a c-myc-tagged scFv derived from variable heavy and light chains from the anti-mouse PD-1-blocking mAb, RMP1–14 30 (Fig. 1a). Primary mouse T cells were transduced to express the CAR constructs (Fig. 1b) and secrete RMP1–14 scFv (25.6 kDa) (Fig. 1c, Supplementary Fig. 1a).
Fig. 1. Mouse CAR-T cells can be co-modified to secrete mouse PD-1-blocking scFv RMP1–14.
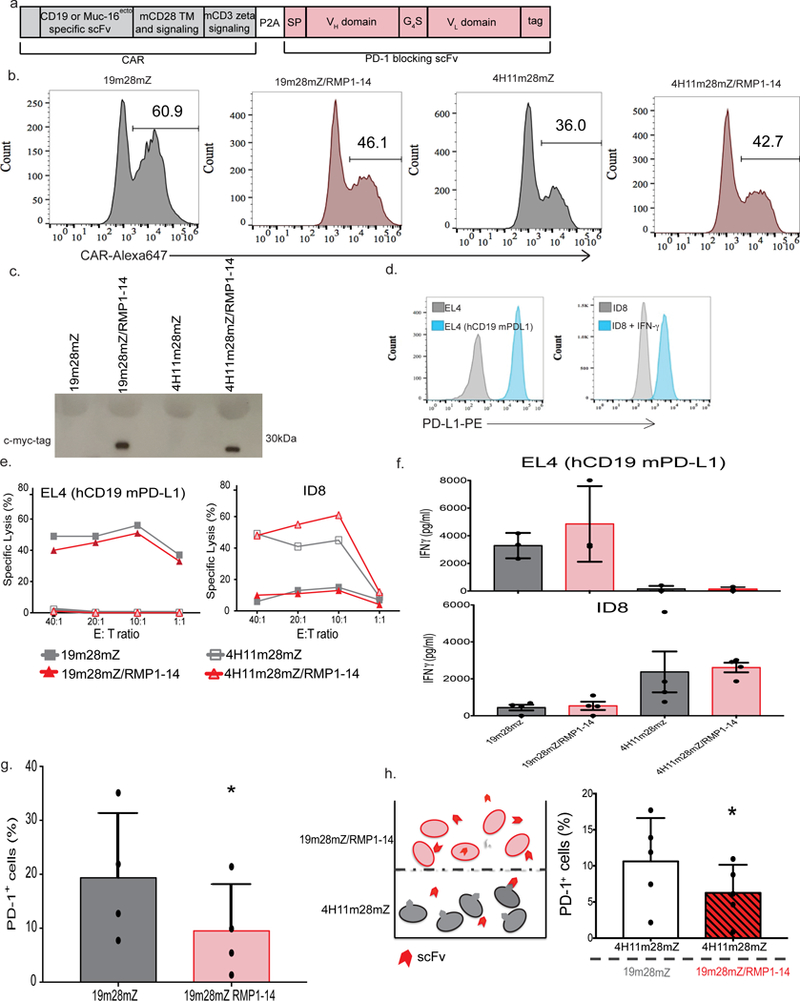
(a) Schematic of the bi-cistronic vectors utilized for syngeneic mouse studies encoding the CD19-targeted 19m28mZ CAR, or ovarian MUC16ecto-targeted 4H11m28mZ CAR, linked with a P2A element to the secretable PD-1-blocking scFv, RMP1–14. A c-myc-tag is included for detection of the scFv. (b) Representative flow cytometry plot demonstrating CAR expression following mouse T cell transduction, detected with fluorescently labeled CAR-specific idiotypic antibodies. Data shown is representative of ≥3 independent experiments. (c) Western blot on supernatant from equivalent numbers of viral packaging cells transduced to express the secretable scFv with the CAR, detected with anti-myc-tag antibody. Data shown is representative of 3 independent experiments. (d) Flow cytometry histograms depicting expression of mouse PD-L1 on EL4 (hCD19 mPD-L1) or ID8 cells. Data shown is representative of 3 independent experiments. (e) 4-hour 51Cr release assay demonstrating lysis of tumor cells. Data shown is representative of 3 independent experiments. (f) All 4 CAR constructs produce antigen-specific IFN-γ after co-culture with tumor cells. Data shown is mean +/− SEM from 4 independent experiments. (g) Quantification of PD-1 detection on CAR-T cells, as measured by flow cytometry. Data shown is mean +/− SEM from 4 independent experiments, *p=0.011 by two-tailed paired t-test. (h) Experimental schematic and quantification of decreased PD-1 detection by flow cytometry on 4H11m28mZ T cells when cultured in a transwell plate with 19m28mZ or 19m28mZ/RMP1–14 T cells. Data shown is mean +/− SEM from 5 separate donors, *p=0.012 by two-tailed paired t-test.
We co-cultured scFv-secreting CAR-T cells with human CD19-expressing mouse lymphoma EL4 cells (hCD19 mPD-L1) or MUC16ecto expressing ovarian tumor ID8 cells, which upregulate PD-L1 after exposure to IFN-γ (Fig. 1d). Conventional and RMP1–14 scFv-secreting CAR-T cells specifically lyse (Fig. 1e) and produce IFN-γ (Fig. 1f) when cultured with tumor targets.
We next evaluated binding of secreted RMP1–14 scFv to PD-1. First, we showed that the scFv binds in an autocrine manner to secreting CAR-T cells by showing that 19m28mZ/RMP1–14 T cells significantly decreased surface detection of PD-1 compared to cells modified to express the CAR alone (p=0.01) (Fig. 1g and Supplementary Fig. 1b). Next, to demonstrate binding of scFv to bystander PD-1 expressing cells, we co-cultured 4H11m28mZ T cells in the bottom well of a transwell plate with 19m28mZ or 19m28mZ/RMP1–14 cells on top. After 24-hours, we detected lower levels of PD-1 on the surface of 4H11m28mZ cells cultured with 19m28mZ/RMP1–14 cells as compared to those cultured with 19m28mZ T cells (p=0.04) (Fig. 1h), indicating binding of secreted scFv to bystander cells.
PD-1-blocking scFv secreted by CAR-T cells in vivo
We next validated the presence of CAR-T cell-secreted PD-1-blocking scFv in the TME. Tumor-bearing mice with palpable ascites were injected intraperitoneally (i.p.). with CAR-T cells (Fig. 2a). Ascites from these mice was harvested 48-hours later and RMP1–14 scFv secreted from 4H11m28mZ/RMP1–14 T cells was detected by immunoprecipitation with an anti-myc-tag antibody and western blot analysis (Fig. 2b, Supplementary Fig. 2), as well as by anti-myc-tag luminex (Fig. 2c) (p=0.0004).
Fig. 2. CAR-T cells secreting RMP1–14 scFv have enhanced anti-tumor function in syngeneic mouse tumor models.
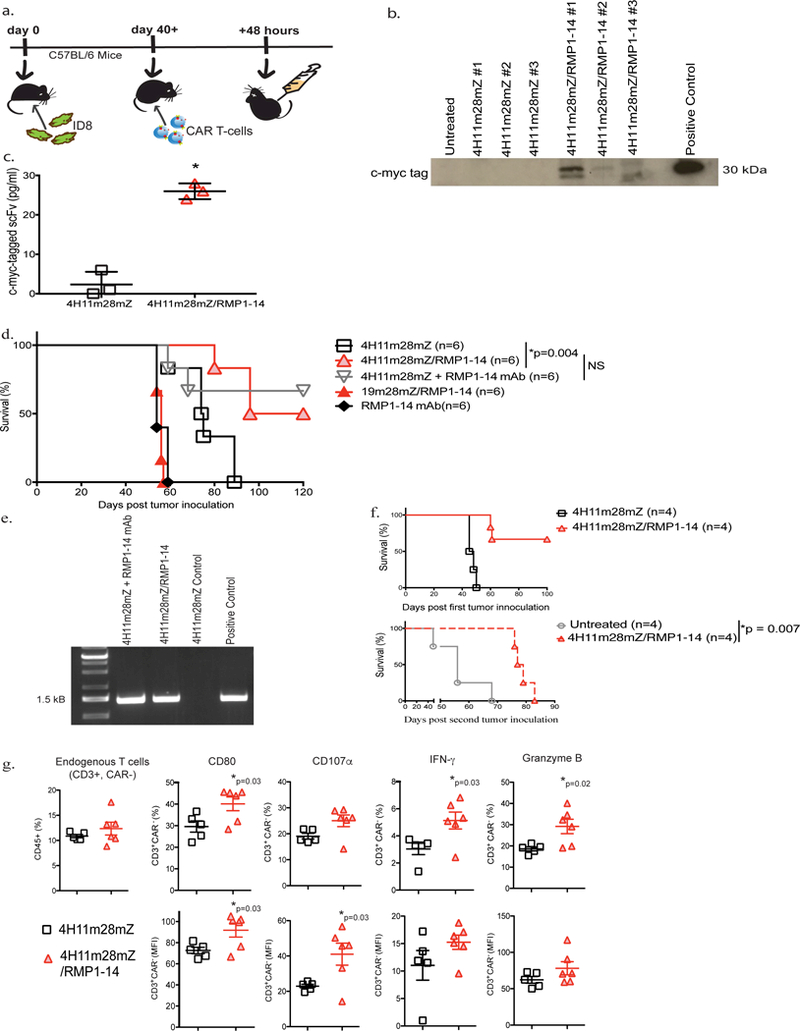
(a) Schematic diagram of experimental setup to detect scFv secretion in vivo. C57BL/6 mice were inoculated with ID8 tumor, monitored until development of ascites and subsequently treated i.p. with 4H11m28mZ or 4H11m28mZ/RMP1–14 T cells. In vivo secretion of RMP1–14 scFv was detected by harvesting ascites from tumor-bearing mice 48 hours later. The ascites was immunoprecipitated with an anti-myc-tag antibody and (b) run on a Western blot using an anti-myc tag antibody or (c) run on Luminex utilizing anti-myc-tag beads (*p<0.0004 using an two-tailed unpaired t test, Mean ± SEM of 4H11m28mZ and 4H11m28mZ/RMP1–14 are 2.3 ± 1.9 and 26 ± 1.2, respectively), Data shown is from 2 independent experiments. (d) C57BL/6 mice were injected i.p. with ID8 tumor cells and treated with CAR-T cells, 250 μg RMP1–14 mAb or a combination of both 7 days later. RMP1–14 mAb was given on days 3, 7 and 14 post-tumor inoculation (*p=0.004 by Log-rank Mantel-Cox Test, with a 95% Confidence Interval (CI) of 0.4 to 0.9). Data shown is from 2 independent experiments. (e) PCR of bone marrow from mice surviving >120 days since tumor inoculation in Fig. 2d. CAR-T cells were detected in the bone marrow of long-term surviving mice treated with 4H11m28mZ T cells + RMP1–14 mAb or 4H11m28mZ/RMP1–14 T cells. Data shown is from 2 independent experiments. (f) C57BL/6 mice were inoculated with ID8 tumor and treated 7 days later with 4H11m28mZ or 4H11m28mZ/RMP1–14 T cells. Long term surviving mice in the 4H11m28mZ/RMP1–14 T cell cohort were re-challenged with a second inoculation of ID8 cells and compared to naïve untreated ID8-innoculated mice (*p=0.007 by Log-rank Mantel-Cox Test, with a 95% Confidence Interval of 0.2 to 0.3). (g) Quantification of flow cytometric analysis demonstrating endogenous, CAR- T cells extracted from C57BL/6 mice bearing B16-F10 mouse melanoma and treated with PD-1-blocking scFv CAR-T cells have enhanced activation and cytokines levels as compared to mice treated with second-generation CAR-T cells (*p values indicated on figure, by two-tailed unpaired t test). Data shown is pooled from 6 mice and 2 independent experiments.
We next tested the efficacy of scFv-secreting CAR-T cells in a syngeneic, immune-competent mouse model of metastatic ovarian carcinoma. C57BL/6 mice were injected i.p with ID8 cells and treated 7 days later with CAR-T cells. 4H11m28mZ/RMP1–14 T cells enhanced survival as compared to mice treated with 4H11m28mZ (p=0.004) and 19m28mZ/RMP1–14 T cell controls (p=0.0006). Treatment with 4H11m28mZ/RMP1–14 T cells or 4H11m28mZ T cells + RMP1–14 antibody demonstrated comparable survival benefit (Fig. 2d) (p=0.051). Tumor-bearing mice treated with RMP1–14 mAb alone had no survival benefit over mice treated with control CAR-T cells.
PD-1 binding to PD-L1 can result in T cell exhaustion, anergy, and/or apoptosis. We found that long-term surviving mice treated with 4H11m28mZ/RMP1–14 T cells or 4H11m28mZ T cells + RMP1–14 antibody had detectable CAR-T cells by PCR in the bone marrow over 120 days post-tumor inoculation (Fig. 2e). In addition, 4H11m18mz/RMP1–14 treated mice were able to mount an anti-tumor response when re-challenged with the initial dose of ID8 tumor cells (Fig. 2f) (p<0.0001), as compared to naïve untreated mice.
We hypothesized that CAR-T cells secreting PD-1-blocking scFv could re-invigorate endogenous tumor-specific T cells. To test this, C57BL/6 mice were injected subcutaneously (s.c.) with the immunogenic melanoma cell line B16-F10. These mice mount an endogenous response to the tumor but cannot eradicate it 30. Endogenous bystander tumor infiltrating lymphocytes (TILs) isolated from mice treated intratumorally with 4H11m28mZ/RMP1–14 CAR-T cells expressed significantly higher levels of CD80, CD107α, IFN-γ, and Granzyme B than those treated with 4H11m28mZ (Fig. 2g and Supplementary Fig. 3) (p<0.05).
Selection of human PD-1-blocking scFv E27
The human PD-1-blocking scFvs E23, E26 and E27 were isolated from a human scFv phage display library (Eureka Therapeutics). The dissociation constant (KD) for the 3 lead candidates E23, E26 and E27 binding to PD-1 was 8.3, 6.3 and 3.6 nM, respectively. The E23, E26 and E27 antibodies were able to block PD-1 from binding to PD-L1 in a dose-dependent manner (Fig. 3a). We collected supernatant from equivalent numbers of transduced viral producer cells and detected anti-PD1 secretable scFvs (Fig. 3b). The most intense band was observed for clone E27, indicating that the scFv was the most stable in supernatant. For this reason, E27 was used in subsequent studies (Fig. 3c and Supplementary Fig. 4a).
Fig. 3. Human CAR-T cells can be co-modified to secrete a novel PD-1-blocking scFv, E27.
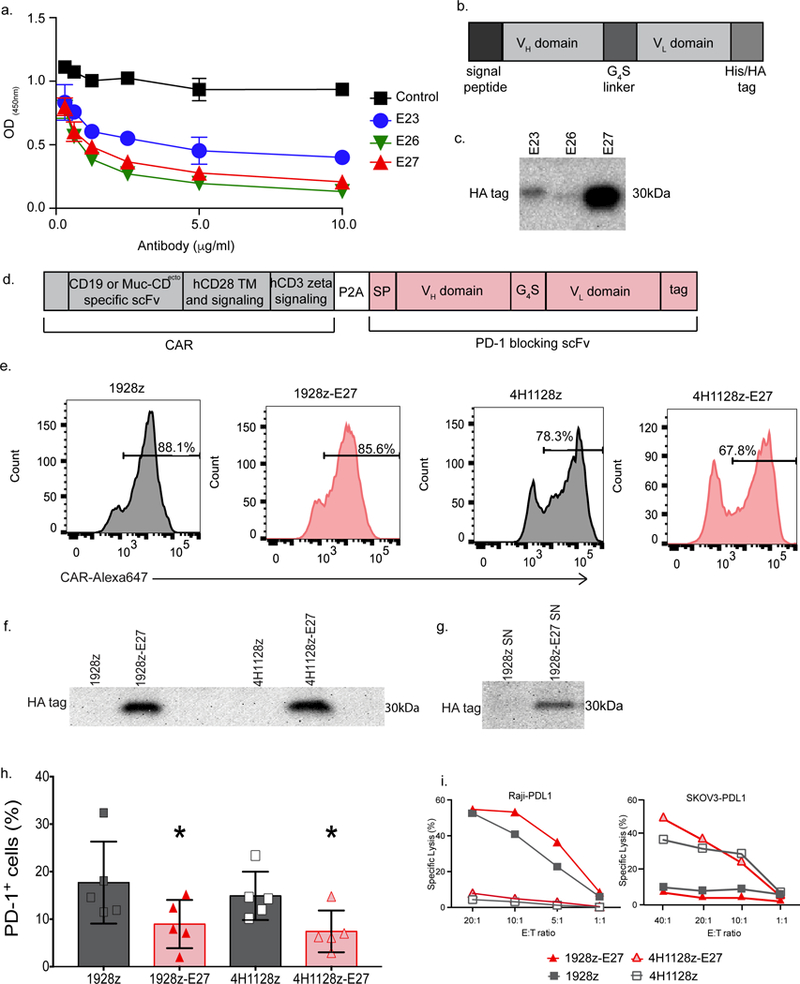
(a) Novel human PD-1-blocking mAb candidates E27, E26 and E23 were utilized in a competitive binding assay to detect interruption of PD-1 binding to PD-L1 at varying concentrations, compared to a human IgG1 isotype control mAb (control). Data shown is mean of three independent experiments. (b) Schematic representation of PD-1-blocking scFv designed from the E23, E26 and E27 mAbs used in (c), where the signal peptide was linked to the variable heavy sequence, serine glycine linker and the variable light chain sequence. The His/HA tag was included for detection of the scFv. (c) Western blot on supernatant from equivalent numbers of 293-Glv9 packaging cells transduced to secrete scFvs with the 1928z CAR, stained with anti-HA antibody. Data shown is representative of 2 independent experiments. (d) Schematic of the bi-cistronic vector encoding the CD19-targeted 1928z CAR, or ovarian MUC16ecto-targeted 4H1128z CAR, linked with a P2A element to the secretable anti-human PD-1-blocking scFv, E27. (e) Representative flow cytometry plot demonstrating CAR expression following donor human T cell transduction, detected with fluorescently labeled CAR-specific idiotypic antibodies. Data shown is representative of ≥3 independent experiments. (f) Western blot on supernatant from CAR-T cells stained with anti-HA mAb, demonstrating a ~30 kDa protein in the 1928z-E27 and 4H1128z-E27 T cells. Data shown is representative of 2 independent experiments. (g) Western blot analysis of 293Glv9-PD-1+ cells incubated in supernatant from 1928z and 1928z-E27 T cells, stained with anti-HA mAb, showing a ~30 kDa protein in the PD-1+ cells incubated with supernatant from 1928z-E27. Data shown is representative of 2 independent experiments. (h) Quantification of PD-1 detection by flow cytometry on 1928z-E27 and 4H1128z-E27 T cell, as compared to second-generation CAR-T cells. Data shown is mean +/− SEM from 5 independent donors. For comparison of 1928z to 1928z-E27 *p=0.05, and 4h1128z to 4H1128z-E27 *p=0.006, both by a two-tailed paired t test. (i) 4-hour 51Cr release assay demonstrating that all 4 CAR constructs have antigen-dependent lysis of tumor cells. Data shown is representative of 3 independent donors and experiments.
Human CAR-T cells modified to secrete an anti-human PD-1-blocking scFv
We generated human CAR constructs encoding the CD19 or MUC16ecto-targeted CAR and the his-tagged E27 scFv (Fig. 3d, 1928z-E27 and 4H1128z-E27, respectively). Transduced T cells expressed CAR on the surface (Fig. 3e) and secreted E27 scFv (Fig. 3f and Supplementary Fig. 4b). Binding of E27 to PD-1 was demonstrated by His-tag detection on 293Glv9-PD1+ cells after incubation in supernatant from 1928z-E27 T cells (Fig. 3g and Supplementary Fig. 4c). Furthermore, 1928z-E27 and 4H1128z-E27 T cells had decreased detection of PD-1 compared to cells modified to express the CAR alone, indicating binding of E27 scFv to T cell PD-1 (Fig. 3h and Supplementary Fig. 4d). We corroborated antigen-dependent lysis by CAR-T cells in the context of tumor cells (Fig. 3i).
We transduced Raji and NALM6 tumor cells to express PD-L1. 1928z and 1928z-E27 T cells were co-cultured with Raji-PD-L1 or NALM6-PDL1 tumor at 1:1 tumor:CAR-T cell ratio. After 72-hours of co-culture, flow cytometry was used to detect remaining tumor cells. 1928z-E27 T cells lysed more Raji-PDL1 and NALM6-PDL1 tumor cells compared to 1928z T cells. (Fig. 4a and 4b). Using this assay, we found that 1928z-E27 T-cells had increased expansion following co-culture with Raji-PDL1 or NALM-PDL1 tumor, compared to 1928z T cells (Fig. 4c), consistent with 1928z-E27 T cells response to PD-L1 mediated suppression. Following co-culture with Raji-PD-L1, 1928z-E27 cells had lower levels of surface PD-1 detection compared to 1928z T cells by both percent positive cells and the mean fluorescence intensity of the staining (Fig. 4d and 4e).
Fig. 4. Co-expression of CAR and E27 scFv protects proliferative and lytic capacity of T cells in the context of PD-L1+ tumor cells.
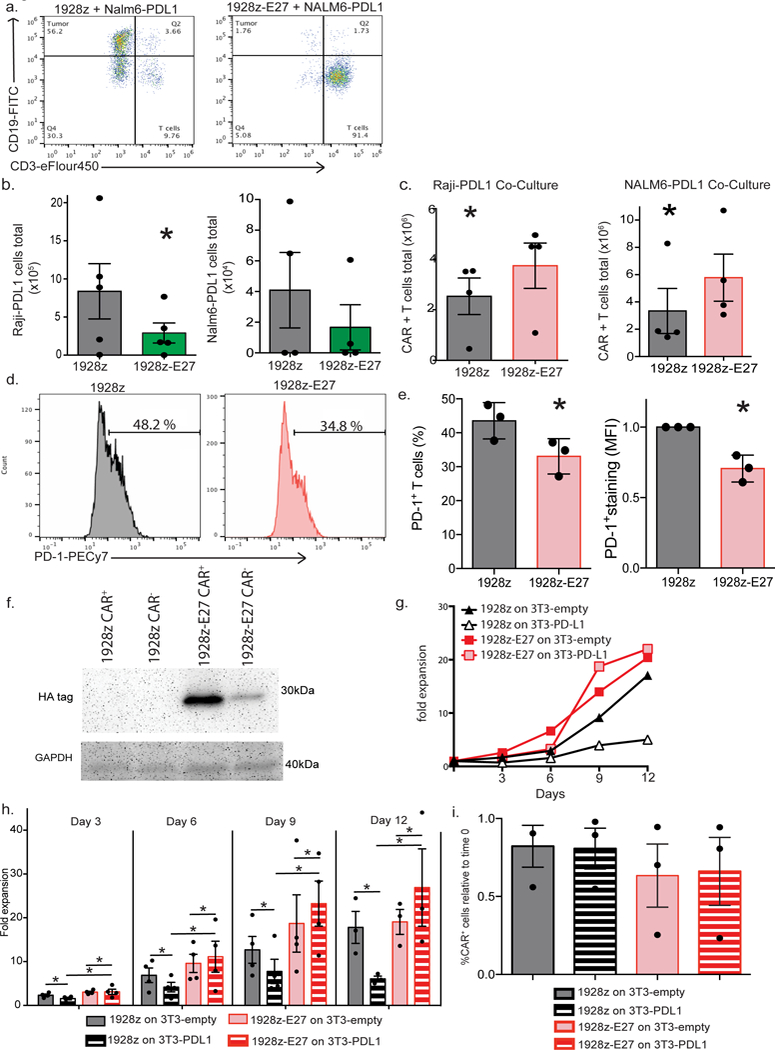
(a) Representative flow cytometry dot plots demonstrating lysis of Raji-PDL1 tumor cells, as determined by flow cytometry following 72 hour co-culture. Data shown is representative of 3 independent donors and experiments. (b). 1928z-E27 T cells lyse significantly more Raji-PDL1 tumor cells compared to 1928z T cells, data shown the mean +/− SEM from 5 independent experiments, *p=0.03 by a one-tailed paired t test. (c) CAR-T cells expansion numbers following co-culture with Raji-PDL1 or NALM6-PDL1 tumor cells as determined by flow cytometry, data shown is the average total number of T cells +/− SEM from 4 independent experiments, *p=0.05 for Raji experiment and *p=0.02 for Nalm6 experiment, both by a two-tailed paired t test. Representative flow cytometry plot (d) and quantification (e) showing increased PD-1 detection on 1928z T-cells compared to 1928z-E27 T-cells following 7 days co-culture with Raji-PDL1 tumor cells. Data shown in the mean +/− SEM from 3 independent experiments. *p=0.03 for percent positive CAR-T cell and MFI of staining, both by two-tailed paired test. (f) 1928z and 1928z-E27 T-cells were co-cultured with human T cells transduced to overexpress PD-1 and after 4 days stimulation with CD3/CD28 beads, the cells were sorted by flow cytometry to separate CAR+ and CAR- cells. Western blot was performed on the sorted populations and probed with anti-HA mAb. Data shown is representative of 3 independent donors and experiments. (g) Representative example of 1928z and 1928z-E27 T cells fold expansion when cultured with 3T3-empty or 3T3-PDL1 cells and stimulated with CD3/CD28 beads. Data shown is representative of 3 independent donors and experiments. (h) Cells were enumerated and re-plated on new 3T3 cells on days 3, 6, 9 and 12. 1928z T cells had reduced expansion when cultured with 3T3-PDL1 cells compared to 3T3- empty cells. 1928z-E27 cells had equivalent expansion when cultured on 3T3- empty or 3T3-PDL1 cells. Data shown is the mean fold expansion +/− SEM from 4 independent experiments, *p<0.05 by two-tailed paired t test. (i) Expansion of 1928z-E27 T cells on 3T3-PDL1 cells was due to an increase in both CAR+ and CAR- cells, comparing populations on day 0 and following expansion on 3T3-PDL1 cells at day 12. Data shown is representative of 3 independent experiments.
E27 binds to bystander cells
To demonstrate that untransduced bystander cells can benefit from the E27 scFv secreted from nearby CAR-T cells, we first co-cultured human healthy donor T cells with 1928z or 1928z-E27 CAR-T cells and stimulated the cells with CD3/CD28 beads. After 4 days co-culture, we performed CAR+ and conventional T cells were sorted and western blot analysis demonstrated E27 binding to PD-1 on both CAR+ and CAR- cells in the 1928z-E27/PD-1 T cell co-culture but not the 1928z/PD-1 T-cell co-culture (Fig. 4f and Supplementary Fig. 5).
Then, we cultured 1928z and 1928z-E27 T cells with 3T3-empty or 3T3-PDL1 cells and stimulated with anti-CD3/CD28 beads. 1928z T cells expanded on 3T3-empty cells had enhanced proliferation compared to 1928z T cells cultured with 3T3-PDL1 cells (Fig. 4g and 4h). However, 1928z-E27 T cells had similar expansion when cultured with 3T3-empty and 3T3-PDL1 cells (Fig. 4h). There was no significant difference between the expansion of 1928z and 1928z-E27 T cells when cultured on 3T3-empty cells (Fig. 4h) (p=0.063).
By day 12, 1928z T cells cultured on 3T3-PDL1 cells had significantly decreased cell numbers as compared to those cultured on 3T3-empty. It is anticipated that the population of CAR+ and CAR- cells in this condition had contracted in a similar ratio and the overall CAR+ percentage remained the same relative to time 0. This is what we observe when the CAR+ cells relative to time 0 is calculated in Fig. 4i. However, since the 1928z-E27 cells cultured on 3T3-PDL1 expanded significantly by day 12, it may be expected the total number of CAR+ cells would increase, hence leading to an increased percentage of CAR+ cells. This would be the case if the PD-1-blocking scFv gave select proliferative advantage only to the cell that was secreting it. However, we observed that the percentage of CAR+ 1928z-E27 T cells following expansion on 3T3-PDL1 cells was not increased at day 12 compared to the day 0, indicating that the E27 scFv protects the expansion of transduced T-cells as well as untransduced bystander cells in the context of PD-L1 (Fig. 4i).
CAR-T cells secreting E27 scFv have enhanced anti-tumor function in vivo
The in vivo anti-tumor efficacy of E27-secreting CAR-T cells was determined by inoculating SCID/Beige mice with Raji-PDL1 or NALM6-PDL1 tumor cells intravenously (i.v.). Infusion of 1928z-E27 T cells significantly enhanced survival of both Raji-PDL1 (Fig. 5a) (p=0.03) or NALM6-PDL1 (Fig. 5b) (p=0.01) tumor-bearing mice compared to infusion of 1928z T-cells.
Fig. 5. CAR-T cells that secrete E27 scFv have enhanced anti-tumor function in vivo.
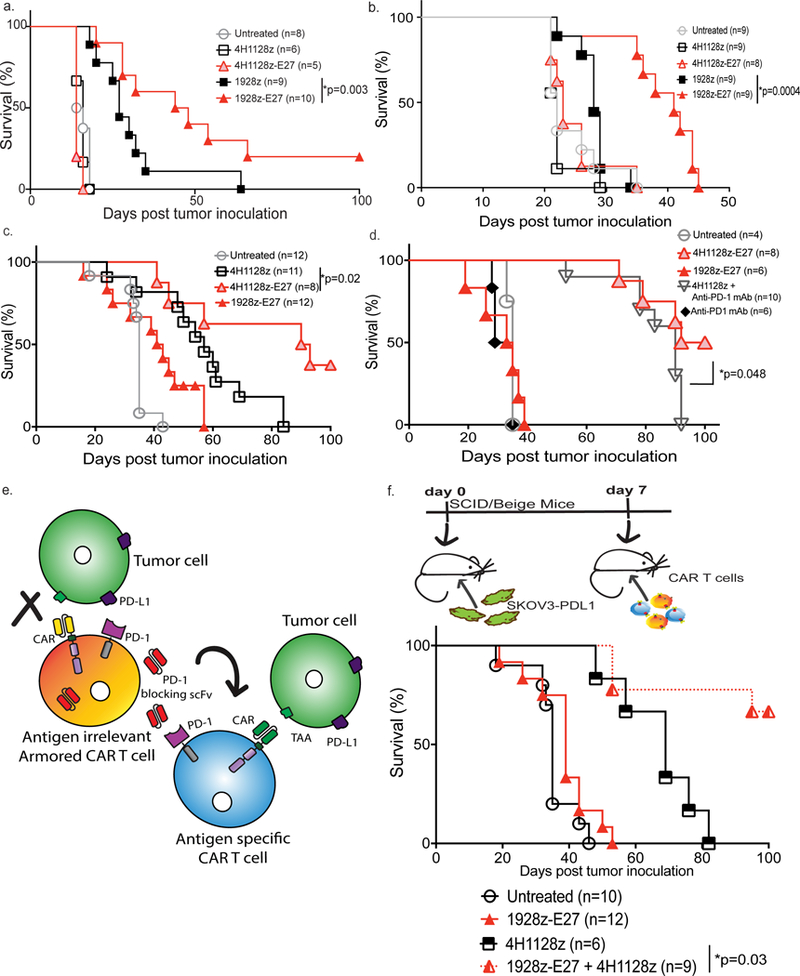
SCID/Beige mice were inoculated with (a) Raji-PDL1 or (b) NALM6-PDL1 tumor cells and treated with 1928z-E27 CAR-T cells have enhanced survival over mice treated with 1928z (*p=0.003 with 95% CI of 0.1–0.9 and *p=0.0004 with 95% CI of 0.02–0.3, respectively by Log-rank Mantel-Cox test). Data shown is pooled from 2 independent experiments. (c) SCID/Beige mice treated with 4H1128z-E27 T cells had enhanced survival compared to mice treated with 4H1128z or irrelevant antigen 1928z-E27 T-cells in mice bearing SKOV3-PDL1 ovarian tumor cells. Data shown is from 2 independent experiments, *p=0.02 by Log-rank Mantel-Cox test with a 95% CI of 0.2–1.5. (d) Survival curve showing mice treated with 4H1128z-E27 T cells had enhanced survival compared to mice treated with 4H1128z + anti-human PD-1 mAb. Data shown is from 2 independent experiments, *p=0.05 by Log-rank Mantel-Cox test with a 95% CI of 0.4–3.1. (e) Schematic illustration of experiment to study bystander effect of scFv-secreting CAR-T cells where SKOV3-PDL1 tumor-bearing mice were treated with a combination of E27-secreting 1928z T-cells, which are antigen irrelevant in this model, together with ovarian-tumor specific 4H1128z T cells. PD-1-blocking scFv secreted by the 1928z-E27 T cells in vivo will bind to PD-1 on 4H1128z T-cells and enhance tumor-specific function. (f) Mice treated 7 days after SKOV3-PDL1 inoculation with a mix of 1928z-E27 + 4H1128z T-cells have enhanced survival as compared to mice treated with 4H1128z (*p=0.007, 95% CI of 0.03–0.6) or 1928z-E27 (*p<0.0001, 95% CI of 0.02–0.2) T cells alone. Data shown is from 2 independent experiments.
Next, we investigated the anti-tumor efficacy of E27-secreting CAR-T cells in a solid tumor model of peritoneal carcinomatosis. SKOV3-PDL1 tumor-bearing SCID/Beige mice treated with 4H1128z-E27 T-cells showed enhanced survival compared to mice treated with 4H1128z T-cells (p=0.02) (Fig. 5c). In addition, utilizing this preclinical xenograft model of metastatic ovarian tumor, mice treated with E27 scFv-secreting 4H1128z T cells had enhanced survival benefit over mice treated with 4H1128z T cells + anti-human PD-1 mAb (p=0.048) (Fig. 5d).
In order to demonstrate in vivo the bystander effect of the E27-secreting CAR-T cells, we treated SKOV3-PD-L1 bearing mice with a combination of antigen-irrelevant 1928z-E27 and tumor-specific 4H1128z CAR-T cells. In this experimental model, only the antigen-irrelevant CAR-T cells express the PD-1-blocking scFv and treatment with 1928z-E27 T cells alone had no survival benefit (Fig. 5c and 5f). However, when injected together, 1928z-E27 enhanced the anti-tumor function of second-generation 4H1128z cells, as compared to mice treated with 4H1128z T cells only (Fig. 5f, p =0.03). This result suggests that the E27 scFv secreted by the 1928z-E27 cells binds to bystander tumor-specific cells in vivo.
E27 scFv secreted by CAR-T cells is only detected in the local TME.
The SKOV3 solid tumor model of peritoneal carcinomatosis was utilized to determine the local and systemic levels of CAR-secreted scFv. First, we used in vivo bioluminescence and fluorescence imaging to visualize the location of scFv or mAb, when given i.p., over time. CAR constructs were generated with E27 scFv fused to Gaussia Princeps Luciferase (GLuc) enzyme, an ATP-independent signal that allowed imaging of E27 location in vivo after being produced by CAR-T cells 31. A commercial anti-human PD-1 mAb was conjugated to VivoTag 680 XL fluorochrome. CAR-T cells secreting E27-GLuc or CAR-T cells + fluorescently-labeled mAb were i.p injected into tumor-bearing mice and imaged over time (Fig. 6a). In as early as 3 hours, the antibody could be found systemically in the mice, whereas the scFv was only detected outside the peritoneal cavity. Following quantification, we did not observe a difference between whole body and local amounts of E27, as E27 remains localized at the tumor site. However, we found changes in the levels of the labeled commercial antibody, as it appeared to circulate out of the tumor area (Fig. 6b).
Fig. 6. The PD-1-blocking E27 scFv secreted by CAR-T cells is only detected in the local tumor microenvironment.
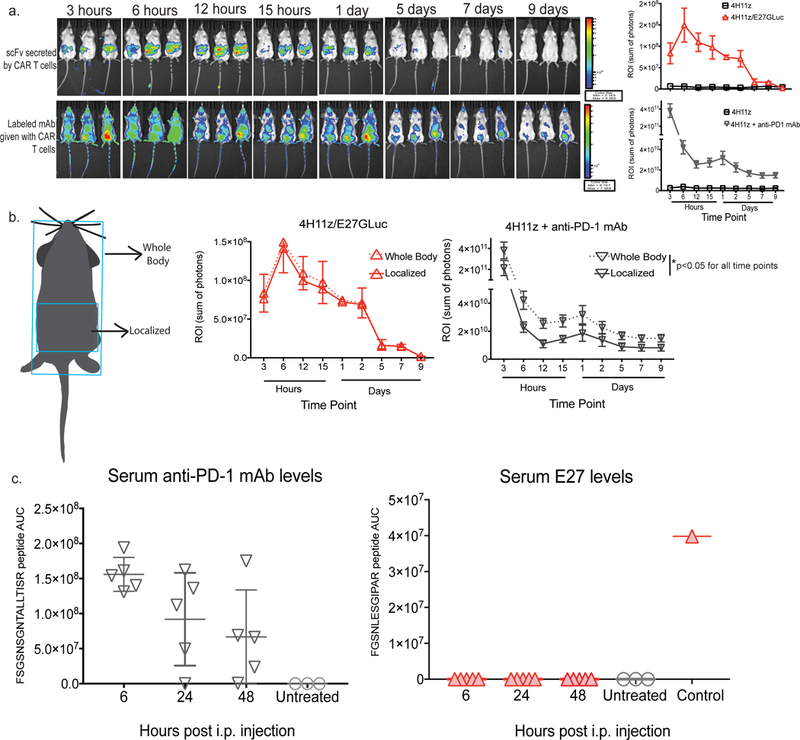
(a) Imaging and quantification over time of E27 scFv tagged with Gaussia Luciferase (GLuc) or fluorescently labeled anti-human PD-1 mAb in SKOV3-PDL1 tumor-bearing SCID/Beige mice treated i.p. with scFv-secreting CAR-T cells or CAR-T cells with anti-PD-1 mAb, 3 animals per group. (b) Schematic representation and quantitation of areas utilized for complete versus local detection of scFv or antibody. *p<0.02 at all time points tested for antibody using multiple T test with 3 animals per group. (c) Quantification utilizing unique peptide sequences by liquid chromatography-tandem mass spectrometry (LC-MS/MS) of serum levels over time of E27 scFv and anti-human PD-1 mAb in in SKOV3-PDL1 tumor-bearing SCID/Beige mice treated i.p. with scFv-secreting CAR-T cells or CAR-T cells with anti-PD-1 mAb (5 animals per group). Systemic infusion of isolated E27 scFv was utilized as a positive control.
To further quantify the systemic levels of antibody or scFv over time, we used targeted mass spectrometry on a high resolution quadrupole orbitrap instrument (Supplementary Fig. 6). SCID/Beige mice were injected i.p. with ovarian SKOV3 tumor cells and 7 days later i.p. injected with E27-secreting CAR-T cells or antibody + CAR-T cells. We harvested serum samples over time and carried out targeted parallel reaction monitoring spectrometry to quantify the amounts of E27 or commercial antibody over time. Two unique tryptic peptides from the complementarity-determining regions of the scFv and mAb not present in other mouse or human proteins were used. Commercial PD-1-blocking mAb was found systemically in the serum and decreased over time as expected (Fig. 6c). However, we did not detect E27 detected in the serum at any time point, indicating that it remains localized in the TME. A control sample consisting of isolated scFv was injected i.v. to demonstrate our assay was capable of detecting E27 in the peripheral serum. These results demonstrate that, unlike checkpoint blockade mAb, E27 secreted by CAR-T cells remains is not present systemically.
Discussion
In multiple in vivo models, we demonstrated that CAR-T cells that secrete PD-1-blocking scFv enhance survival of mice in syngeneic and preclinical xenogeneic hematologic and solid tumor models. PD-1-blocking scFv secreting CAR-T cells are equally effective or superior to therapy with CAR-T cells + PD-1-blocking mAb, indicating that localized delivery of PD-1 blockade is effective at enhancing CAR-T cell anti-tumor function. PD-1 blockade on CAR-T cells results in long-term surviving mice that have detectable CAR-T cells in the bone marrow and can mount an anti-tumor response when re-challenged with tumor. PD-1-blocking scFv produced by CAR-T cells can enhance the function of tumor-specific bystander T cells in vivo in the TME. Finally, unlike systemic checkpoint blockade therapy, scFv secreted by CAR-T cells remain locally in the TME.
Preclinical studies have previously demonstrated that CAR-T cells are susceptible to suppressed effector function mediated through the PD-1 receptor and subsequent combination with PD-1/PDL-1-blocking antibodies, given exogenous or produced by CAR-T cells, resulted in enhanced CAR-T cell-mediated anti-tumor response 32,33. However, these studies were limited to xenograft models that do not recapitulate the endogenous immunosuppressive TME. Specifically, Suarez et al. (Suarez, 2016) used CAR-T cells secreting an antibody (not scFv) and did not compare to antibody alone. ScFv are smaller, less stable molecules than antibodies and successful checkpoint blockade had not been demonstrated previously with them. Furthermore, the smaller size of scFvs enables the engineering of CAR constructs that are smaller, allowing for the possibility of tri-cistronic vectors with a combination of scFv in the future. In addition, the authors did not demonstrate bystander effects or localized delivery, two important advantages of utilizing this system.
Alternative methods to prevent PD-1-related dampening of CAR-T cell responses include co-expression of a chimeric switch receptor 33, with the extracellular domain of PD-1 linked to activation signaling domains 34 or co-modifying CAR-T cells to express a dominant negative PD-1 receptor 35. Both of these methods have been shown to increase the levels of cytolysis, cytokine secretion and enhanced in vivo anti-tumor efficacy in response to PD-L1+ tumor targets compared to T cells expressing a CAR alone. However, this protective effect is limited to the CAR-T cells themselves. Endogenous TILs within the TME are still subject to PD-1-mediated suppression. Given that the PD-1-blocking scFv secreted by CAR-T cells are able to bind to bystander cells, we hypothesize that local secretion of the PD-1-blocking scFv described in this study may protect endogenous anti-tumor immune cells and reduce outgrowth of antigen-negative tumors.
Systemic administration of checkpoint blockade can result in immune related adverse events (IRAEs) 36. IRAEs are frequent in patients treated with checkpoint blockade 11,13,36. Given that CAR-T cells traffic to and expand at the site of tumor, delivery of the PD-1-blocking scFv is primarily localized to this area in our models. Therefore, we hypothesize that the strategy presented here, demonstrating localized and not systemic delivery, may decrease the IRAEs associated with systemic checkpoint blockade.
The work presented here validates that CAR-T cells can be utilized to deliver immune modulatory scFvs within the TME in the absence of systemic blockade. With the rapid development of human phage display libraries, it is feasible to expand this immune modulating CAR-T cell approach by utilizing scFvs targeting other molecules such as LAG-3 37,38, TIM-3 39, or CLTA-4 40, as well as combination strategies.
This proof-of-principle study provides support for the strategy of utilizing armored CAR-T cells for targeted delivery of immune modulatory scFvs to the TME. This approach could potentially improve the clinical outcome in response to CAR-T cell therapy and improve the safety of checkpoint blockade therapy.
Materials and methods
For additional information on our Methods, please refer to the Life Sciences Reporting Summary accompanying this manuscript.
Selection of PD-1-blocking scFv
The RMP1–14 scFv was engineered from the RMP1–14 hybridoma. The PD-1-blocking scFv, termed E27, was identified from a human antibody scFv phage library (Eureka Therapeutics Inc., Emeryville CA). The scFv library was screened for binding to human PD-1. Briefly, biotinylated PD-1-Fc fusion protein was mixed with the human scFv phage library. The antigen-scFv complexes were pulled down by streptavidin-conjugated beads and bound clones were eluted and transformed into bacteria. The binding and affinity of scFv to PD-1 was determined using ForteBio Octet QK (Pall Life Sciences, Menlo Park, CA, USA). The ability of the scFv to block PD-1 from interacting with PD-L1 was determined using an ELISA assay. Briefly, PDL1-Fc (R&D Systems, Minneapolis, MN, USA), was loaded onto a plate. PD-1-Fc was mixed with dilutions of antibody derived from the E27 scFv clones or human IgG1 isotype control mAb (Eureka Therapeutics Inc.) and then added to the PDL1-Fc-coated plate. Binding of the clones was determined by a standard ELISA method against biotinylated PD-1. Streptavidin-conjugated HRP (Vector Laboratories, Burlingame, CA, USA) was used to detect binding on an Epoch-2 microplate reader (BioTek Instruments, Winooski, VT, USA).
Generation of retroviral vectors
The RMP1–14 scFv was cloned into the SFG gammaretroviral vector encoding CD19-directed or MUC16ecto-directed CARs with mouse signaling domains. The E27 scFv was cloned into the SFG-retroviral vector encoding the CD19-targeted CAR, termed 1928z, to generate SFG-1928z-E27 or DNA encoding the MUC16ecto -targeted CAR and E27 scFv, termed SFG-4H1128z- E27.
Cell Culture
Retroviral producer cells were maintained in DMEM and human T cells were maintained in RPMI-1640 medium. Tumor cells and mouse T cells were maintained in RMPI-1640 medium supplemented with nonessential amino acids, sodium pyruvate, HEPES (N-2-hydroxyethylpiperazine-N-2-ethane sulfonic acid) buffer, and 2-Mercaptoethanol (all from Invitrogen, Carlsbad, CA, USA). All media was supplemented with 10% fetal bovine serum (Atlanta Biological Flowery Branch, GA, USA), 2mMol L-Glutamine, 100 IU/ml penicillin and 100 μg/ml streptomycin (Invitrogen). Retroviral producer cell lines were generated by using CaPO4 (Promega, Madison WI, USA) to transiently transfect H29 cells with retroviral constructs encoding the CAR or the CAR and the PD-1-blocking E27scFv. Supernatant from the H29 cells was used to transduce Phoenix-ECO or 293Glv9 cells to produce stable retroviral producer cells lines. DNA encoding mouse and human PD-L1 and PD-1 was purchased (Integrated DNA technologies, Coralville, IA, USA) and separately cloned into the SFG retroviral backbone (SFG-hPD-L1, SFG-hPD-1). This was transiently transfected into H29 cells using CaPO4 transfection (Promega) and the H29 supernatant was used to transduce tumor or 293Glv9 packaging cells. Tumor cells lines were sorted by FACS based on high expression of PD-L1 following staining with PE-conjugated anti-PD-L1 (BD Biosciences, San Jose, CA, USA, clone MIH5). ID8 and SKOV3 cells were validated by karyotyping and all cells were routinely checked for mycoplasma.
T cell isolation and modification
Human T cells were activated and transduced as described previously 25. Briefly, peripheral blood mononuclear cells (PBMCs) were isolated from healthy donor peripheral blood or leukopacks (New York Blood Center, New York, NY, USA). All experiments were performed in compliance with all relevant ethical regulations and in accordance with IRB 095091. PBMCs were activated with 2 μg/ml phytohemagglutinin and 100 IU/ml of IL-2 for 2 days prior to transduction. Mouse T cells were mechanically isolated from spleens and activated using IL-2 and 4 μg/ml of concanavalin A (Millipore Sigma, St. Louis, MO). Transduction was achieved by centrifugation of activated PBMCs and retroviral supernatant on RetroNectin-coated plates on 3 consecutive days (TakaraBio, Kusatsu, Shiga, Japan).
Flow cytometry
Flow cytometry was used to determine the transduction efficiency of transduced cells following staining with Alexa-Fluor 647 conjugated anti-idiotype antibodies that detect the CD19-targeted CAR (clone 19E3), 1928z, as well as the ovarian tumor-antigen targeted CAR (clone 22G3), 4H1128z (both generated at the Memorial Sloan Kettering Cancer Center Antibody and Bioresource Core Facility). The following antibodies were used in flow cytometry experiments: FITC conjugated anti-PD-1 (BD Biosciences, clone MIH4, Cat#557860 CD279 Mouse anti-Human, FITC), anti-mouse PD-1 (BD Biosciences, clone J43, Cat#561788 CD279 Hamster anti-Mouse, PE), anti-mouse PD-L1 from BD Biosciences (clone MIH5, Cat#558091), Mouse Anti-Human CD19 mAb (clone SJ25-C1), FITC conjugate, Cat#MHCD1901 from Invitrogen and Anti-Human CD3 eFluor450, Cat#48–0037-42 from eBioscience. Data from cells was collected using a Beckman Coulter Gallios flow cytometer and Kaluza 1.2 and analyzed using FlowJo vX.07 software (FlowJo, Ashland, OR, USA).
For analysis of syngeneic bystander in vivo experiment, the following antibodies and reagents were used for flow cytometry: CD45-PE (clone 30-F11, Cat#103106), CD3-APC/Cy7 (clone 17A2, Cat#100222), CD80-PE (clone 16–10A1, Cat#104708), IFN-γ-FITC (clone XMG1.2, Cat#11–7311-41 from eBioscience), CD107a (LAMP-1)-PE-Cy7 (clone 1D4B, Cat#121619), Granzyme B-FITC (clone GB11, Cat#515403) and Zombie UV Fixable Viability Kit (Cat#423107) were purchased from BioLegend. After 20 minute incubation with Zombie UV Fixable stain at room temperature, all samples were washed with ice-cold BD FACS Buffer, and stained with the appropriate surface antibodies. Intracellular staining for IFN-γ and Granzyme B was performed according to Foxp3/Transcription Factor Staining Buffer Set (eBioscience). Data acquisition was performed on FACSCalibur (BC Biosciences) and analyzed via FlowJo. For analysis intratumoral CD3+ CAR- T cell levels were assessed with phenotypic criteria of CD45+CD3+CAR-, and total CD45+ cells were used as a common denominator.
Western blot analysis
Supernatant was collected and filtered from either 293Glv9 packaging cells or transduced T cells as indicated. To demonstrate E27 binding to PD-1, 293Glv9-PD1 packaging cells were incubated for 24 hours in filtered supernatant from 1928z or 1928z-E27 transduced T cells. To demonstrate that E27 binds to untransduced, bystander cells, 1928z and 1928z-E27 T cells were co-cultured with human T cells transduced to overexpress PD-1, after 4 days stimulation with CD3/CD28 beads, the cells were sorted by flow cytometry following staining with 19E3 mAb to separate CAR+ and CAR- cells. Supernatant or whole cell lysates was loaded onto mini protean TGX gels (BioRad, Hercules, CA, USA) and then transferred to Immun-blot PVDF membranes (BioRad). The was probed with mouse-anti-HA antibody (Cell Signaling,6E2, Cat#2367S) and then goat anti-mouse HRP conjugated antibody (Millipore, GT X MS AP127 Cat#6C0112) for E27. RMP1–14 was detected using a HRP-conjugated mouse-anti-myc tag antibody (Cell Signaling, Cat#2040S). Detection of antibody was achieved with Pierce ECL western blot substrate (Thermo Scientific, Waltham, MA, USA). RMP1–14 scFv co-immunoprecipitation was performed using the Peirce c-Myc Tag IP/Co-IP Kit (Thermo Scientific) according to the manufacturer’s instructions and detected by western blot as described above.
NIH/3T3 expansion assay
NIH/3T3 (3T3-Empty) or NIH/3T3 cells transduced to express human PD-L1 (3T3-PDL1) were plated and transduced T cells were added and incubated for 24 hours. CD3/CD28 beads (Invitrogen) were then added at a 1:2 bead:T cell ratio. Every 3 days, the T cells were moved to fresh 3T3 coated plates to ensure continuous stimulation with PD-L1. At 6 days, the T cells were debeaded before being plated on fresh 3T3 cells, and left without beads for 24 hours. The cells were then re-stimulated with CD3/CD28 beads at the same ratio (second stimulation). Trypan blue exclusion was used to enumerate cells following removal from 3T3 cells.
Tumor expansion assay
Transduced T cells were co-cultured with Raji-PDL1+eGFP+ or NALM6-PDL1+eGFP+ tumor cells at 1:1 tumor:CAR+ T-cell ratio. Following 72 hours co-culture, flow cytometry was used to detect tumor and T cells following staining with anti-human CD19-FITC (Invitrogen). Cells were enumerated using 123 count ebeads (eBiosciences, San Diego, CA, USA) according to the manufacturer’s instructions.
In vivo experiments
All experiments were performed in compliance with all relevant ethical regulations and in accordance with an Institutional Animal Care and Use Committee-approved protocol (protocol 00–05-065).
For syngeneic survival experiments, C57BL/6 mice (The Jackson Laboratory) of 6–8 weeks were inoculated i.p. with 10 × 106 ID8 tumor cells and treated 7 days later with 2 × 106 CAR-T cells, as indicated, with or without 250 μg RMP1–14 antibody (Bio X Cell). Mice were given RMP1–14 antibody i.p. on days 7, 10, and 14 post tumor inoculation, for a total of 3 doses.
For syngeneic bystander experiments, 6–8-week-old female C57BL/6 mice (Jackson Lab) were injected subcutaneously with 1 × 106 B16-F10 mouse melanoma cells transduced to express the MUC16 cell surface antigen. On day 10 after tumor implantation, 2 × 106 CAR-T cells were injected intratumorally. The mice were sacrificed 48 hours after T cell injection. Tumor cells were mechanically dissociated and digested in collagenase-based buffer. The samples were subsequently stained for flow cytometric analysis, as described above.
Fox Chase CB17 (CB17.Cg-PrkdcscidLystbg-J/Crl, SCID/Beige mice (Charles River Laboratories) of 6–8 weeks of age were inoculated i.v. with 1 × 106 Raji-PDL1 or NALM6-PDL1 tumor cells. The following day, mice received one systemic infusion of 1 × 107 CAR+ T cells. For xenograft solid tumor studies, SCID/Beige mice were inoculated i.p. with 10 × 106 SKOV3-PDL1 cells and treated 7 days later with 5 × 106 total CAR+ T cells, as indicated, with or without anti-human PD-1–blocking antibody (clone EH12.2H7, BioLegend). Mice were given 250 μg of antibody i.p. on days 7, 10, and 14 post tumor inoculation, for a total of 3 doses.
The PCR primers used to detect CAR isolated from splenocytes or bone marrow span mouse CD3 and the gamma-retroviral SFG backbone and are as follows: 5’AGAACCTAGAACCTCGCTGGAAAG 3’ and 5’GTGCATTGTATACGCCTTCCTGGGGGT 3’.
Bioluminescent imaging of scFv-GLuc or anti-human PD-1 antibody (clone EH12.2H7, BioLegend) labeled with VivoTag 680 XL Fluorochrome (PerkinElmer, Waltham, MA, USA) in SCID/Beige mice i.p. inoculated with SKOV3-PDL1 and treated 7 days later i.p with 5 × 106 scFv-secreting CAR-T cells or CAR-T cells with 250 μg of anti-human PD-1 antibody (clone EH12.2H7, Biolegend), was performed using Xenogen IVIS imaging system with Living Image software (Xenogen Biosciences, Cranbury, NJ, USA). Image acquisition was done on a 25 cm field of view at medium binning level at various exposure times.
Targeted LC-MS/MS (PRM) analysis of scFv and mAb in serum
To quantitate the levels of the E27 scFv and anti-human PD-1 mAb (clone EH12.2H7, Biolegend) proteins in serum a bioinformatics search was utilized to identify peptide sequences unique to the therapeutic proteins but not found in other proteins in human or mouse proteome. We then quantitated those peptides using targeted parallel reaction monitoring (PRM) on a high resolution and accurate mass quadrupole-orbitrap mass spectrometer 41. To accomplish this goal, we first used Skyline 4.1 42, a freely available software tool (online at http://skyline.maccosslab.org). Tryptic peptides that contained cysteine or methionine residues were excluded and peptides that were from the complementarity-determining region of the therapeutics were prioritized. Two tryptic peptides, one from the scFv (FSGSNSGNTATLTISR, m/z = 806. 8999 (M+2H)2+ ions) and one from the anti-PD-1 mAb (FGSNLEGIPAR, m/z = 624.3226 (M+2H)2+ ions), met our criteria and were used for subsequent analysis.
LC-MS/MS of scFv and mAb biotherapeutics
To obtain the tandem mass spectra for the unique peptides in the biotherapeutics (m/z 806.89 and 624.32), the scFv and anti-PD-1 mAb proteins were separated by SDS/PAGE, stained with Simply Blue (Life Technologies Scientific), enzymatically digested in situ with trypsin 43, and desalted 44. The purified peptides were diluted to 0.1% formic acid and analyzed separately by high resolution LC-MS/MS in data dependent mode. We used a Waters NanoAcquity system (with a 100-μm inner diameter × 10-cm length C18 column (1.7 μm BEH130; Waters) configured with a 180-μm × 2-cm trap column coupled to a Thermo Q-Exactive Plus orbitrap mass spectrometer. Trapping was performed at 15 μL/min buffer A for 1 minute and elution with a 50% linear acetonitrile gradient over 120 minutes. MS data were collected in data dependent acquisition mode. Full scan MS1 spectra were acquired over 380–1600 m/z at a resolution of 70,000 (m/z 400) with automatic gain control (AGC) at 3 × 106 ions. The top 15 most intense precursor ions were selected for HCD fragmentation performed at normalized collision energy (NCE) 25% with target ion accumulation value of 5 X 10(4). MS/MS spectra were collected with resolution of 17,500. See Fig. S3 for MS/MS spectra.
Proteomic analysis of serum
For in solution digestion of mouse serum a previously published protocol was slightly modified 45. Briefly, frozen serum samples were thawed on ice, 10 μL was transferred to a new tube then diluted with 40 μL 50 mM ammonium bicarbonate. 10 μL of the diluted serum was transferred to a new tube and further diluted with 190 μL ammonium bicarbonate containing 0.5% deoxycholate and 10 mM DTT then vortexed at 300 rpm at 560C for 60 minutes. The samples were cooled to room temperature then alkylated with the addition of 4 μL of 0.5 M iodoacetamide in 50 mM ammonium bicarbonate and incubated for 30 minutes in the dark. Sequencing grade trypsin (Promega Gold) was added 2 μg/sample then incubated at 370C for 18 hours with gentle vortexing (300rpm). To quench the reaction, 10 μL formic acid was added, DOC precipitated, then spun at 20,000 g for 4 minutes. 50 μL was removed to a clean tube then 4 μL of the digest taken into an autosampler vial and diluted with 46 μL buffer A prior to targeted LC-MS/MS (PRM) analysis.
The LC-MS/MS PRM analysis was performed on the identical LC-MS system as above but adjusted for PRM mode. The LC gradient was 2% to 50% B over 20 minutes followed by 90% B for 5 minutes with a 14 minute equilibration at 300 nL/minute. Full MS scans were performed with the following parameters: Resolution: 70,000; AGC target: 3e6; Maximum IT: 200ms; Scan Range: 150 to 2000 m/z. Targeted MS2 scans (PRM) were performed on m/z of 806.8999, 1117.5342, and 624.3226 with the following parameters: Resolution: 17,500; AGC target: 2e5; Maximum IT: 100ms; Isolation window: 1.5 m/z; nce: 27; Charge state: +2.
For each peptide, 6 singly charged y-type fragment ions near or above the precursor mass were used for the PRM assay as the signals were robust and showed no interfering background ions in the untreated plasma samples. Thermo Xcalibur version 2.2 was used to identify the presence of EH12/2H7 antibody and E27 scFv antibody. Chromatographic peaks of EH12/2H7 peptide (FSGSNSGNTALLTISR) and E27 scFv peptide (FGSNLESGIPAR) were identified by searching (M+2H)2+=m/z 624.3226 and (M+2H)2+=m/z 806.8999, respectively, with 10 ppm mass tolerance and retention time within 60 seconds. Peaks of y-type fragment ions from both peptides were identified by searching corresponding m/z values with the same criteria and retention time based on the analysis of the recombinant scFv or antibody. ICIS integration algorithm was used to detect and integrate the area of each peak. The summed areas of all peaks (6 y-type ions) associated with EH12/2H7 peptide and E27 scFv peptide from each time point (6 hour, 24 hours and 48 hour) were calculated separately then plotted by time point using Prism. Chromatograms of untreated and scFv i.v. samples were integrated and plotted using the same method.
Statistical analysis
Log-rank, unpaired or paired t tests were performed using GraphPad Prism 7 software, La Jolla, CA, USA) where appropriate.
Supplementary Material
Acknowledgements:
The authors thank the following for financial support: National Institutes of Health Grants (PO1CA190174–01 (R.J.B.), P50 CA192937 (R.J.B.)), The Annual Terry Fox Run for Cancer Research (New York, NY) organized by the Canada Club of New York (R.J.B.), Kate’s Team (R.J.B.), Carson Family Charitable Trust (R.J.B.), the Leukemia & Lymphoma Society Specialized Center of Research Program (7014) (R.J.B.), William Lawrence and Blanche Hughes Foundation (R.J.B.), the Experimental Therapeutics Center of Memorial Sloan Kettering Cancer Center (Innovations in the structures, functions and targets of monoclonal antibody-based drugs for cancer) (R.J.B.).
We would like to acknowledge Y. Iragashi and A. Rookard for technical assistance with in vivo experiments and the MSKCC Molecular Cytogenetics Core and NIH Cancer Center support grant P30 CA008748 for karyotyping of the ID8 and SKOV3 cells.
Footnotes
Author contributions: S.R., O.O.Y., H.J.J., and R.J.B. designed the experiments, interpreted the results and wrote the manuscript. T.J.P., D.G.vL., D.J.D, M.S., M.M.M., Z.L., P.W., S.Y. J.X. X.M., R.C.H., C.L designed, performed, and/or analyzed experiments. V.E.S. performed the statistical analysis.
Competing Financial Interests Statement: R.J.B. is a co-founder and receives royalties from Juno Therapeutics. R.J.B, H.J.J., and C.L. have submitted a patent related to this work.
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the paper (and its supplementary information files).
References
- 1.Jackson HJ, Rafiq S & Brentjens RJ Driving CAR-T cells forward. Nature reviews. Clinical oncology 13, 370–383 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang M, et al. A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J Ovarian Res. 7:19 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knutson KL, et al. Regulatory T cells, inherited variation, and clinical outcome in epithelial ovarian cancer. Cancer Immunol Immunother 12, 1495–504 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Erfani N, et al. FoxP3+ regulatory T cells in peripheral blood of patients with epithelial ovarian cancer. Iran J Immunol 2, 105–12 (2014). [PubMed] [Google Scholar]
- 5.Shevach EM Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity 30(5), 636–45 (2009). [DOI] [PubMed] [Google Scholar]
- 6.Noy R & Pollard JW Tumor-associated macrophages: from mechanisms to therapy. Immunity 41(1), 49–61 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abiko K, et al. IFN-γ from lymphocytes induces PD-L1 expression and promotes progression of ovarian cancer. Br J Cancer 112 (9), 1501–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yeku OO, Purdon TJ, Koneru M, Spriggs D, & Brentjens RJ Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci Rep 7(1) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frey AB Suppression of T cell responses in the tumor microenvironment. Vaccine 33, 7393–7400 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Kershaw MH, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res 12(20 Pt 1), 6106–15 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brahmer JR et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. The New England journal of medicine 366, 2455–2465 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Powles T et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 515, 558–562 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Topalian SL et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. The New England journal of medicine 366, 2443–2454 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Armand P Immune checkpoint blockade in hematologic malignancies. Blood 125, 3393–3400 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Rizvi NA et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tumeh PC et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cha E et al. Improved survival with T cell clonotype stability after anti-CTLA-4 treatment in cancer patients. Science translational medicine 6, 238ra270 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gajewski TF, Louahed J & Brichard VG Gene signature in melanoma associated with clinical activity: a potential clue to unlock cancer immunotherapy. Cancer journal 16, 399–403 (2010). [DOI] [PubMed] [Google Scholar]
- 19.Ku GY et al. Single-institution experience with ipilimumab in advanced melanoma patients in the compassionate use setting: lymphocyte count after 2 doses correlates with survival. Cancer 116, 1767–1775 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Snyder A et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. The New England journal of medicine 371, 2189–2199 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Curran KJ, et al. Enhancing antitumor efficacy of chimeric antigen receptor T cells through constitutive CD40L expression. Mol Ther 23(4), 769–78 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pegram HJ, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood 119(8), 4133–41 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koneru M, Purdon TJ, Spriggs D, Koneru S, & Brentjens RJ IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology 4(3) (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pegram HJ, et al. IL-12-secreting CD19-targeted cord blood-derived T cells for the immunotherapy of B-cell acute lymphoblastic leukemia. Leukemia 29, 415–22 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rafiq S et al. Optimized T-cell receptor-mimic chimeric antigen receptor T cells directed toward the intracellular Wilms Tumor 1 antigen. Leukemia 8, 1788–1797 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Avanzi MP, et al. Engineered Tumor-Targeted T Cells Mediate Enhanced Anti-Tumor Efficacy Both Directly and through Activation of the Endogenous Immune System. Cell Rep 23, 2130–2141 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.John LB et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clinical cancer research : an official journal of the American Association for Cancer Research 19, 5636–5646 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Rosewell SA, et al. Adenovirotherapy Delivering Cytokine and Checkpoint Inhibitor Augments CAR T Cells against Metastatic Head and Neck Cancer. Mol Ther 25, 2440–2451 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chekmasova AA et al. Successful eradication of established peritoneal ovarian tumors in SCID-Beige mice following adoptive transfer of T cells genetically targeted to the MUC16 antigen. Clinical cancer research : an official journal of the American Association for Cancer Research 16, 3594–3606 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Curran MA, Montalvo W, Yagita H & Allison JP PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proceedings of the National Academy of Sciences of the United States of America 107, 4275–4280 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Santos EB et al. Sensitive in vivo imaging of T cells using a membrane-bound Gaussia princeps luciferase. Nature medicine 15, 338–344 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suarez ER et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget 7, 34341–34355 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prosser ME, Brown CE, Shami AF, Forman SJ & Jensen MC Tumor PD-L1 co-stimulates primary human CD8(+) cytotoxic T cells modified to express a PD1:CD28 chimeric receptor. Molecular immunology 51, 263–272 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Liu X et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer research 76, 1578–1590 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cherkassky L et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. The Journal of clinical investigation 126, 3130–3144 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Michot JM et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. European journal of cancer 54, 139–148 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Grosso JF et al. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. The Journal of clinical investigation 117, 3383–3392 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Workman CJ et al. Lymphocyte activation gene-3 (CD223) regulates the size of the expanding T cell population following antigen activation in vivo. Journal of immunology 172, 5450–5455 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Sabatos CA et al. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nature immunology 4, 1102–1110 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Hodi FS et al. Improved survival with ipilimumab in patients with metastatic melanoma. The New England journal of medicine 363, 711–723 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only References
- 41.Peterson AC, Russell JD, Bailey DJ, Westphall MS & Coon JJ Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol Cell Proteomics 11, 1475–1488 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Egertson JD, MacLean B, Johnson R, Xuan Y & MacCoss MJ Multiplexed peptide analysis using data-independent acquisition and Skyline. Nat Protoc 10, 887–903 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shevchenko A, Tomas H, Havlis J, Olsen JV & Mann M In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc 1, 2856–2860 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Rappsilber J, Mann M & Ishihama Y Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc 2, 1896–1906 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Lee AY et al. Measurement of fractional synthetic rates of multiple protein analytes by triple quadrupole mass spectrometry. Clin Chem 58, 619–627 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.