

Abstract
We report a genome-editing strategy to correct compound heterozygous mutations, a common genotype in patients with recessive genetic disorders. Adeno-associated viral vector delivery of Cas9 and guide RNA induces allelic exchange and rescues the disease phenotype in mouse models of hereditary tyrosinemia type I and mucopolysaccharidosis type I. This approach recombines non-mutated genetic information present in two heterozygous alleles into one functional allele without using donor DNA templates.
Cas9-mediated gene editing promises to correct DNA mutations underlying human diseases. In principle, many mutations can be individually corrected by homology-directed repair (HDR) using an exogenous DNA template. However, monogenic recessive genetic diseases typically involve many distinct mutations present across the gene. For example, more than 95 known mutations in the FAH gene can cause hereditary tyrosinemia type 1 (HT1)1,2. Consequently, the rapid development of gene-editing as a human therapy requires a mutation-independent gene repair strategy. A strategy that avoids the need to deliver an exogenous DNA template would also substantially simplify the design and delivery of the necessary gene editing components.
In compound heterozygotes, each allele of the mutant gene harbors a different genetic lesion. A sizable fraction of human autosomal recessive genetic disorders—including HT1 and lysosomal diseases—are caused by such compound heterozygous mutations1,3. We hypothesized that reconstituting the correct genetic information present on both compound heterozygous mutant alleles into one allele could create a functional copy of the disease gene. (Fig. 1a). Targeted nucleases4–8 have been used to induce reciprocal translocation between non-homologous chromosomes in vitro and in vivo, aiming to study the mechanisms of chromosome translocation or model certain types of cancer. Here, we used Cas9 to create double-stranded DNA breaks in both chromosomal homologs in a compound heterozygous mouse model of HT1, thereby inducing allelic exchange between two different mutant alleles and rescuing the disease phenotype.
Figure 1. Cas9-mediated allelic exchange in compound heterozygous HT1 mice.
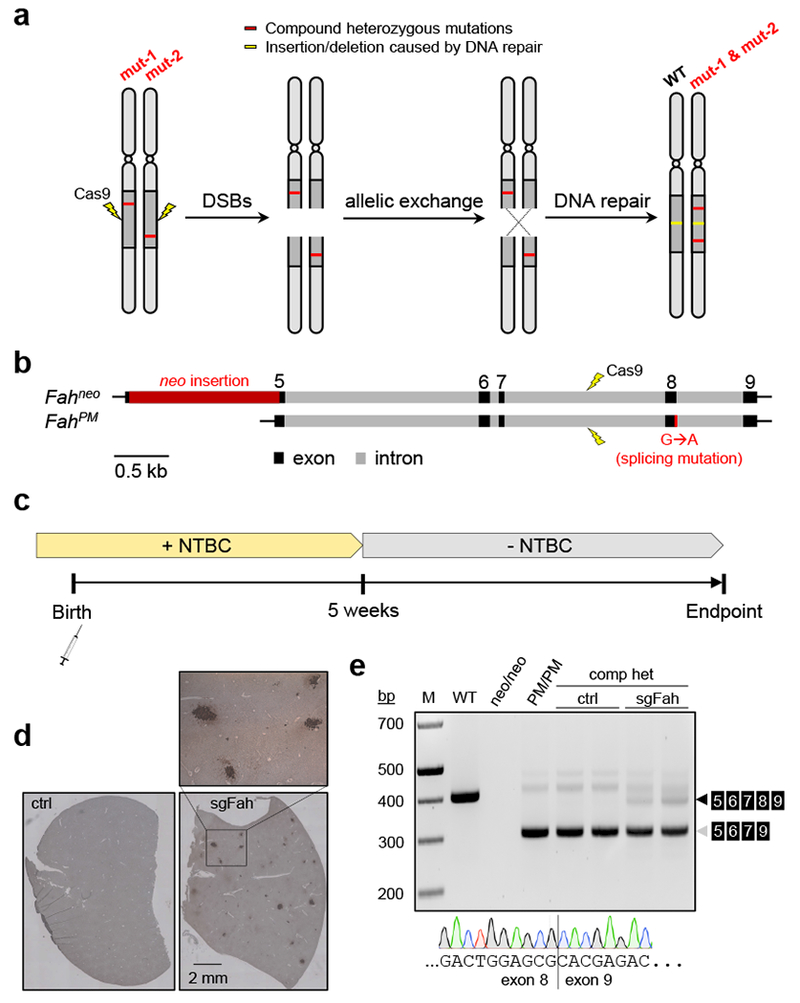
(a) Cartoon showing the concept of repairing recessive compound heterozygous mutations by Cas9-mediated allelic exchange. (b) Genomic structure of the Fahneo and FahPM alleles and strategy to induce allelic exchange between the two alleles (drawn to scale). (c) Experimental timeline. See text and Methods for details. (d) Representative images of FAH immunohistochemistry (IHC) in liver sections from a mouse treated with rAAV9-SpCas9 and scAAV8-sgAspa (ctrl hereafter), and a mouse treated with rAAV9-SpCas9 and scAAV8-sgFah (sgFah hereafter). N=2 mice per group. These mice were treated at postnatal day 1 (P1), maintained on NTBC water, and euthanized at five weeks old. The boxed area is enlarged and shown to visualize the FAH-positive cells (dark brown color). (e) Agarose gel image showing the detection of reverse-transcription (RT) PCR products of Fah messenger RNA in liver lysate. Samples include (from left to right): Wild-type (WT) mouse; Homozygous Fahneo/neo mouse (neo/neo); Homozygous FahPM/PM mouse (PM/PM); Compound heterozygous (comp het) mice treated with ctrl rAAV at P1 (ctrl, n=2 mice); Compound heterozygous mice treated with sgFah rAAV at P1 (sgFah, n=2 mice). All mice were euthanized at five weeks old. A representative Sanger sequencing chromatogram is shown at the bottom, revealing the junction between exon 8 and exon 9 in the RT-PCR product (black arrowhead) of a sgFah-treated mouse. N=2 mice. Note that the last nucleotide in exon 8 by Sanger sequencing is the wild-type guanine (G), but is mutated to adenine (A) in the FahPM allele.
HT1 is caused by loss-of-function mutations in the FAH gene, which encodes fumarylacetoacetate hydrolase (FAH), an enzyme required for tyrosine catabolism. Loss of FAH causes accumulation of toxic tyrosine metabolites in the liver and other organs. The toxicity can be relieved by oral administration of 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC), which blocks tyrosine catabolism at early step and thus prevents the formation of toxic intermediates9. To test our strategy, we used two HT1 mouse models that carry different mutations in the Fah gene. The Fahneo/neo mouse carries an insertion of a neomycin-resistance gene (neo) in exon 5, while the FahPM/PM mouse carries a G-to-A transition that causes exon 8 skipping during splicing. Neither Fah mutant produces FAH protein detectable by immunohistochemistry (IHC). By crossing homozygotes of each mutation, we generated compound heterozygous Fahneo/PM mice (Fig. 1b).
To generate genomic DNA breaks promoting allelic exchange, newborn Fahneo/PM mice were systematically treated with a pair of liver-tropic recombinant adeno-associated virus (rAAV) vectors: one expressing S. pyogenes Cas9 (rAAV9-SpCas9) and the other producing a single guide RNA (sgRNA) targeting Fah intron 7 (scAAV8-sgFah; Supplementary Fig. 1a-c). As a control, we replaced sgFah with an sgRNA targeting intron 2 of the Aspa gene (scAAV8-sgAspa). To allow sufficient time for genome editing to occur, mice were maintained on drinking water containing NTBC until five weeks old (Fig. 1c). We euthanized two mice from each group at five weeks old, and confirmed that both the Fah and Aspa sgRNAs had induced insertions or deletions (indels) at the targeted genomic sites in the liver (Supplementary Fig. 1d-f). FAH IHC of liver tissue sections detected clusters of FAH-positive hepatocytes in the sgFah-treated mice but not the sgAspa control mice (Fig. 1d). Reverse-transcription PCR (RT-PCR) of total liver RNA revealed the presence of wild-type Fah mRNA spanning exon 5 to exon 9 in the sgFah but not the sgAspa control mice (Fig. 1e).
To determine whether sufficient functional FAH protein was produced by the Cas9/sgFah-treated mice, NTBC was withdrawn from a subset of treated mice five weeks after birth. All control mice lost >20% body weight within one month after NTBC withdrawal due to liver failure and were euthanized (Fig. 2a). Naïve compound heterozygous mice receiving no rAAV treatment suffered similar weight loss within one month (Supplementary Fig. 2). In contrast, the sgFah-treated mice initially lost weight, but eventually achieved normal, stable body weight (Fig. 2a). FAH IHC showed that FAH-positive hepatocytes repopulated majority of the liver in the sgFah-treated mice, reflecting the growth advantage of corrected hepatocytes over FAH-null cells (Fig. 2b)9. The FAH-positive hepatocytes were morphologically normal as assessed by hematoxylin and eosin staining (Fig. 2c). Three additional sgFah-treated mice were not initially rescued after NTBC withdrawal, but after returning them to NTBC for one more week, showed rescue comparable to the other rescued mice and no longer required NTBC (Supplementary Fig. 3). The variation of rescue efficacy in individual mice may be due to the efficiency of FAH restoration in the liver, which ranges from 0.4% to 1.6% (median=0.61%) and is not correlated with the Cas9 delivery efficiency (Supplementary Fig. 4). Whereas the control mice showed elevated serum transaminase levels reflecting liver injury, the sgFah-treated mice had significantly reduced serum transaminase levels, suggesting recovery from liver injury (Fig. 2d). Reverse-transcription–quantitative PCR (RT-qPCR) for Fah mRNA containing exons 8 and 9 showed that mutation-free Fah gene expression was restored to 40% of normal level in the sgFah-treated mice (Fig. 2e). High-throughput RNA sequencing showed a similar level of Fah mRNA with exon 8 inclusion (Fig. 2f). Cas9/sgFah-treatment regimen did not rescue mice homozygous for either mutation (Supplementary Fig. 5). In another set of experiments, rAAV delivery of Cas9 and sgFah in young adult compound heterozygous mice also rescued the disease phenotype (Supplementary Fig. 6), suggesting that allelic exchange can occur in hepatocytes beyond the fast-proliferating, postnatal stage.
Figure 2. Rescue of compound heterozygous HT1 mice by allelic exchange.
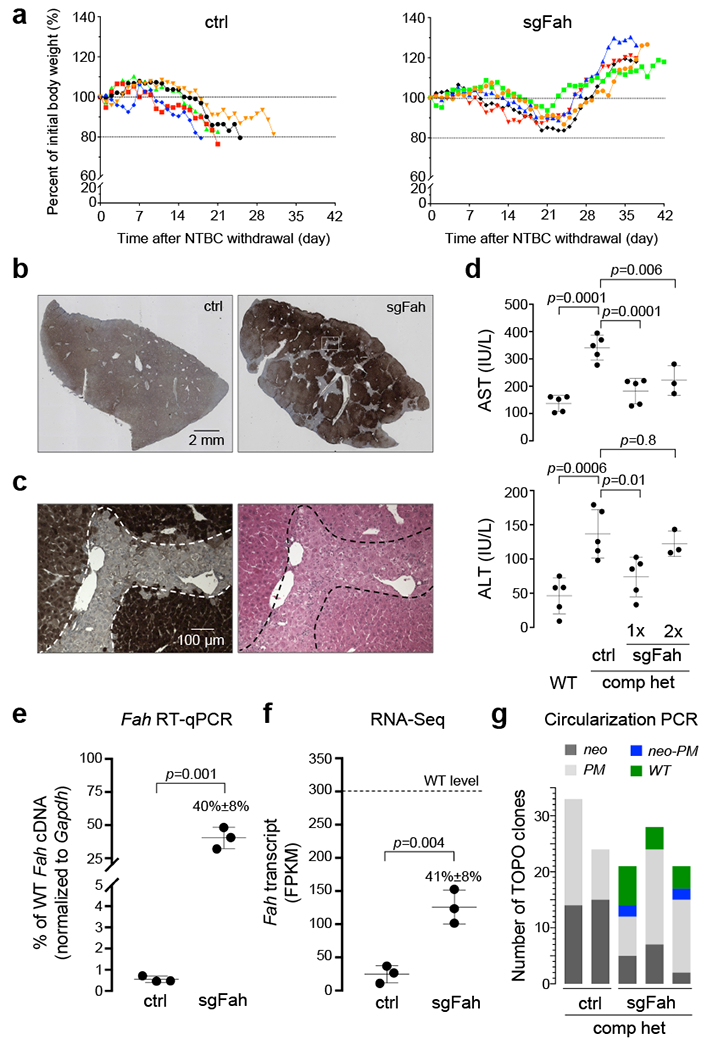
(a) Body weight curves after NTBC withdrawal at five weeks old. The body weight measured immediately prior to NTBC withdrawal was set as 100%, and relative weight in percentage over time was measured daily until euthanasia. N=5 mice per group. (b) Representative images of Fah IHC in liver sections from a mouse treated with ctrl rAAV, and a mouse treated with sgFah rAAV. These mice were treated at P1, maintained on NTBC water until five weeks old, and since then fed with regular water until euthanasia. N=5 mice per group. (c) Neighboring liver sections were used in FAH IHC and hematoxylin and eosin (HE) staining. The dashed lines mark the approximate border separating the corrected and non-corrected cells. N=5 mice per group. (d) Serum aspartate transaminase (AST) (top panel) and alanine transaminase (ALT) (bottom panel) levels in WT mice and compound heterozygous (comp het) mice treated with ctrl rAAV or sgFah rAAV. 1×: After one round of NTBC withdrawal; 2×: After two rounds of NTBC withdrawal. One-way ANOVA was performed (p<0.001), followed by multiple comparisons. P values are adjusted for multiple comparisons. (e) Reverse-transcription, quantitative PCR (RT-qPCR) to measure Fah mRNA in the liver, normalized to Gapdh mRNA. N=3 mice per group. P=0.001 by Student’s t-test (2-sided). Full-length image is shown in Supplementary Fig. 13a. (f) Fah mRNA abundance determined by Illumina sequencing of polyadenylated RNA. FPKM: fragments per kilobase of transcript per million mapped reads. N=3 mice per group. P=0.001 by Student’s t-test (2-sided). (g) Stacked histogram showing the number of TOPO clones obtained from circularization PCR and TOPO sequencing (see Supplementary Fig. 4 for detailed procedure). Each bar represents one mouse, either treated with ctrl rAAV and euthanized due to >20% body weight loss after NTBC withdrawal (ctrl), or treated with sgFah rAAV and euthanized after initial body weight loss was rescued (sgFah). TOPO reads indicative of different Fah alleles are color-coded. Note that the Fahneo-PM and FahWT alleles resulting from allelic exchange were detected only in the sgFah group, but not in the ctrl group. In all dot plots, each dot represents one mouse. Horizontal lines depict the mean values. Error bars represent SD.
We further tested the therapeutic efficacy of allelic exchange in a compound heterozygous mouse model of mucopolysaccharidosis type I (MPS-I) (Supplementary Fig. 7a-b), a lysosomal disease caused by the lack of α-L-iduronidase (IDUA) and the resulting accumulation of certain types of glycosaminoglycans (GAGs). Residual IDUA activity as low as 0.2% of normal level has been shown to attenuate the disease severity in MPS-I patients10. We detected allelic exchange in the heart at both the genomic DNA level (Supplementary Fig. 7c) and the cDNA level (Supplementary Fig. 7d). The apparent high level of restoration of the mutation-free Idua cDNA is likely due to the nonsense-mediated mRNA decay of the IduaW392X transcript11. In the compound heterozygous MPS-I mice, rAAV treatment at young adult age restored IDUA activity to ~0.5% of the wild-type level, and substantially reduced GAG accumulation in the heart (Supplementary Fig. 7e-f). Thus, Cas9-induced, allelic exchange has the potential for gene correction in post-mitotic tissues.
To test whether allelic exchange was the mechanism for disease correction in the compound heterozygous Fah mutant mice, we used a circularization PCR strategy to generate short amplicons containing the two original mutations; sequencing the amplicons readily differentiates between the original compound heterozygous and the exchanged alleles that carry either both or neither of the original mutations (Supplementary Fig. 8a). We detected exchanged alleles only in the sgFah-treated mice and not in the control mice (Fig. 2g and Supplementary Fig. 8b). The Fahneo/PM allele detected in the sgFah-treated mice surviving NTBC withdrawal is unlikely to have been generated by gene conversion (Supplementary Fig. 9a), because cells harboring such a Fah double-mutation allele would be lost during selection without the co-existence of a Fah functional allele. Indeed, a gene conversion experiment targeting Fah intron 9 did not restore FAH production (Supplementary Fig. 9b-e). Using a fluorescent reporter and a LIG4−/− cell line, we found that LIG4 (a key NHEJ enzyme) plays a role in DNA translocation in human cells (Supplementary Fig. 10). Although these experiments were performed with episomal plasmid DNA and in cancer cell lines, the results are consistent with previous reports that chromosomal translocation relies on an end-joining DNA repair mechanism12,13.
The mice homozygous for either Fah mutation were maintained on different genetic backgrounds that harbor different tyrosinase (Tyr) alleles in linkage with Fah (see Animal use section in Methods), allowing us to examine whether allelic exchange can occur between homologous chromosomes during cell division by analyzing the segregation pattern of the Tyr and Fah mutations (Supplementary Fig. 11a). This analysis indicates that allelic exchange may occur between homologous chromosomes during G2 phase, followed by X segregation during mitosis (i.e., the two exchanged alleles are segregated into different daughter cells) (Supplementary Fig. 11b). However, we cannot rule out that allelic exchange may also precede Z segregation (i.e., the two exchanged alleles segregate into the same daughter cell) or at G0/G1 phase.
Unlike allele-specific approaches that target specific disease-causing mutations, our allelic exchange approach has the potential to treat a large population of patients bearing different mutations of each disease-gene allele. Of course, more rare homozygous mutations must still be corrected by HDR14,15.
Several advantages distinguish our Cas9/sgRNA-mediated allelic exchange from other therapeutic in vivo genome-editing approaches such as HDR. First, a single validated sgRNA suffices to correct multiple distinct mutations. Second, the sgRNA can target an intronic site, avoiding the potentially disrupting effect of out-of-frame indels in the protein-coding sequence16 and increasing the range of potential target sites. In contrast, correction of a specific mutation by HDR requires that the sgRNA targeting site be close to the mutation, limiting sgRNA selection. Finally, allelic exchange uses the correct genetic information already present in the diploid genome, so no exogenous DNA repair template is needed. The polyploidy nature of some hepatocytes 17 may potentially enhance the exchange frequency due to the presence of multiple copies of homologous chromosomes and concurrent Cas9-mediated cleavages.
Although the exact DNA repair mechanism involved in Cas9/sgRNA-mediated allelic exchange is beyond the scope of this study, we postulate that it is likely the same as in general chromosomal translocation. Non-homologous end joining (NHEJ) has been shown to mediate chromosomal translocation in both human and mouse cells12,13,18. One limitation of our approach is that allelic exchange occurs at low frequency. For diseases such as HT1 in which corrected hepatocytes have a growth advantage allowing them to repopulate nearly the entire liver, the low frequency of allelic exchange poses no barrier to successful therapy. Such growth advantage of gene-modified cells has been recently reported for other diseases afflicting hepatocytes19. For diseases where genetic correction confers no growth advantage, an engineered system allowing selection of gene-modified cells could be used20.
METHODS
Animal use
The mice homozygous for the Fahneo or FahPM allele21,22 were kindly provided to us by Dr. Markus Grompe, and maintained on 129 background and C57 background, respectively. The C57 background harbors the TyrC mutation23 that causes albinism. Mice were fed with 10 mg/L NTBC [2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione] (Sigma-Aldrich, Cat. No. PHR1731-1G) in drinking water when indicated. HT1 mice of both sexes were used in experiments. The mice homozygous for the Iduaneo or IduaW392X allele11,24 were purchased from the Jackson Laboratory (Stock No. 004068 and 017681, respectively). Only male MPS-I mice were used in experiments. All animal procedures were reviewed and approved by The Institutional Animal Care and Use Committee (IACUC) at University of Massachusetts Medical School, and performed in compliance with all relevant ethical regulations.
Recombinant adeno-associated viral (rAAV) vectors
The mouse U1a promoter25 drives the expression of S. pyogenes Cas9 (SpCas9). This cassette was packaged into a single-stranded rAAV9 vector. The single guide RNAs (sgRNAs) targeting the mouse Fah intron 7 (sgFah), Fah intron 9 (sgFah.Intron9), Aspa intron 2 (sgAspa), and Idua intron 9 (sgIdua) are driven by U6 promoter. The sgRNA sequences are shown in Supplementary Fig. 12a. The U6-sgRNA cassette is followed by another cassette expressing enhanced green fluorescent protein (EGFP) driven by the cytomegalovirus-enhancer/chicken beta-actin promoter. These constructs were packaged into self-complementary vectors of AAV8 (scAAV8, for sgFah, sgFah.Intron9 and sgAspa) or scAAV9 (for sgIdua). rAAV vectors were produced by a triple-transfection method, purified by CsCl sedimentation and dialysis26. Titer of rAAV was determined by droplet digital PCR (for rAAV genome) and gel electrophoresis followed by silver staining (for rAAV capsid). Postnatal day 1 (P1) pups were injected with a mixture of rAAV-SpCas9 and scAAV-sgRNA at 1:1 ratio [4.0E+11 genome copies (GCs) in total] by facial vein injection. Six weeks old mice were injected with the same mixture of rAAV vectors of 2.0E+12 GCs in total by tail vein injection.
T7 endonuclease I (T7EI) assay
Tissues were powdered in liquid nitrogen, and an aliquot of tissue powder (~30 milligrams) was used to extract total DNA using the QIAamp DNA Mini Kit (Qiagen, Cat. No. 51306). PCR was carried out with the primers shown in Supplementary Fig. 12b using KOD Hot Start Master Mix (EMD Millipore, Cat. No. 71842-4) or KOD Xtreme Hot Start DNA Polymerase (Sigma-Aldrich, Cat. No. 71975-3). PCR products were purified using the QIAquick PCR Purification Kit (Qiagen, Cat. No. 28106). T7EI assay was performed accordingly to manufacturer’s instruction (NEB, Cat. No. M0302L).
TOPO sequencing
Purified PCR products were cloned into a TOPO vector (Life Technologies, Cat. No. 450245) to transform DH5α E. coli cells. Individual clones were expanded and subjected to plasmid extraction using QIAcube (Qiagen, Cat. No. 9001292), and Sanger sequencing using the M13F primer (Supplementary Fig. 12b).
Histological analysis
Mice were transcardially perfused with ice-cold PBS. Liver was dissected and fixed in 10% buffered formalin overnight. Immunohistochemistry (IHC) and hematoxylin and eosin (HE) stain were performed by the Morphology Core at University of Massachusetts Medical School using standard procedures. The FAH antibody used in IHC (Abcam, Cat. No. ab81087) was 1:400 diluted.
Reverse transcription PCR
An aliquot of tissue powder was used to extract total RNA using the Direct-zol RNA Miniprep kit (Zymo Research, Cat. No. R2052). RNA was reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Fisher Scientific, Cat. No. 43-688-13), and subjected to PCR with primers DW871 and DW874 (Supplementary Fig. 12b) using KOD Hot Start Master Mix (EMD Millipore, Cat. No. 71842-4). PCR products were resolved on agarose gel with ethidium bromide.
Droplet Digital PCR (ddPCR)
Mouse liver genomic DNA or cDNA was used in ddPCR to quantify rAAV-SpCas9 genome copy (GC) number, SpCas9 cDNA level, and neo gene copy number. SpCas9 GC number was determined in a multiplexed reaction using a customized TaqMan reagent targeting SpCas9 (ThermoFisher Scientific, Assay ID: AIAA0SW) and a commercial TaqMan reagent targeting Tfrc (ThermoFisher Scientific, Cat. No. 4458367). SpCas9 cDNA was determined in a multiplexed reaction using the same SpCas9 TaqMan reagent and a TaqMan reagent targeting Gusb (ThermoFisher Scientific, Assay ID: Mm01197698_m1). The neo gene copy number was quantified using an EvaGreen-based PCR (primers shown in Supplementary Fig. 12), and normalized to the copy number of Tfrc. DdPCR was performed with a QX200 ddPCR system (Bio-Rad).
Serum aspartate transaminase (AST), alanine transaminase (ALT) assay
Blood (~300 microliters) was drawn from facial vein immediately prior to euthanasia, and serum was isolated using a serum separator (BD, Cat. No. 365967) and stored under −80 °C until assay. AST and ALT levels were determined using commercial test kits per manufacturer’s instructions (TECO DIAGNOSTICS, Cat. No. A559150 and A524150).
Reverse transcription-quantitative PCR (RT-qPCR)
Liver cDNA was prepared as mentioned above, and used for qPCR. Two separate reactions were performed for each sample. One reaction is for Fah with primers DW884 and DW885 (Supplementary Fig. 12b) and iTaq Universal SYBR Green Supermix (Bio-Rad, Cat. No. 1725125). The other reaction is for Gapdh as normalization control using a pre-designed TaqMan reagent (Life Technologies, Cat. No. 4352339E).
Next generation RNA sequencing (RNA-Seq)
Liver total RNA was extracted using the mirVana miRNA Isolation Kit (Life Technologies, Cat. No. AM1561). Ribosomal RNA (rRNA) was depleted using a mixture of 186 DNA oligos targeting mouse rRNA and RNase H (Epicentre, Cat. No. H39500). RNA-Seq library was prepared using a method previously described27, and quantified using the KAPA Library Quantification Kit (Kapa Biosystems, Cat. No. KK4873). Equal amount of library material derived from each mouse was pooled and subjected to sequencing. Next generation sequencing was carried out using the Illumina NextSeq 500 System.
Iduronidase activity assay
Mice were transcardially perfused with ice-cold PBS, and tissues were immediately dissected, snap frozen in liquid nitrogen, and stored under −80 °C until assay. The iduronidase activity assay was performed using a protocol previously described11,28 with minor modifications. Tissues were homogenized in ice-cold T-PER protein extraction reagent (ThermoFisher Scientific, Cat. No. 78510) with protease inhibitor (Roche, Cat. No. 4693159001) using TissueLyser II (Qiagen). Supernatant was used to quantify total protein concentration using the BCA method (Pierce, Cat. No. 23225). No more than 80 μg of total protein was used in the enzymatic reaction (100 μL of total reaction volume), which includes sodium formate buffer, pH 3.5 (130 mM), D-saccharic acid 1,4-lactone monohydrate (0.42 mg/mL, Sigma-Aldrich, Cat. No. S0375), and 4MU-iduronic acid (0.12 mM, Gold Biotechnology, Cat. No. M-570-5). The reaction was incubated under 37°C for 24 to 48 hours, and quenched with glycine buffer, pH 10.8. The fluorescence of released 4MU (excitation wavelength: 365 nm; emission wavelength: 450 nm) was detected using a fluorescence plate reader (BioTek), and compared against a standard curve generated using 4MU (Sigma-Aldrich, Cat. No. M1381). The iduronidase specific activity was calculated as 4MU released (pmole) per milligram of total protein per hour.
Glycosaminoglycan (GAG) assay
Tissues were homogenized in a mixture of chloroform and methanol (2:1) using TissueLyser II (Qiagen), and dried in a Vacufuge (Eppendorf) to remove fat. The dried and defatted tissue was weighed, and digested using papain (Sigma-Aldrich, Cat. No. P3125) at 60°C overnight. The supernatant was used in the Blyscan assay to quantify GAG content using chondroitin-4-sulfate as standard (Accurate Chemical, Cat. No. CLRB1000). The GAG level was calculated as GAGs (microgram) per milligram of dried, defatted tissue.
Circularization PCR
Liver total DNA (5 micrograms) was digested with SphI-HF (NEB, Cat. No. R3182L) and ScaI-HF (NEB, Cat. No. R3122L) for one hour. The resulting DNA fragments were resolved on 0.8% agarose gel, and the DNA from the 3.0-4.0 kbp region was excised from the gel and purified using the QIAquick Gel Extraction Kit (Qiagen, Cat. No. 28706). DNA was end-blunted using DNA polymerase I, large (Klenow) fragment (NEB, Cat. No. M0210L), and underwent self-ligation using T4 DNA ligase (NEB, Cat. No. M0202T). Ligation product was purified using the QIAquick PCR Purification Kit (Qiagen, Cat. No. 28106), and subjected to PCR with primers DW888 and DW889 (Supplementary Fig. 12b) using KOD Hot Start Master Mix (EMD Millipore, Cat. No. 71842-4). PCR products were TOPO cloned and sequenced as mentioned above.
Cell culture and transfection
GreenGo29, HCT116 and LIG4−/− cells were cultured in Dulbecco’s Modified Eagle Media (DMEM, ThermoFisher Scientific, Cat. No. 11965084) with 10% fetal bovine serum (FBS, ThermoFisher Scientific, Cat. No. 10438034) under standard cell culture condition. Cells were transfected using Lipofectamine 2000 (ThermoFisher Scientific, Cat. No. 11668-019) according to manufacturer’s instruction.
Flow cytometry
HCT116 and LIG4−/− cells were suspended in Hank’s balanced salt solution (Gibco, Cat. No. 14175-079) containing 3% FBS, 1 mM EDTA, and 0.1 μg/mL DAPI (ThermoFisher Scientific, Cat. No. D1306). Flow cytometry was performed on a LSR II flow cytometer (BD Biosciences). 50,000 live cells were analyzed for each sample.
Quantification of mutation frequency
The neo gene copy number was determined by ddPCR as described above. Normalization to Tfrc gene copy number (two copies per diploid genome) allows for the calculation of neo frequency (i.e. neo copy number / Tfrc copy number). Short amplicons spanning the TyrC or FahPM mutation were generated by PCR using KOD Hot Start Master Mix (EMD Millipore, Cat. No. 71842-4). Amplicons generated from individual mice were labeled by using barcoded PCR primers (Supplementary Fig. 12). PCR products were purified using the QIAquick PCR Purification Kit (Qiagen, Cat. No. 28106). Purified amplicons from multiple mice were pooled, and the mixture was subjected to Single Molecule, Real-Time (SMRT) sequencing using an RSII Instrument (Pacific Biosciences), running the SMRT Analysis v2.3 software packages at the Deep Sequencing Core Facility at University of Massachusetts Medical School. Reads were de-multiplexed and aligned to custom reference sequences representing the Tyr or Fah amplicons using BWA-MEM on the Galaxy web-based platform for genome data analysis30–32. “Mutation frequencies” at each locus were reported as the percentage of aligned reads carrying the TyrC allele (G-to-C mutation) or the FahPM allele (G-to-A mutation) among all reads that correctly map to the respective Tyr or Fah references, as assessed on the Integrative Genomes Viewer (IGV, version 2.3.98)33.
Idua amplicon sequencing
To quantify the allelic exchange frequency at the genomic DNA level, a nested PCR was used to amplify a portion of the Idua gene spanning the loxP insertion and the W392X mutation site (primers are listed in Supplementary Fig. 12) (Supplementary Fig. 7). The bar-coded amplicons from six samples were pooled together and subjected to SMRT sequencing as mentioned above. Amplicons that contain both the loxP insertion and the W392X mutation are derived from the original IduaW392X allele, whereas the amplicons that contain the loxP insertion but not the W392X mutation represent the exchanged mutation-free allele. To examine the restoration of mutation-free Idua mRNA, a gene-specific primer was used to reverse-transcribe Idua mRNA into cDNA using the SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific, Cat. No. 18080051). A nested PCR was used to amplify a portion of the Idua cDNA spanning the neo insertion in exon 7 and the W392X mutation in exon 10 (primers are listed in Supplementary Fig. 12) (Supplementary Fig. 7). The bar-coded amplicons from six samples were pooled together and subjected to SMRT sequencing. Amplicons that contain the W392X mutation represent the cDNA expressed from the original IduaW392X allele, whereas the amplicons that contain neither the neo insertion nor the W392X mutation represent the cDNA expressed from the exchanged mutation-free allele.
Statistical analysis
Comparison between two groups were analyzed by Student’s t-test (2-sided). Comparison among three or more groups were analyzed by one-way ANOVA, followed by pair-wise comparison that was corrected for multiple comparisons. All statistical analysis was performed using Prism 7. More information about data analysis can be found in the Life Sciences Reporting Summary.
Data availability
SMRT sequencing data and RNA sequencing data can be found in the NCBI sequence read archive (SRA) under BioProject ID PRJNA472739. Other data supporting the findings of this study are available within the paper, or from the corresponding authors upon reasonable request. More information about data collection can be found in the Life Sciences Reporting Summary.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Markus Grompe (Oregon Health & Science University) for providing the homozygous Fahneo/neo and FahPM/PM mice, Dr. Liqun Luo (Stanford University) for providing the pGT and pTG plasmids (Addgene, #36885 and #36887), and Dr. Eric Hendrickson (University of Minnesota) for providing the HCT116 and LIG4−/− cells. This work was supported by grants from the National Institute of Health to G.G. (1P01AI100263, 1R01NS076991, 5P01HD080642, R01AI12135) and to W.X. (DP2HL137167, P01HL131471), a grant from the National High Technology Research and Development Program (“863” Program) of China to G.G. (2012AA020810), and a grant to W.X. from Hyundai Hope on Wheels. The authors thank Dr. Hao Yin and the members of Gao Lab and Xue Lab for helpful discussions.
Footnotes
COMPETING FINANCIAL INTERESTS
D.W. and G.G. have submitted a patent application concerning the methodology described in this study. P.D.Z. and G.G. are scientific co-founders of Voyager Therapeutics and hold equity in the company. P.D.Z. and G.G. are inventors on patents with potential royalties licensed to Voyager Therapeutics and other biopharmaceuticals.
REFERENCES
- 1.Bergman AJ, van den Berg IE, Brink W et al. Spectrum of mutations in the fumarylacetoacetate hydrolase gene of tyrosinemia type 1 patients in northwestern Europe and Mediterranean countries. Hum Mutat 12, 19–26 (1998). [DOI] [PubMed] [Google Scholar]
- 2.St-Louis M & Tanguay RM Mutations in the fumarylacetoacetate hydrolase gene causing hereditary tyrosinemia type I: overview. Hum Mutat 9, 291–299 (1997). [DOI] [PubMed] [Google Scholar]
- 3.Li P, Wood T & Thompson JN Diversity of mutations and distribution of single nucleotide polymorphic alleles in the human alpha-L-iduronidase (IDUA) gene. Genet Med 4, 420–426 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Choi PS & Meyerson M Targeted genomic rearrangements using CRISPR/Cas technology. Nature communications 5, 3728 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blasco RB, Karaca E, Ambrogio C et al. Simple and rapid in vivo generation of chromosomal rearrangements using CRISPR/Cas9 technology. Cell reports 9, 1219–1227 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Torres R, Martin MC, Garcia A et al. Engineering human tumour-associated chromosomal translocations with the RNA-guided CRISPR-Cas9 system. Nature communications 5, 3964 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Vanoli F, Tomishima M, Feng W et al. CRISPR-Cas9-guided oncogenic chromosomal translocations with conditional fusion protein expression in human mesenchymal cells. Proc Natl Acad Sci U S A 114, 3696–3701 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spraggon L, Martelotto LG, Hmeljak J et al. Generation of conditional oncogenic chromosomal translocations using CRISPR-Cas9 genomic editing and homology-directed repair. J Pathol 242, 102–112 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grompe M The pathophysiology and treatment of hereditary tyrosinemia type 1. Semin Liver Dis 21, 563–571 (2001). [DOI] [PubMed] [Google Scholar]
- 10.Bunge S, Clements PR, Byers S et al. Genotype-phenotype correlations in mucopolysaccharidosis type I using enzyme kinetics, immunoquantification and in vitro turnover studies. Biochim Biophys Acta 1407, 249–256 (1998). [DOI] [PubMed] [Google Scholar]
- 11.Wang D, Shukla C, Liu X et al. Characterization of an MPS I-H knock-in mouse that carries a nonsense mutation analogous to the human IDUA-W402X mutation. Mol Genet Metab 99, 62–71 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghezraoui H, Piganeau M, Renouf B et al. Chromosomal translocations in human cells are generated by canonical nonhomologous end-joining. Molecular cell 55, 829–842 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simsek D, Brunet E, Wong SY et al. DNA ligase III promotes alternative nonhomologous end-joining during chromosomal translocation formation. PLoS Genet 7, e1002080 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yin H, Xue W, Chen S et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol 32, 551–553 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yin H, Song CQ, Dorkin JR et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat Biotechnol 34, 328–333 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang Y, Wang L, Bell P et al. A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat Biotechnol (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang MJ, Chen F, Lau JTY & Hu YP Hepatocyte polyploidization and its association with pathophysiological processes. Cell Death Dis 8, e2805 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nussenzweig A & Nussenzweig MC Origin of chromosomal translocations in lymphoid cancer. Cell 141, 27–38 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borel F, Tang Q, Gernoux G et al. Survival Advantage of Both Human Hepatocyte Xenografts and Genome-Edited Hepatocytes for Treatment of alpha-1 Antitrypsin Deficiency. Mol Ther 25, 2477–2489 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nygaard S, Barzel A, Haft A et al. A universal system to select gene-modified hepatocytes in vivo. Sci Transl Med 8, 342ra379 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grompe M, al-Dhalimy M, Finegold M et al. Loss of fumarylacetoacetate hydrolase is responsible for the neonatal hepatic dysfunction phenotype of lethal albino mice. Genes Dev 7, 2298–2307 (1993). [DOI] [PubMed] [Google Scholar]
- 22.Aponte JL, Sega GA, Hauser LJ et al. Point mutations in the murine fumarylacetoacetate hydrolase gene: Animal models for the human genetic disorder hereditary tyrosinemia type 1. Proc Natl Acad Sci U S A 98, 641–645 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yokoyama T, Silversides DW, Waymire KG et al. Conserved cysteine to serine mutation in tyrosinase is responsible for the classical albino mutation in laboratory mice. Nucleic Acids Res 18, 7293–7298 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clarke LA, Russell CS, Pownall S et al. Murine mucopolysaccharidosis type I: targeted disruption of the murine alpha-L-iduronidase gene. Hum Mol Genet 6, 503–511 (1997). [DOI] [PubMed] [Google Scholar]
- 25.Bartlett JS, Sethna M, Ramamurthy L et al. Efficient expression of protein coding genes from the murine U1 small nuclear RNA promoters. Proc Natl Acad Sci U S A 93, 8852–8857 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao G & Sena-Esteves M in Molecular Cloning, Edn. 4th (eds. Green MR & Sambrook JR) 1209–1330 (Cold Spring Harbor Laboratory Press, Cold Spring Harbor; 2012). [Google Scholar]
- 27.Zhang Z, Theurkauf WE, Weng Z & Zamore PD Strand-specific libraries for high throughput RNA sequencing (RNA-Seq) prepared without poly(A) selection. Silence 3, 9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang D, Belakhov V, Kandasamy J et al. The designer aminoglycoside NB84 significantly reduces glycosaminoglycan accumulation associated with MPS I-H in the Idua-W392X mouse. Mol Genet Metab 105, 116–125 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanchez-Rivera FJ, Papagiannakopoulos T, Romero R et al. Rapid modelling of cooperating genetic events in cancer through somatic genome editing. Nature 516, 428–431 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goecks J, Nekrutenko A, Taylor J & Galaxy T Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol 11, R86 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giardine B, Riemer C, Hardison RC et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res 15, 1451–1455 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blankenberg D, Von Kuster G, Coraor N et al. Galaxy: a web-based genome analysis tool for experimentalists Current protocols in molecular biology / edited by Frederick M. Ausubel … [et al.] Chapter 19, Unit 19 10 11–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robinson JT, Thorvaldsdottir H, Winckler W et al. Integrative genomics viewer. Nat Biotechnol 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
SMRT sequencing data and RNA sequencing data can be found in the NCBI sequence read archive (SRA) under BioProject ID PRJNA472739. Other data supporting the findings of this study are available within the paper, or from the corresponding authors upon reasonable request. More information about data collection can be found in the Life Sciences Reporting Summary.