

Abstract
We have previously demonstrated that methylene blue (MB) counteracts the effects of hydrogen sulfide (H2S) cardiotoxicity by improving cardiomyocyte contractility and intracellular Ca2+ homeostasis disrupted by H2S poisoning. In vivo, MB restores cardiac contractility severely depressed by sulfide and protects against arrhythmias, ranging from bundle branch block to ventricular tachycardia or fibrillation. To dissect the cellular mechanisms by which MB reduces arrhythmogenesis and improves bioenergetics in myocytes intoxicated with H2S, we evaluated the effects of H2S on resting membrane potential (Em), action potential (AP), Na+/Ca2+ exchange current (INaCa), depolarization-activated K+ currents and ATP levels in adult mouse cardiac myocytes and determined if MB could counteract the toxic effects of H2S on myocyte electrophysiology and ATP. Exposure to toxic concentrations of H2S (100 μM) significantly depolarized Em, reduced AP amplitude, prolonged AP duration at 90% repolarization (APD90), suppressed INaCa and depolarization-activated K+ currents, and reduced ATP levels in adult mouse cardiac myocytes. Treating cardiomyocytes with MB (20 μg/ml) 3 min after H2S exposure restored Em, APD90, INaCa, depolarization-activated K+ currents, and ATP levels towards normal. MB improved mitochondrial membrane potential (Δψm) and oxygen consumption rate (OCR) in myocytes in which Complex I was blocked by rotenone. We conclude that MB ameliorated H2S-induced cardiomyocyte toxicity at multiple levels: (i) reversing excitation-contraction coupling defects (Ca2+ homeostasis and L-type Ca2+ channels); (ii) reducing risks of arrhythmias (Em, APD, INaCa and depolarization-activated K+ currents); and (iii) improving cellular bioenergetics (ATP, Δψm).
Keywords: sulfide toxicity, arrhythmogenesis, ion currents, patch-clamp
Introduction
The justification of the present study is the persistent risk of accidental, environmental and industrial H2S exposure (3, 21), and the use of sulfide as a method of suicide (56). One of the major toxic effects of H2S resides in its ability to dramatically depress ventricular contractility that can lead to pulseless electrical activity (PEA) within a few minutes (37, 43, 73). In addition, we have recently demonstrated in rats that H2S infusion led to frequent premature ventricular complexes, severe bradycardia, and in some instances, ventricular tachycardia or fibrillation (43). Finally, when a patient or an animal recovers from H2S-induced coma, the continued presence of cardiogenic shock has been shown to contribute to the development of diffuse cortical and subcortical neuronal necrosis. This effect is mediated by a reduction in cerebral blood flow often associated with a decrease in arterial partial pressure of O2 due to H2S-induced ventilatory depression (6), which potentiates the direct toxicity of sulfide on neurons.
We have recently reported that methylene blue (MB) exerts a significant salutatory effect on the immediate outcome of sulfide intoxication (70) ; largely accounted for by the very rapid counteraction by MB of H2S-induced depression in cardiac contractility in sedated or un-sedated rats (70) and in isolated cardiomyocytes (43).
MB is a redox molecule, which is reduced to leucomethylene blue (LMB) in the blood and in cells by NADH, NADPH (59) or reduced glutathione (44). LMB can provide electrons to a variety of oxidizing molecules (including O2 and metallo-compounds) and is re-oxidized back to MB in the process, allowing for a new cycle of reduction-oxidation to occur. The theoretical mechanisms by which MB/LMB counteract H2S toxicity include: (i) MB/LMB oxidation of ferrous iron into ferric iron present in various metallo-compounds. This occurs when high concentrations of MB (87) trap H2S and catalyzes its oxidation in a cyclic manner. Of note, LMB acts as a reducing agent on ferric iron, an effect typically observed with low concentrations of MB, a property used for the treatment of methemoglobinemia (91); (ii) direct oxidation of H2S by MB, a condition where H2S could become a reducing agent allowing MB to form LMB (58); (iii) restoration by LMB of the redox environment of cardiac ion channels altered directly or indirectly, i.e., though ROS production (99) or protein sulfhydration (93); and (iv) a direct effect of MB/LMB on the mitochondrial electron chain transport complexes (89, 94).
The purpose of the present study was to determine the cellular mechanisms by which MB reduces the risks of arrhythmogenesis in animals exposed to H2S; and to evaluate if MB confers beneficial effects on cellular bioenergetics in myocytes. Using adult mouse cardiomyocytes, we determined the effects of H2S on resting membrane potential (Em), action potential (AP), Na+/Ca+ exchange current (INaCa) and depolarization-activated K+ currents, and whether H2S-induced alterations in electrophysiological parameters can be ameliorated by MB, thereby accounting for the beneficial anti-arrhythmic effects of MB observed in vivo (43). To simulate a clinically relevant scenario, MB was administered after H2S exposure to isolated cardiomyocytes, at a time when signs of toxicity were already manifest. The effects of H2S, with and without MB, on myocyte ATP levels were then measured. Finally, as a first step to dissect the mechanisms by which MB improves cellular bioenergetics, we measured mitochondrial membrane potential (Δψm) and oxygen consumption rate (OCR) in adult myocytes in which the electron chain transport activity was inhibited and tested whether MB could restore this function.
Methods
Isolation of adult murine cardiac myocytes
Cardiac myocytes were isolated from the LV free wall and septum of mice according to the protocol of Zhou et al. (98) and modified by us (68, 69, 79, 82–84). Myocytes were used within 2-8 h of isolation.
Myocyte shortening measurements
Myocytes adherent to coverslips were bathed in 0.7 ml of air- and temperature-equilibrated (37°C), HEPES-buffered (20 mM, pH 7.4) medium 199 containing 1.8 [Ca2+]o. Myocytes were field stimulated to contract (2 Hz) between platinum wire electrodes spaced 2 mm apart. Images of myocytes viewed through an Olympus DApoUV x40/1.30 numerical aperture (NA) oil objective situated in a Zeiss IM35 inverted microscope were captured by a charge-coupled device video camera (Myocam; Ionoptix, Milton, MA). Edge detection algorithm was used to measure myocyte motion, and data were analyzed off-line by Ionoptix software as previously described (68, 69, 79, 82–84).
Electrophysiological measurements
Na+/Ca2+ exchanger current (INaCa)(68, 69, 82, 83, 95) and action potential (1 Hz)(68, 79, 82, 83) were measured in isolated LV myocytes (30°C) with whole cell patch-clamp. Fire-polished pipettes (tip diameter 4-6 μm) with resistances of 0.8-1.4 MΩ when filled with pipette solutions were used. Compositions of solutions and voltage protocols are given in Figure Legends.
For measurement of depolarization-activated K+ currents, only myocytes isolated from the LV free wall were used since the slow component of transient outward current (Ito,s) is absent in this subset of mouse myocytes (92). Pipette solution contained (in mM): 135 KCl, 1 CaCl2, 14 EGTA, 10 HEPES and 5 MgATP, pH 7.1. ATP was included to block ATP-sensitive outward K+ current. External solution contained (in mM): 132 NaCl, 5.4 KCl, 1.8 CaCl2, 1.8 MgCl2, 0.6 NaH2PO4, 10 HEPES, 0.5 CdCl2 and 10 glucose, pH 7.4. CdCl2 was added to block L-type Ca2+ current and INaCa. Holding potential was at −70 mV. Voltage-gated K+ currents (30°C) were evoked during 5 s depolarizing voltage steps (from −40 to +60 mV; 10 mV increments). Myocytes were returned to −70 mV for 200 ms prior to next voltage step (68). After identifying peak currents, the decaying phases of the currents were fitted with 2 exponentials of the form: A1e−t/τ1 + A2e−t/τ2 + Ass; where A1, A2 and Ass are the amplitudes of fast component of the rapidly inactivating transient outward current (Ito,f), slowly inactivating K+ current (IK,slow), and non-inactivating steady-state current (Iss), respectively; and τ1 and τ2 are the time constants of decay of Ito,f and IK,slow, respectively (92). Only values derived from curve fits with correlation coefficients of ≥ 0.97 were reported.
In vitro study protocol
For contraction experiments, at time zero, myocytes were exposed to either MB (0 to 500 μg/ml) or MB (0 to 500 μg/ml) + H2S (100 μM; prepared from NaHS) and contraction from 3-5 myocytes per glass coverslip was measured at 10 min. For electrophysiological experiments, either saline or NaHS (100 μM) was added at time zero. At 3 min., MB (20 μg/ml) or saline was added and INaCa, depolarization-activated K+ currents and AP were measured at 7 min.
Measurement of mitochondrial membrane potential (ΔΨm)
LV cardiac myocytes were suspended in an intracellular-like medium (ICM) containing (in mM): KCl 120, NaCl 10, KH2PO4 1, HEPES–Tris 20, thapsigargin (2 μg/ml), digitonin (80 μg/ml), pH 7.2; and protease inhibitors (EDTA-free complete tablets, Roche Applied Science)(40, 52). Permeabilized myocytes were supplemented with succinate (10 mM) and gently stirred. JC-1 (800 nM; Molecular Probes) was added to measure Δψm. Fluorescence signals were monitored in a temperature-controlled (37 °C) multiwavelength-excitation and dual wavelength-emission spectrofluorometer (Delta RAM, Photon Technology International), using 490-nm ex/535-nm em for the monomer and 570-nm ex/595-nm em for the J-aggregate of JC-1. Δψm was calculated as the ratio of the fluorescence of the JC-1 oligomeric to monomeric forms.
Measurement of mitochondrial O2 consumption and ATP levels
The oxygen consumption rate in intact adult LV myocytes was measured at 37°C in an XF96 extracellular flux analyzer (Seahorse Bioscience). Myocytes were seeded in laminin-coated wells and sequentially exposed to oligomycin, FCCP, and rotenone plus antimycin A, using the XF Cell Mito Stress Kit (Seahorse Bioscience) according to the manufacturer’s instructions (52). Preliminary experiments were performed to select optimal seeding density (104 cells/well) and compound concentrations, according to manufacturer’s instructions.
To measure ATP levels, isolated myocytes were lysed and ATP (luminescence) levels were measured using the CellTiter-Glo luminescent cell viability assay kit as described previously (41).
Statistics
All results are expressed as means ± SE. For analysis of INaCa and depolarization-activated K+ currents as a function of group (control, MB, H2S, and MB + H2S) and voltage, 2-way ANOVA was used. For analysis of action potential parameters, contraction amplitudes, ATP levels, ΔΨm and OCR, 1-way ANOVA was used. A commercially available software package (JMP version 12, SAS Institute, Cary, NC) was used. In all analyses, P<0.05 was taken to be statistically significant.
Ethics Statement
All protocols and procedures applied to the mice in this study were approved by the Institutional Animal Care and Use Committees of Temple University and The Pennsylvania State University.
Results
Rescue of H2S-induced myocyte contractile dysfunction by MB: dose response
We have previously demonstrated that MB (20 μg/ml) ameliorated myocyte contractile dysfunction after exposure to H2S (100 μM)(43). To determine the effective dose and to evaluate potential toxicity of MB, a dose response curve for MB was performed. MB alone at concentrations as high as 500 μg/ml had no negative inotropic effects after 10 min of exposure (Fig. 1). By contrast, myocyte contraction amplitudes decreased by ~50% (p<0.0001, control vs. H2S) 10 min after addition of H2S (100 μM)(Fig. 1), in agreement with our previous report (43). MB at 5 (p=0.0004, H2S vs. H2S + MB) but not at 1 μg/ml (p=0.2068, H2S vs. H2S + MB) significantly improved contractile dysfunction measured in H2S-treated myocytes, with full therapeutic benefit (p<0.11; control vs. H2S + MB) achieved at 10 μg/ml of MB (Fig, 1, inset).
Figure 1.
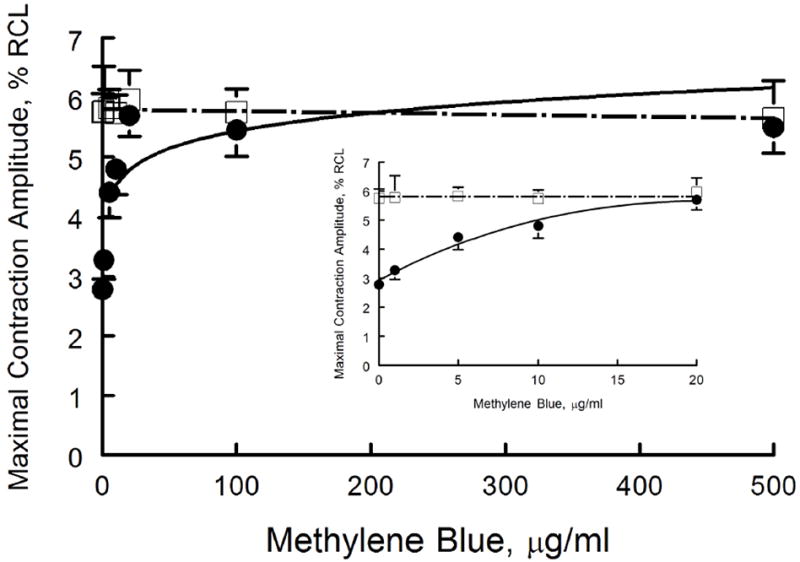
MB rescues contractile dysfunction of cardiomyocytes exposed to H2S: MB dose response. Freshly isolated myocytes from mouse LV and septum were plated on laminin-coated coverslips, bathed in medium 199 ([Ca2+]o 1.8 mM) and paced (2 Hz) to contract (37°C)(Methods). MB (0 to 500 μg/ml) or NaHS (100 μM) + MB (0 to 500 μg/ml) were added at time 0, and contractions were measured at 10 min. Maximal contraction amplitudes (% of resting cell length, %RCL) are shown for MB (□) and NaHS + MB (●) myocytes. For MB alone, there were 20, 7, 10, 6, 10, 8 and 6 myocytes at 0, 1, 5, 10, 20, 100 and 500 μg/ml, respectively. For NaHS + MB, there were 25, 8, 9, 8, 9, 7, and 8 myocytes at 0, 1, 5, 10, 20, 100 and 500 μg/ml of MB, respectively. Inset: expanded view of contractile response at MB doses from 0 to 20 μg/ml.
Effects of H2S on action potential: rescue by MB
Various abnormal cardiac rhythms were observed in rat (45) and sheep within minutes of exposure to H2S (72). These include frequent premature ventricular contractions, bradycardias, and less frequently, ventricular tachycardia and ventricular fibrillation. Since alterations in action potential morphology underlie the cellular basis of increased arrhythmogenesis, AP morphology and parameters in ventricular myocytes exposed to H2S ± MB were measured. Compared to control myocytes, exposure to H2S (100 μM) for 7 min resulted in Em depolarization from −76.1 ± 1.5 to −67.2 ± 1.1 mV (p<0.0015), reduction in AP amplitude from 130.9 ± 3.3 to 110.1 ± 2.5 mV (p<0.0015), and prolongation of APD90 from 43.6 ±3.8 to 81.3 ± 9.1 ms (p=0.005)(Fig. 2). APD50 tended to be prolonged by H2S although the differences did not reach statistical significance. Addition of MB (20 μg/ml) at 3 min had no significant effect on Em, AP amplitude, APD50 and APD90 compared to myocytes treated with saline (Fig. 2). Compared to myocytes exposed to H2S only, MB added 3 min after H2S addition significantly restored Em (p<0.01) and APD90 (p<0.02) but not AP amplitude (p<0.37) toward values observed in control myocytes (Fig. 2).
Figure 2.
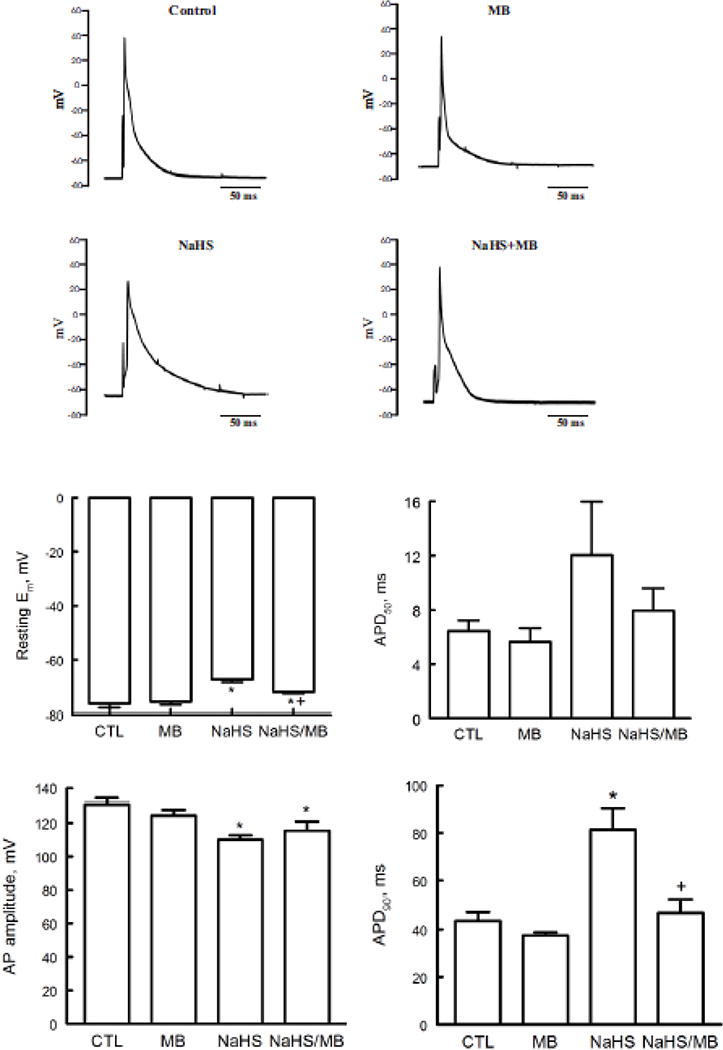
H2S depolarizes resting membrane potential (Em) and prolongs action potential duration (APD): rescue by MB. Em and APD were measured in myocytes isolated from mouse LV and septum with whole cell patch-clamp. Myocytes were paced at 1 Hz. Pipette solution consisted of (in mM) 125 KCl, 4 MgCl2, 0.06 CaCl2, 10 HEPES, 5 K+-EGTA, 3 Na2ATP, and 5 Na2-creatine phosphate (pH 7.2). External solution consisted of (in mM) 132 NaCl, 5.4 KCl, 1.8 CaCl2, 1.8 MgCl2, 0.6 NaH2PO4, 7.5 HEPES, 7.5 Na+-HEPES, and 5 glucose, pH 7.4. At time 0, either saline or NaHS (100 μM) was added followed by MB (20 μg/ml) or saline at 3 min before action potential was measured at 7 min. Top. Representative action potentials from myocytes treated with saline (control), MB alone, NaHS alone, and NaHS + MB recorded using current-clamp configuration at 1.5x threshold stimulus, 4-ms duration and at 30°C (75, 79, 96, 97). Bottom: Means ± SE of resting Em, action potential amplitude, action potential duration at 50% (APD50) and at 90% repolarization (APD90) from 5 control, 4 MB, 5 NaHS and 4 NaHS + MB myocytes are shown. * P<0.045, control vs. NaHS or NaHS + MB; + P<0.02, NaHS vs. NaHS + MB.
Effects of H2S on INaCa and depolarization-activated K+ currents: rescue by MB
Action potential duration is predominantly determined by L-type Ca2+ current (ICa), INaCa and depolarization-activated K+ currents. We have previously demonstrated that H2S significantly depressed ICa and that MB added 3 min after H2S exposure was effective in restoring ICa towards normal (43). Exposure to H2S (100 μM) for 7 min significantly (p<0.0001; group × voltage interaction effect) suppressed INaCa when compared to control myocytes treated with saline (Fig. 3). Addition of MB 3 min after H2S exposure significantly (p<0.0001) ameliorated the toxic effects of H2S on INaCa (H2S vs. H2S + MB; Fig. 3) although not to the normal control values (p<0.0012; control vs. H2S + MB).
Figure 3.
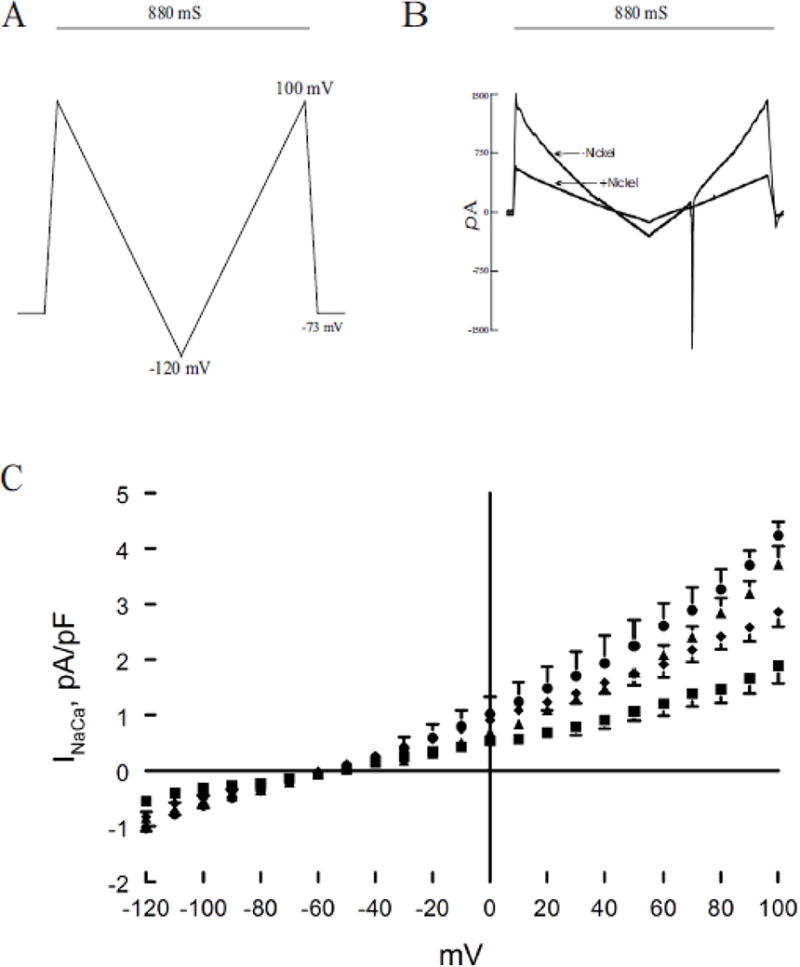
H2S inhibits Na+/Ca2+ exchanger current (INaCa): reversal by MB. Pipette solution contained (in mM) 100 Cs+ glutamate, 7.25 NaCl, 1 MgCl2, 20 HEPES, 2.5 Na2ATP, 10 EGTA and 6 CaCl2, pH 7.2. Free Ca2+ in the pipette solution was 205 nM, measured fluorimetrically with fura-2. External solution contained (in mM) 130 NaCl, 5 CsCl, 1.2 MgSO4, 1.2 NaH2PO4, 5 CaCl2, 10 HEPES, 10 Na+ HEPES and 10 glucose, pH 7.4. Verapamil (1 μM) was used to block ICa,L. Our measurement conditions were biased towards measuring outward (3 Na+ out: 1 Ca2+ in) INaCa. (A). After holding the myocyte at the calculated reversal potential (−73 mV) of INaCa for 5 min (to minimize fluxes through Na+/Ca2+ exchanger and thus allowed [Na+]i and [Ca2+]i to equilibrate with those in pipette solution), INaCa (30°C) was measured in myocytes using a descending (from +100 to −120 mV; 500 mV/s) – ascending (from −120 to +100 mV; 500 mV/s) voltage ramp, first in the absence and then in the presence of 1 mM NiCl2. (B). Raw currents measured in a WT myocyte. INaCa was defined as the difference current measured in the absence and presence of Ni+ during the descending voltage ramp. Note that with the exception of small contamination of the ascending ramp by the cardiac Na+ current, there were little to no differences in INaCa measured between the descending and ascending voltage ramps. This suggests that [Ca2+]i and [Na+]i sensed by Na+/Ca2+ exchanger did not appreciably change by ion fluxes during the brief (880 ms) voltage ramp. INaCa was divided by Cm prior to comparisons. (C). At time 0, either saline or NaHS (100 μM) was added followed by MB (20 μg/ml) or saline at 3 min before INaCa was measured at 7 min. Current-voltage relationships of INaCa (means ± SE) from control (▲; n=6), MB (●; n=3), NaHS (■; n=5) and NaHS + MB (◆; n=5) myocytes are shown. The reversal potential of INaCa was ~ −60 mV, close to the theoretical reversal potential of −73 mV. Error bars are not shown if they fall within the boundaries of the symbol.
Peak amplitudes of depolarization-activated K+ currents (Ipeak) were significantly (p<0.0001; H2S vs. control, group × voltage interaction effect) depressed by H2S but were rescued by addition of MB 3 min post H2S exposure (p<0.0003; H2S vs. H2S + MB) (Fig. 4). When depolarization-activated K+ currents were segregated into their respective components (68, 92), H2S consistently decreased Ito,f, IK slow, and Iss (Fig. 4; p<0.025, control vs. H2S). MB added 3 min after H2S exposure restored Ipeak, Ito,f, IK,slow and Iss towards values measured in control myocytes exposed to saline (Fig. 4; p<0.05, H2S vs. H2S + MB). MB alone had no effects on Ipeak, Ito,f, IK,slow and Iss (Fig. 4; p<0.85; control vs. MB). The time constants of decay (at +40 mV) for Ito,f (p<0.45) and IK,slow (p<0.40) were not affected by either H2S or MB.
Figure 4.
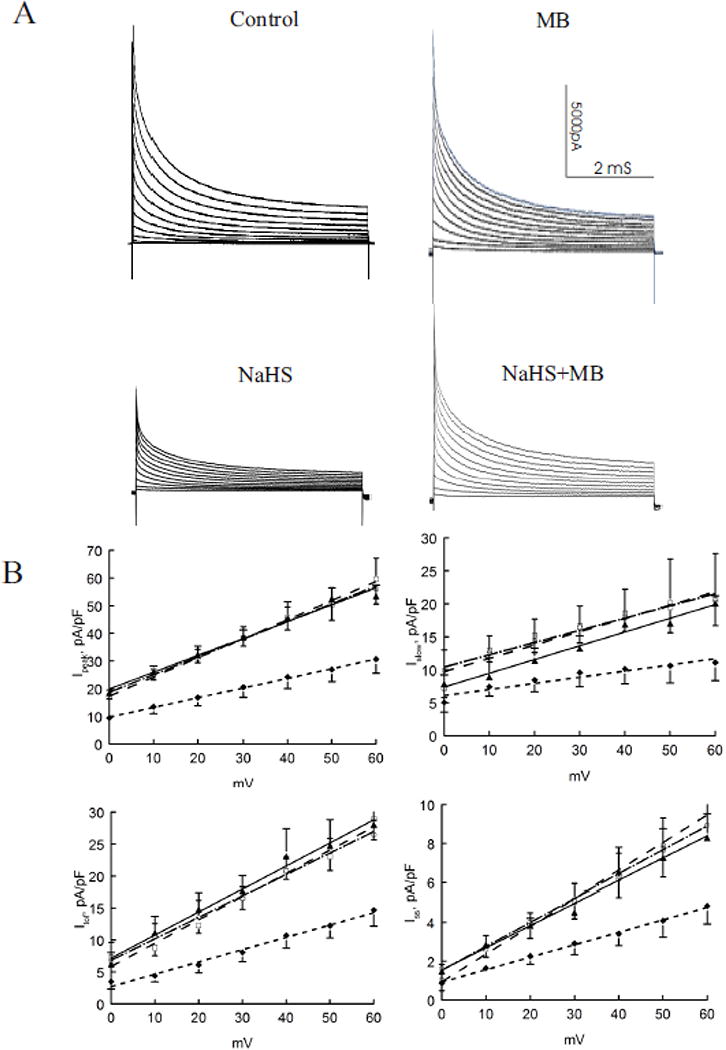
H2S decreases depolarization-activated K+ currents: rescue by MB. Depolarization-activated K+ currents (30°C, 135 mM [K+]i) were measured in control (□; n=3), MB (▲; n=4), NaHS (◆; n=4) and NaHS + MB (○; n=3) myocytes isolated from the LV free wall (Methods). Top. Raw tracings of depolarization-activated K+ currents from control, MB, NaHS and NaHS + MB myocytes. K+ currents were separated into 3 components (Methods). Bottom. Current-voltage relationships of peak currents, fast component of transient outward currents (Ito,f), slowly inactivating K+ currents (IK,slow) and steady-state non-inactivating K+ currents (Iss) are shown. Values are means ± SE. Error bars are not shown if they fall within the boundaries of a symbol. Data for K+ currents are fitted by linear regression.
Effects of MB on cellular ATP and cardiac mitochondrial respiration
Improved cellular bioenergetics is a potential mechanism by which MB ameliorated contractile dysfunction in myocytes exposed to H2S (Fig. 1). Indeed, ATP levels were depressed after 10 min of exposure to H2S but rescued by MB (Fig. 5A). Since ATP is primarily synthesized via oxidative phosphorylation in cardiac myocytes, the effects of MB on mitochondrial function were measured. As shown in Fig. 5B, collapse of mitochondrial membrane potential (Δψm) by the Complex I inhibitor rotenone (10 nM) was partially restored by MB (20 μg/ml) although addition of MB alone had no effect on Δψm. MB prevented the decrease in mitochondrial oxygen consumption rate (OCR) induced by rotenone and the Complex III inhibitor antimycin A (1 μM)(Fig. 5C). One interesting observation is that OCR was higher in myocytes exposed to MB, regardless of presence or absence of inhibitors of mitochondrial electron transport (Fig. 5C). In the absence of cardiomyocytes, neither MB alone nor MB + NaHS had any significant OCR (data not shown).
Figure 5.
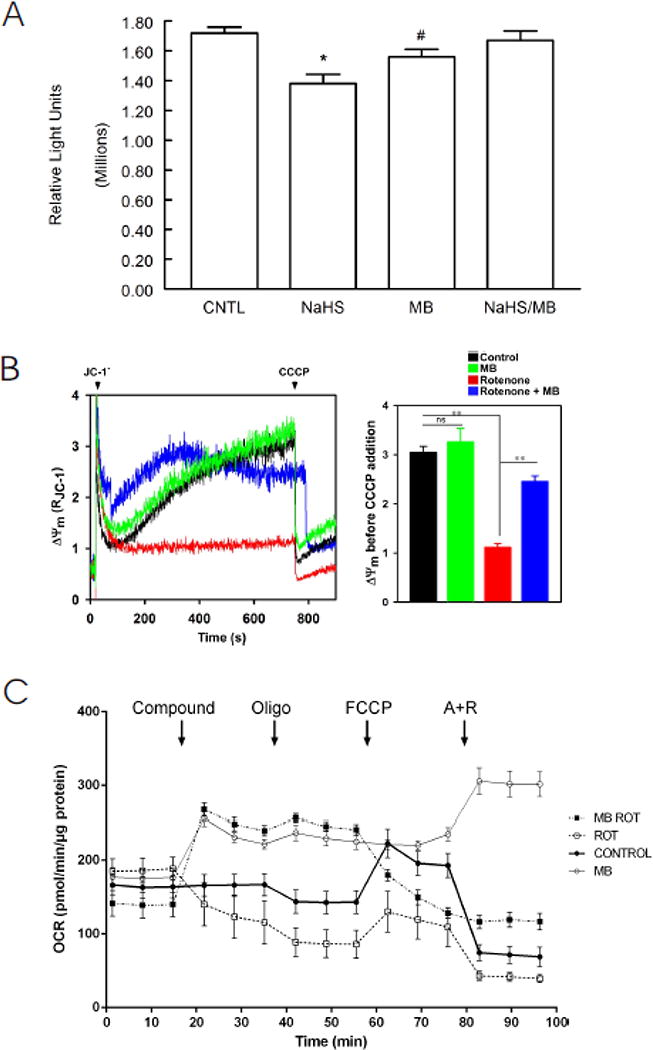
Effects on MB on cardiomyocyte bioenergetics: ATP levels, mitochondrial membrane potential (Δψm), and O2 consumption rate (OCR). (A). ATP (luminescence, relative light units) levels from LV myocytes treated with saline (CNTL, n=8), MB (n=8), NaHS (n=7) and MB + NaHS (n=7) for 10 min were determined with CellTiter-Glo luminescent cell viability kit (Methods). *p<0.0005, CNTL vs. NaHS; #p<0.03; CNTL vs. MB. There are no differences in ATP levels between CNTL and NaHS + MB myocytes (p=0.53). (B). LV myocytes were permeabilized with digitonin and supplemented with succinate. Left: the ratiometric indicator JC-1 was added at 20s and used to monitor Δψm. Arrows indicate addition of JC-1 and the mitochondrial uncoupler CCCP (2 μM), respectively. Rotenone (10 nM) and/or MB (20 μg/ml) were added at time 0. Right: Summary of Δψm before CCCP addition (n=3 each). **p<0.01; ns, not significant. (C). OCR was measured in intact myocytes (Methods). After basal OCR was obtained, either saline control (●), MB alone (20 μg/ml; ○), rotenone (1 μM; □), or MB (20 μg/ml) + rotenone (■) were added (indicated by “Compound”). At times indicated, oligomycin (1 μM) was added to inhibit F0F1ATPase (Complex V) followed by addition of the uncoupler FCCP (1 μM) to measure maximal OCR. Finally, antimycin A + rotenone (1 μM each) were added to inhibit cytochrome bc1 complex (Complex III) and NADH dehydrogenase (Complex I), respectively. Each point in the traces represents the average of 8 different wells. This experiment was repeated 3 times with similar results.
Discussion
The major finding of the present study is that H2S greatly affected the electrophysiology of isolated cardiomyocytes beyond the inhibition of L-type Ca2+ currents (ICa,L) that we and others have previously reported (43, 74, 93). A significant alteration of Na+/Ca2+ exchange and depolarization-activated K+ currents was effected by toxic level of H2S, i.e., at concentrations that in vivo lead to lethal cardiogenic shock and several types of arrhythmias (73). MB was capable of restoring, for most part, normal cardiomyocyte contractile and electrophysiological activity.
On the use of high μM H2S in solution to study toxicity in isolated cardiac myocytes
Typically, whenever solutions of H2S at concentrations ≥ 50 μM were used in isolated hearts or isolated cells, the effect was a measurable depression in cardiac contractility (43, 74). This depression is an obvious pathological and toxic change in the function at the level of both individual myocytes and the intact heart. In vitro, the activity of the mitochondrial cytochrome c oxidase is abolished by a solution of H2S at concentrations of H2S/HS− ranging from 10 to 30 μM (17, 49). In vivo, severe depression in cardiac contractility can be produced in rodents and in large mammals by infusing or inhaling H2S at levels yielding blood concentrations of gaseous H2S between 2–5 μM (38, 46, 71, 73), corresponding to level of total dissolved/free sulfide of < 20 μM. In a recent study, we found that the ejection fraction of the left ventricle started to decrease for an average level of gaseous H2S of 3 μM, while blood pressure was decreased ~ 5 μM. This corresponds to a total level dissolved H2S/HS− of about 15–20 μM (assuming that 2/3 of total dissolved sulfide present is in the form of HS− at physiological pH). Therefore, the H2S concentration used in our present study was consistent with previous studies to produce toxic effects on the cardiomyocyte.
Early studies reported H2S concentrations in the blood or in the tissues in the high μM range and was assumed to be due to endogenous H2S production only (24). Subsequent work (50, 88) demonstrated that the high μM concentrations of free/soluble H2S are unrealistically high by several orders of magnitude. Two main hypotheses have been advanced to explain why high μM levels of “endogenous” H2S were initially (and are still) reported in the blood and tissues. The first reason relates to the nature of the pools of sulfide present in the blood and in tissues found in post-mortem conditions. No relevant (or trivial) levels of free/dissolved H2S, which should smell like rotten eggs, can be found in the blood, the brain (24, 50) or heart homogenates (50). However, significant amount of H2S can be mobilized after exposing these tissues to a strong acid (lowering the pH of the tissues to <2) and can be measured after evaporating from the brain or the heart (50, 55, 80). This pool of H2S represents several micromoles of sulfide per kg of tissue (26, 42, 50, 55, 80), but is “trapped”, literally fossilized, in the form of metallo-sulfides (including Fe2+, Zn2+, Co2+, etc.). Similarly, strong reducing agents can release H2S from a solution of proteins or from various tissues, where H2S is present under the form protein-bound thiols (55, 85). The presence of H2S evaporating after such reactions should not be confused with the presence of dissolved H2S, able to freely diffuse and to potentially act as a gaseous-transmitter. We have discussed the complex issues regarding solutions containing H2S (prepared from NaHS) and H2S in tissues in a recent review (33).
H2S poisoning induced cardiac toxicity
H2S is commonly referred to as a mitochondrial Complex IV inhibitor, mimicking the effects of cyanide or sodium azide, by impeding the activity of the mitochondrial cytochrome C oxidase (17, 19, 45). Our results that H2S inhibited various cardiac ionic currents (ICa,L (43), INaCa and depolarization-activated K+ currents) suggest that blockade of the mitochondrial cytochrome C oxidase may not be the sole mechanism of sulfide toxicity. H2S depressed the rapidly activating and inactivating transient outward current (Ito,f), the rapidly activating but slowly inactivating delayed rectifier K+ current (IK,slow), and the fast activating and non-inactivating steady-state K+ current (Iss). In addition, the depolarized resting membrane potential and reduced action potential amplitude in ventricular cells exposed to H2S are consistent with suppression of the inwardly rectifying Kir currents (IK1) and fast Na+ current INa, respectively. The effects of H2S on APD50 are complicated since it suppresses both ICa,L (shortens APD50) and Ito,f (prolongs APD50). Our observation that H2S tended to prolong APD50 suggests that its effects on Ito,f are dominant. Similar considerations apply to the toxic effects of H2S on INaCa (shortens APD90) and IK,slow (prolongs APD90). Our observation that APD90 was significantly prolonged supports the view that the effects of H2S on depolarization-activated K+ currents on APD90 are dominant. Wei and al. (86), using cardiomyocytes of ventricular, atrial and nodal subtypes differentiated from H9 embryonic stem cells and human induced pluripotent stem cells, have reported similar effects of high concentrations of H2S (100 to 300 μM) on APD, ICa,L, IK,slow, the rapidly activating and inactivating delayed rectifier K+ current (IK,rapid), and the hyperpolarization-activated inward current (If) found in sinoatrial nodal cells.
Since one of the primary functions of Na+/Ca2+ exchanger in adult cardiomyocytes is to extrude during diastole the amount of Ca2+ that has entered during systole via the L-type Ca2+ channel, thereby maintaining steady-state beat-to-beat Ca2+ balance (9), inhibition of Na+/Ca2+ exchanger by H2S would be expected to result in Ca2+ overload and progressive elevation of diastolic [Ca2+]i. However, diastolic [Ca2+]i was similar between control and H2S-toxic myocytes (43). Absence of overt Ca2+ overload in H2S intoxicated cardiomyocytes is likely due to the simultaneous inhibition of L-type Ca2+ channels by H2S, thereby limiting Ca2+ influx during systole and maintaining steady-state Ca2+ balance. This parallels the situation in adult cardiac-specific Na+/Ca2+ exchanger knockout ventricular myocytes in which L-type Ca2+ channel activity was substantially reduced when compared to wild-type myocytes (39).
The mechanisms by which most channels are affected at the same time remain undetermined. As recently proposed for Ca2+ channels, H2S could react with cysteine residues not only in L-type Ca2+ channels (74, 93), but also the sarcoplasmic reticulum ryanodine receptors (RyR), thereby altering the 3D configurations and function of the channels. Whether other cardiac channels can be affected through a similar mechanism remains unknown. H2S induced inhibition of the mitochondrial electron transport chain has been shown to result in an increase in ROS production (12) which could also contribute to this effect (99) through alterations in the redox environment of these ion channels.
MB as a treatment of H2S intoxication
H2S is the second most common cause of death by gas exposure in the workplace after carbon monoxide (8, 23, 28, 57). H2S is a primary chemical hazard in oil and gas production, well drilling and gas refining industries (3, 21), and a significant occupational hazard during various farming activities (14). H2S can be “weaponized”, as demonstrated by British troops during World War I (22). It is also used as a method of suicide (11). This form of suicide has increased in an alarming manner in the US (30) and is accomplished by mixing a source of sulfide and various types of acidic solutions readily available in most household chemicals. This has created major challenges for first responders (56, 78).
As soon as it enters the blood, only a very small portion of sulfide remains in a “free/soluble” or diffusible form, comprising the gaseous form H2S (7, 13, 18, 20) and the sulfhydryl anion HS− (2, 53). A larger pool of sulfide will combine with metallo-proteins such as the ferrous iron in hemoglobin or will react with cysteine residues present in proteins (29, 63, 66, 90), creating a large sink for H2S. While free/soluble H2S is the only form able to diffuse into the cells, bound H2S represents the toxic pool (38, 46). The most remarkable feature of H2S metabolism is that H2S disappears from the blood and the tissues (29, 46, 76) at a very rapid rate. Indeed H2S is almost immediately oxidized in the mitochondria, a reaction catalyzed by various enzymes such as the sulfide quinone oxido-reductase, sulfur dioxigenase and the sulfur transferase enzyme rhodenase (12, 48, 49, 54). We found that this oxidation can account for the disappearance of soluble H2S following sulfide poisoning, within one minute in large and small mammals (38, 46), while the form “hidden” in disulfide bonds persists for a much longer period of time (46) in the tissues (85). As a consequence, the toxic effects of H2S persist beyond the phase of exposure without being accessible to antidotes, which primarily act on the soluble form.
The challenge is therefore to find compounds antagonizing the effects of H2S toxicity while the pool of exchangeable sulfide is already gone. The treatment of H2S poisoning has been traditionally aimed at trapping free H2S using metallo-compounds, e.g. ferric iron contained in methemoglobin (15, 36, 81) or cobalt in hydroxycobalamin (HyCo) (35, 62, 64, 77, 81). Other antidotes are based on empirical observations, such as sodium bicarbonate (1, 27) and hyperoxia (10, 34, 60, 67) with no proven efficacy. Nitrite-induced methemoglobinemia (47, 61–63, 66, 81) however can only trap H2S outside the cells and has little or no effects on the combined forms after exposure (38). Sodium nitrite further decreases arterial blood pressure in subjects already in shock (31, 32) and affects oxygen transport (4, 65, 66). Cobalt contained in HyCo (81) has several theoretical advantages over methemoglobinemia (51, 77). Whether this very large molecule can penetrate cells at sufficient concentrations and rapidly enough (5) to neutralize the effects of H2S remains to be demonstrated.
Although methylene blue (MB) is an old compound (16, 25), its use in H2S intoxication represents a novel and very promising paradigm, as it counteracts the consequences of H2S intoxication. Our data demonstrate that MB is effective even when given after H2S exposure provide support for its use as an emergency treatment in the most severe forms of H2S-induced coma with shock but also in non-life threatening conditions.
CONCLUSIONS
MB ameliorated changes in Em, APD90, INaCa and depolarization-activated K+ currents in cardiac myocytes exposed to H2S. In addition, MB restored depressed ATP levels after H2S exposure. Our data support the hypothesis that MB exerts a potent antidotal effect during H2S intoxication through restoration of cardiac function depressed by sulfide. The redox properties of the couple MB/LMB may exert widespread effects ranging from modification of the activity of metallo-proteins or sulfhydrated proteins, to the alteration of essential metabolic pathways.
Acknowledgments
This work has been partially supported by NIH RO1-HL123093, RO1-HL137426, UO1-NS097162, R21-NS098991, and American Heart Association Grant-in-Aid 15GRNT25680042.
Footnotes
Disclosures
No conflict of interest, financial and otherwise, is declared by the authors.
References
- 1.Almeida AF, Guidotti TL. Differential sensitivity of lung and brain to sulfide exposure: a peripheral mechanism for apnea. Toxicol Sci. 1999;50:287–293. doi: 10.1093/toxsci/50.2.287. [DOI] [PubMed] [Google Scholar]
- 2.Almgren T, Dyrssen D, Elgquist B, Johannsson O. Dissociation of hydrogen sulfide in seawater and comparison of pH scales. Marine Chemistry. 1976;4:289–297. [Google Scholar]
- 3.Arnold IM, Dufresne RM, Alleyne BC, Stuart PJ. Health implication of occupational exposures to hydrogen sulfide. J Occup Med. 1985;27:373–376. doi: 10.1097/00043764-198505000-00018. [DOI] [PubMed] [Google Scholar]
- 4.Ash-Bernal R, Wise R, Wright SM. Acquired methemoglobinemia: a retrospective series of 138 cases at 2 teaching hospitals. Medicine (Baltimore) 2004;83:265–273. doi: 10.1097/01.md.0000141096.00377.3f. [DOI] [PubMed] [Google Scholar]
- 5.Astier A, Baud FJ. Complexation of intracellular cyanide by hydroxocobalamin using a human cellular model. Human & experimental toxicology. 1996;15:19–25. doi: 10.1177/096032719601500104. [DOI] [PubMed] [Google Scholar]
- 6.Baldelli RJ, Green FH, Auer RN. Sulfide toxicity: mechanical ventilation and hypotension determine survival rate and brain necrosis. Journal of applied physiology. 1993;75:1348–1353. doi: 10.1152/jappl.1993.75.3.1348. [DOI] [PubMed] [Google Scholar]
- 7.Barrett TJ, Anderson GM, Lugowski JT. The solubility of hydrogen sulphide in 0-5 m NaCl solutions at 25-95 C and one atmosphere. Geochimica et Cosmochimica Acta. 1988;52:807–811. [Google Scholar]
- 8.Beauchamp RO, Jr, Bus JS, Popp JA, Boreiko CJ, Andjelkovich DA. A critical review of the literature on hydrogen sulfide toxicity. Crit Rev Toxicol. 1984;13:25–97. doi: 10.3109/10408448409029321. [DOI] [PubMed] [Google Scholar]
- 9.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 10.Bitterman N, Talmi Y, Lerman A, Melamed Y, Taitelman U. The effect of hyperbaric oxygen on acute experimental sulfide poisoning in the rat. Toxicol Appl Pharmacol. 1986;84:325–328. doi: 10.1016/0041-008x(86)90140-7. [DOI] [PubMed] [Google Scholar]
- 11.Bott E, Dodd M. Suicide by hydrogen sulfide inhalation. The American journal of forensic medicine and pathology. 2013;34:23–25. doi: 10.1097/PAF.0b013e31827ab5ad. [DOI] [PubMed] [Google Scholar]
- 12.Bouillaud F, Blachier F. Mitochondria and sulfide: a very old story of poisoning, feeding, and signaling? Antioxid Redox Signal. 2011;15:379–391. doi: 10.1089/ars.2010.3678. [DOI] [PubMed] [Google Scholar]
- 13.Carroll JJ, Mather AE. The solubility of hydrogen sulfide in water from 0 to 90°C and pressures to 1 MPa. Geochim Cosmochim Acta. 1989;53:1163–1170. [Google Scholar]
- 14.Chenard L, Lemay SP, Lague C. Hydrogen sulfide assessment in shallow-pit swine housing and outside manure storage. Journal of agricultural safety and health. 2003;9:285–302. doi: 10.13031/2013.15458. [DOI] [PubMed] [Google Scholar]
- 15.Chenuel B, Sonobe T, Haouzi P. Effects of infusion of human methemoglobin solution following hydrogen sulfide poisoning. Clin Toxicol (Phila) 2015;53:93–101. doi: 10.3109/15563650.2014.996570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clifton J, 2nd, Leikin JB. Methylene blue. American journal of therapeutics. 2003;10:289–291. doi: 10.1097/00045391-200307000-00009. [DOI] [PubMed] [Google Scholar]
- 17.Cooper CE, Brown GC. The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: chemical mechanism and physiological significance. J Bioenerg Biomembr. 2008;40:533–539. doi: 10.1007/s10863-008-9166-6. [DOI] [PubMed] [Google Scholar]
- 18.De Bruyn WJ, Swartz E, Hu JH, Shorter JA, Davidovits P, Worsnop DR, Zahniser S, Kolb CE. Henry’s law solubilities and Setchenow coefficients for biogenic reduced sulfur species obtained from gas-liquid uptake measurements. Journal of Geophysical research. 1995;100:7245–7251. [Google Scholar]
- 19.Dorman DC, Moulin FJ, McManus BE, Mahle KC, James RA, Struve MF. Cytochrome oxidase inhibition induced by acute hydrogen sulfide inhalation: correlation with tissue sulfide concentrations in the rat brain, liver, lung, and nasal epithelium. Toxicol Sci. 2002;65:18–25. doi: 10.1093/toxsci/65.1.18. [DOI] [PubMed] [Google Scholar]
- 20.Douabul AA, Riley JP. The solubility of gases in distilled water and seawater – V. Hydrogen sulphide. Deep-Sea Research. 1979;26A:259–268. [Google Scholar]
- 21.EPA. Toxicological Review of Hydrogen Sulfide (CAC No 7783-06-04) Washington DC: United States Environmental Protection Agency; 2003. [Google Scholar]
- 22.Foulkes CH. Gas!” The story of the special brigade. Blackwood & Sons. 1934 [Google Scholar]
- 23.Fuller DC, Suruda AJ. Occupationally related hydrogen sulfide deaths in the United States from 1984 to 1994. J Occup Environ Med. 2000;42:939–942. doi: 10.1097/00043764-200009000-00019. [DOI] [PubMed] [Google Scholar]
- 24.Furne J, Saeed A, Levitt MD. Whole tissue hydrogen sulfide concentrations are orders of magnitude lower than presently accepted values. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1479–1485. doi: 10.1152/ajpregu.90566.2008. [DOI] [PubMed] [Google Scholar]
- 25.Ginimuge PR, Jyothi SD. Methylene blue: revisited. Journal of anaesthesiology, clinical pharmacology. 2010;26:517–520. [PMC free article] [PubMed] [Google Scholar]
- 26.Goodwin LR, Francom D, Dieken FP, Taylor JD, Warenycia MW, Reiffenstein RJ, Dowling G. Determination of sulfide in brain tissue by gas dialysis/ion chromatography: postmortem studies and two case reports. J Anal Toxicol. 1989;13:105–109. doi: 10.1093/jat/13.2.105. [DOI] [PubMed] [Google Scholar]
- 27.Guidotti TL. Hydrogen sulfide: advances in understanding human toxicity. Int J Toxicol. 2010;29:569–581. doi: 10.1177/1091581810384882. [DOI] [PubMed] [Google Scholar]
- 28.Guidotti TL. Hydrogen sulphide. Occup Med (Lond) 1996;46:367–371. doi: 10.1093/occmed/46.5.367. [DOI] [PubMed] [Google Scholar]
- 29.Haggard HW. The fate of sulfides in the blood. J Biol Chem. 1921;49:519–529. [Google Scholar]
- 30.Hagihara A, Abe T, Omagari M, Motoi M, Nabeshima Y. The impact of newspaper reporting of hydrogen sulfide suicide on imitative suicide attempts in Japan. Soc Psychiatry Psychiatr Epidemiol. 2014;49:221–229. doi: 10.1007/s00127-013-0741-8. [DOI] [PubMed] [Google Scholar]
- 31.Hall AH, Rumack BH. Hydrogen sulfide poisoning: an antidotal role for sodium nitrite? Vet Hum Toxicol. 1997;39:152–154. [PubMed] [Google Scholar]
- 32.Hall AH, Saiers J, Baud F. Which cyanide antidote? Crit Rev Toxicol. 2009;39:541–552. doi: 10.1080/10408440802304944. [DOI] [PubMed] [Google Scholar]
- 33.Haouzi P. Is exogenous hydrogen sulfide a relevant tool to address physiological questions on hydrogen sulfide? Respir Physiol Neurobiol. 2016;229:5–10. doi: 10.1016/j.resp.2016.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haouzi P, Bell H, Philmon M. Hydrogen sulfide oxidation and the arterial chemoreflex: effect of methemoglobin. Respir Physiol Neurobiol. 2011;177:273–283. doi: 10.1016/j.resp.2011.04.025. [DOI] [PubMed] [Google Scholar]
- 35.Haouzi P, Bell H, Van de Louw A. Hypoxia-induced arterial chemoreceptor stimulation and hydrogen sulfide: too much or too little? Respir Physiol Neurobiol. 2011;179:97–102. doi: 10.1016/j.resp.2011.09.009. [DOI] [PubMed] [Google Scholar]
- 36.Haouzi P, Klingerman CM. Fate of intracellular H2S/HS− and metallo-proteins. Respir Physiol Neurobiol. 2013;188:229–230. doi: 10.1016/j.resp.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haouzi P, Sonobe T, Judenherc-Haouzi A. Developing effective countermeasures against acute hydrogen sulfide intoxication: challenges and limitations. Ann N Y Acad Sci. 2016;1374:29–40. doi: 10.1111/nyas.13015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haouzi P, Sonobe T, Torsell-Tubbs N, Prokopczyk B, Chenuel B, Klingerman CM. In vivo interactions between cobalt or ferric compounds and the pools of sulphide in the blood during and after H2S poisoning. Toxicol Sci. 2014;141:493–504. doi: 10.1093/toxsci/kfu140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Henderson SA, Goldhaber JI, So JM, Han T, Motter C, Ngo A, Chantawansri C, Ritter MR, Friedlander M, Nicoll DA, Frank JS, Jordan MC, Roos KP, Ross RS, Philipson KD. Functional adult myocardium in the absence of Na+-Ca2+ exchange: cardiac-specific knockout of NCX1. Circ Res. 2004;95:604–611. doi: 10.1161/01.RES.0000142316.08250.68. [DOI] [PubMed] [Google Scholar]
- 40.Hoffman NE, Miller BA, Wang J, Elrod JW, Rajan S, Gao E, Song J, Zhang XQ, Hirschler-Laszkiewicz I, Shanmughapriya S, Koch WJ, Feldman AM, Madesh M, Cheung JY. Ca2+ entry via Trpm2 is essential for cardiac myocyte bioenergetics maintenance. Am J Physiol Heart Circ Physiol. 2015;308:H637–650. doi: 10.1152/ajpheart.00720.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Irrinki KM, Mallilankaraman K, Thapa RJ, Chandramoorthy HC, Smith FJ, Jog NR, Gandhirajan RK, Kelsen SG, Houser SR, May MJ, Balachandran S, Madesh M. Requirement of FADD, NEMO, and BAX/BAK for aberrant mitochondrial function in tumor necrosis factor alpha-induced necrosis. Molecular and cellular biology. 2011;31:3745–3758. doi: 10.1128/MCB.05303-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ishigami M, Hiraki K, Umemura K, Ogasawara Y, Ishii K, Kimura H. A source of hydrogen sulfide and a mechanism of its release in the brain. Antioxidants & redox signaling. 2009;11:205–214. doi: 10.1089/ars.2008.2132. [DOI] [PubMed] [Google Scholar]
- 43.Judenherc-Haouzi A, Zhang XQ, Sonobe T, Song J, Rannals MD, Wang J, Tubbs N, Cheung JY, Haouzi P. Methylene blue counteracts H2S toxicity-induced cardiac depression by restoring L-type Ca channel activity. Am J Physiol Regul Integr Comp Physiol. 2016;310:R1030–1044. doi: 10.1152/ajpregu.00527.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kelner MJ, Alexander NM. Methylene blue directly oxidizes glutathione without the intermediate formation of hydrogen peroxide. J Biol Chem. 1985;260:15168–15171. [PubMed] [Google Scholar]
- 45.Khan AA, Schuler MM, Prior MG, Yong S, Coppock RW, Florence LZ, Lillie LE. Effects of hydrogen sulfide exposure on lung mitochondrial respiratory chain enzymes in rats. Toxicol Appl Pharmacol. 1990;103:482–490. doi: 10.1016/0041-008x(90)90321-k. [DOI] [PubMed] [Google Scholar]
- 46.Klingerman CM, Trushin N, Prokopczyk B, Haouzi P. H2S concentrations in the arterial blood during H2S administration in relation to its toxicity and effects on breathing. Am J Physiol Regul Integr Comp Physiol. 2013;305:R630–638. doi: 10.1152/ajpregu.00218.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kohn MC, Melnick RL, Ye F, Portier CJ. Pharmacokinetics of sodium nitrite-induced methemoglobinemia in the rat. Drug Metab Dispos. 2002;30:676–683. doi: 10.1124/dmd.30.6.676. [DOI] [PubMed] [Google Scholar]
- 48.Lagoutte E, Mimoun S, Andriamihaja M, Chaumontet C, Blachier F, Bouillaud F. Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim Biophys Acta. 2010;1797:1500–1511. doi: 10.1016/j.bbabio.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 49.Leschelle X, Goubern M, Andriamihaja M, Blottiere HM, Couplan E, Gonzalez-Barroso MD, Petit C, Pagniez A, Chaumontet C, Mignotte B, Bouillaud F, Blachier F. Adaptative metabolic response of human colonic epithelial cells to the adverse effects of the luminal compound sulfide. Biochim Biophys Acta. 2005;1725:201–212. doi: 10.1016/j.bbagen.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 50.Levitt MD, Abdel-Rehim MS, Furne J. Free and acid-labile hydrogen sulfide concentrations in mouse tissues: anomalously high free hydrogen sulfide in aortic tissue. Antioxidants & redox signaling. 2011;15:373–378. doi: 10.1089/ars.2010.3525. [DOI] [PubMed] [Google Scholar]
- 51.Mihajlovic A. Antidotal mechanisms for hydrogen sulfide toxicity. Toronto. 1999:69. [Google Scholar]
- 52.Miller BA, Hoffman NE, Merali S, Zhang XQ, Wang J, Rajan S, Shanmughapriya S, Gao E, Barrero CA, Mallilankaraman K, Song J, Gu T, Hirschler-Laszkiewicz I, Koch WJ, Feldman AM, Madesh M, Cheung JY. Trpm2 channels protect against cardiac ischemia-reperfusion injury: role of mitochondria. J Biol Chem. 2014;289:7615–7629. doi: 10.1074/jbc.M113.533851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Millero FJ. The thermodynamics and kinetics of hydrogen sulfide system in natural waters. Marine Chemistry. 1986;18:121–147. [Google Scholar]
- 54.Modis K, Bos EM, Calzia E, van Goor H, Coletta C, Papapetropoulos A, Hellmich MR, Radermacher P, Bouillaud F, Szabo C. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part II. Pathophysiological and therapeutic aspects. British journal of pharmacology. 2014;171:2123–2146. doi: 10.1111/bph.12368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ogasawara Y, Isoda S, Tanabe S. Tissue and subcellular distribution of bound and acid-labile sulfur, and the enzymic capacity for sulfide production in the rat. Biological & pharmaceutical bulletin. 1994;17:1535–1542. doi: 10.1248/bpb.17.1535. [DOI] [PubMed] [Google Scholar]
- 56.Reedy SJ, Schwartz MD, Morgan BW. Suicide fads: frequency and characteristics of hydrogen sulfide suicides in the United States. West J Emerg Med. 2011;12:300–304. [PMC free article] [PubMed] [Google Scholar]
- 57.Reiffenstein RJ, Hulbert WC, Roth SH. Toxicology of hydrogen sulfide. Annu Rev Pharmacol Toxicol. 1992;32:109–134. doi: 10.1146/annurev.pa.32.040192.000545. [DOI] [PubMed] [Google Scholar]
- 58.Resch P, Field RJ, Schneider W, Burger M. Reduction of methylene blue by sulfide ion in the presence and absence of oxygen: Simulation of the methylene blue-Op-HS- CSTR Oscillations. J Phys Chem. 1989;93:8181–8186. [Google Scholar]
- 59.Sevcikt P, Dunford H. Kinetics of the oxidation of NADH by methylene blue In a closed system. J Phys Chem. 1991;95:2411–2415. [Google Scholar]
- 60.Smilkstein MJ, Bronstein AC, Pickett HM, Rumack BH. Hyperbaric oxygen therapy for severe hydrogen sulfide poisoning. J Emerg Med. 1985;3:27–30. doi: 10.1016/0736-4679(85)90216-1. [DOI] [PubMed] [Google Scholar]
- 61.Smith L, Kruszyna H, Smith RP. The effect of methemoglobin on the inhibition of cytochrome c oxidase by cyanide, sulfide or azide. Biochem Pharmacol. 1977;26:2247–2250. doi: 10.1016/0006-2952(77)90287-8. [DOI] [PubMed] [Google Scholar]
- 62.Smith RP. Cobalt salts: effects in cyanide and sulfide poisoning and on methemoglobinemia. Toxicol Appl Pharmacol. 1969;15:505–516. doi: 10.1016/0041-008x(69)90052-0. [DOI] [PubMed] [Google Scholar]
- 63.Smith RP. The oxygen and sulfide binding characteristics of hemoglobins generated from methemoglobin by two erythrocytic systems. Mol Pharmacol. 1967;3:378–385. [PubMed] [Google Scholar]
- 64.Smith RP, Gosselin RE. Current concepts about the treatment of selected poisonings: nitrite, cyanide, sulfide, barium, and quinidine. Annu Rev Pharmacol Toxicol. 1976;16:189–199. doi: 10.1146/annurev.pa.16.040176.001201. [DOI] [PubMed] [Google Scholar]
- 65.Smith RP, Gosselin RE. Hydrogen sulfide poisoning. J Occup Med. 1979;21:93–97. doi: 10.1097/00043764-197902000-00008. [DOI] [PubMed] [Google Scholar]
- 66.Smith RP, Gosselin RE. On the mechanism of sulfide inactivation by methemoglobin. Toxicol Appl Pharmacol. 1966;8:159–172. doi: 10.1016/0041-008x(66)90112-8. [DOI] [PubMed] [Google Scholar]
- 67.Smith RP, Kruszyna R, Kruszyna H. Management of acute sulfide poisoning. Effects of oxygen, thiosulfate, and nitrite. Arch Environ Health. 1976;31:166–169. doi: 10.1080/00039896.1976.10667212. [DOI] [PubMed] [Google Scholar]
- 68.Song J, Gao E, Wang J, Zhang XQ, Chan TO, Koch WJ, Shang X, Joseph JI, Peterson BZ, Feldman AM, Cheung JY. Constitutive overexpression of phospholemman S68E mutant results in arrhythmias, early mortality and heart failure: Potenial involvement of Na+/Ca2+ exchanger. Am J Physiol Heart Circ Physiol. 2012;302:H770–H781. doi: 10.1152/ajpheart.00733.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Song J, Zhang XQ, Wang J, Cheskis E, Chan TO, Feldman AM, Tucker AL, Cheung JY. Regulation of cardiac myocyte contractility by phospholemman: Na+/Ca2+ exchange vs. Na+-K+-ATPase. Am J Physiol Heart Circ Physiol. 2008;295:H1615–H1625. doi: 10.1152/ajpheart.00287.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sonobe T, Chenuel B, Cooper TK, Haouzi P. Immediate and long-term outcome of acute H2S intoxication induced coma in unanesthetized rats: effects of methylene blue. PLoS One. 2015;10:e0131340. doi: 10.1371/journal.pone.0131340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sonobe T, Haouzi P. H2S concentrations in the heart after acute H2S administration: methodological and physiological considerations. Am J Physiol Heart Circ Physiol. 2016;311:H1445–H1458. doi: 10.1152/ajpheart.00464.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sonobe T, Haouzi P. H2S induced coma and cardiogenic shock in the rat: effects of phenothiazinium chromophores. Clin Toxicol (Phila) 2015;53:525–539. doi: 10.3109/15563650.2015.1043440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sonobe T, Haouzi P. Sulfide intoxication-induced circulatory failure is mediated by a depression in cardiac contractility. Cardiovascular toxicology. 2016;16:67–78. doi: 10.1007/s12012-015-9309-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sun YG, Cao YX, Wang WW, Ma SF, Yao T, Zhu YC. Hydrogen sulphide is an inhibitor of L-type calcium channels and mechanical contraction in rat cardiomyocytes. Cardiovasc Res. 2008;79:632–641. doi: 10.1093/cvr/cvn140. [DOI] [PubMed] [Google Scholar]
- 75.Tadros GM, Zhang XQ, Song J, Carl LL, Rothblum LI, Tian Q, Dunn J, Lytton J, Cheung JY. Effects of Na+/Ca2+ exchanger downregulation on contractility and [Ca2+]i transients in adult rat myocytes. Am J Physiol Heart Circ Physiol. 2002;283:H1616–1626. doi: 10.1152/ajpheart.00186.2002. [DOI] [PubMed] [Google Scholar]
- 76.Toombs CF, Insko MA, Wintner EA, Deckwerth TL, Usansky H, Jamil K, Goldstein B, Cooreman M, Szabo C. Detection of exhaled hydrogen sulphide gas in healthy human volunteers during intravenous administration of sodium sulphide. Br J Clin Pharmacol. 2010;69:626–636. doi: 10.1111/j.1365-2125.2010.03636.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Truong DH, Mihajlovic A, Gunness P, Hindmarsh W, O’Brien PJ. Prevention of hydrogen sulfide (H2S)-induced mouse lethality and cytotoxicity by hydroxocobalamin (vitamin B(12a)) Toxicology. 2007;242:16–22. doi: 10.1016/j.tox.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 78.Truscott A. Suicide fad threatens neighbours, rescuers. CMAJ. 2008;179:312–313. doi: 10.1503/cmaj.080878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tucker AL, Song J, Zhang XQ, Wang J, Ahlers BA, Carl LL, Mounsey JP, Moorman JR, Rothblum LI, Cheung JY. Altered contractility and [Ca2+]i homeostasis in phospholemman-deficient murine myocytes: Role of Na+/Ca2+ exchange. Am J Physiol Heart Circ Physiol. 2006;291:H2199–H2209. doi: 10.1152/ajpheart.01181.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ubuka T, Abe T, Kajikawa R, Morino K. Determination of hydrogen sulfide and acid-labile sulfur in animal tissues by gas chromatography and ion chromatography. J Chromatogr B Biomed Sci Appl. 2001;757:31–37. doi: 10.1016/s0378-4347(01)00046-9. [DOI] [PubMed] [Google Scholar]
- 81.Van de Louw A, Haouzi P. Ferric Iron and Cobalt (III) compounds to safely decrease hydrogen sulfide in the body? Antioxid Redox Signal. 2013;19:510–516. doi: 10.1089/ars.2012.4513. [DOI] [PubMed] [Google Scholar]
- 82.Wang J, Chan TO, Zhang XQ, Gao E, Song J, Koch WJ, Feldman AM, Cheung JY. Induced overexpression of Na+/Ca2+ exchanger transgene: Altered myocyte contractility, [Ca2+]i transients, SR Ca2+ contents and action potential duration. Am J Physiol Heart Circ Physiol. 2009;297:H590–H601. doi: 10.1152/ajpheart.00190.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang J, Gao E, Rabinowitz J, Song J, Zhang XQ, Koch WJ, Tucker AL, Chan TO, Feldman AM, Cheung JY. Regulation of in vivo cardiac contractility by phospholemman: role of Na+/Ca2+ exchange. Am J Physiol Heart Circ Physiol. 2011;300:H859–868. doi: 10.1152/ajpheart.00894.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang J, Gao E, Song J, Zhang XQ, Li J, Koch WJ, Tucker AL, Philipson KD, Chan TO, Feldman AM, Cheung JY. Phospholemman and β-adrenergic stimulation in the heart. Am J Physiol Heart Circ Physiol. 2010;298:H807–815. doi: 10.1152/ajpheart.00877.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Warenycia MW, Goodwin LR, Francom DM, Dieken FP, Kombian SB, Reiffenstein RJ. Dithiothreitol liberates non-acid labile sulfide from brain tissue of H2S-poisoned animals. Arch Toxicol. 1990;64:650–655. doi: 10.1007/BF01974693. [DOI] [PubMed] [Google Scholar]
- 86.Wei H, Zhang G, Qiu S, Lu J, Sheng J, Manasi, Tan G, Wong P, Gan SU, Shim W. Hydrogen sulfide suppresses outward rectifier potassium currents in human pluripotent stem cell-derived cardiomyocytes. PLoS One. 2012;7:e50641. doi: 10.1371/journal.pone.0050641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wendel WB. The Mechanism of antidotal action of methylene blue in cyanide poisoning. Science. 1934;80:381–382. [Google Scholar]
- 88.Whitfield NL, Kreimier EL, Verdial FC, Skovgaard N, Olson KR. Reappraisal of H2S/sulfide concentration in vertebrate blood and its potential significance in ischemic preconditioning and vascular signaling. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1930–1937. doi: 10.1152/ajpregu.00025.2008. [DOI] [PubMed] [Google Scholar]
- 89.Wiklund L, Basu S, Miclescu A, Wiklund P, Ronquist G, Sharma HS. Neuro- and cardioprotective effects of blockade of nitric oxide action by administration of methylene blue. Ann N Y Acad Sci. 2007;1122:231–244. doi: 10.1196/annals.1403.016. [DOI] [PubMed] [Google Scholar]
- 90.Wintner EA, Deckwerth TL, Langston W, Bengtsson A, Leviten D, Hill P, Insko MA, Dumpit R, VandenEkart E, Toombs CF, Szabo C. A monobromobimane-based assay to measure the pharmacokinetic profile of reactive sulphide species in blood. Br J Pharmacol. 2010;160:941–957. doi: 10.1111/j.1476-5381.2010.00704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wright RO, Lewander WJ, Woolf AD. Methemoglobinemia: etiology, pharmacology, and clinical management. Ann Emerg Med. 1999;34:646–656. doi: 10.1016/s0196-0644(99)70167-8. [DOI] [PubMed] [Google Scholar]
- 92.Xu H, Guo W, Nerbonne JM. Four kinetically distinct depolarization-activated K+ currents in adult mouse ventricular myocytes. J Gen Physiol. 1999;113:661–678. doi: 10.1085/jgp.113.5.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang R, Sun Y, Tsai H, Tang C, Jin H, Du J. Hydrogen sulfide inhibits L-type calcium currents depending upon the protein sulfhydryl state in rat cardiomyocytes. PloS one. 2012;7:e37073. doi: 10.1371/journal.pone.0037073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang X, Rojas JC, Gonzalez-Lima F. Methylene blue prevents neurodegeneration caused by rotenone in the retina. Neurotox Res. 2006;9:47–57. doi: 10.1007/BF03033307. [DOI] [PubMed] [Google Scholar]
- 95.Zhang XQ, Ahlers BA, Tucker AL, Song J, Wang J, Moorman JR, Mounsey JP, Carl LL, Rothblum LI, Cheung JY. Phospholemman inhibition of the cardiac Na+/Ca2+ exchanger. Role of phosphorylation. J Biol Chem. 2006;281:7784–7792. doi: 10.1074/jbc.M512092200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang XQ, Qureshi A, Song J, Carl LL, Tian Q, Stahl RC, Carey DJ, Rothblum LI, Cheung JY. Phospholemman modulates Na+/Ca2+ exchange in adult rat cardiac myocytes. Am J Physiol Heart Circ Physiol. 2003;284:H225–233. doi: 10.1152/ajpheart.00698.2002. [DOI] [PubMed] [Google Scholar]
- 97.Zhang XQ, Zhang LQ, Palmer BM, Ng YC, Musch TI, Moore RL, Cheung JY. Sprint training shortens prolonged action potential duration in postinfarction rat myocyte: mechanisms. J Appl Physiol. 2001;90:1720–1728. doi: 10.1152/jappl.2001.90.5.1720. [DOI] [PubMed] [Google Scholar]
- 98.Zhou YY, Wang SQ, Zhu WZ, Chruscinski A, Kobilka BK, Ziman B, Wang S, Lakatta EG, Cheng H, Xiao RP. Culture and adenoviral infection of adult mouse cardiac myocytes: methods for cellular genetic physiology. Am J Physiol Heart Circ Physiol. 2000;279:H429–436. doi: 10.1152/ajpheart.2000.279.1.H429. [DOI] [PubMed] [Google Scholar]
- 99.Zima AV, Blatter LA. Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res. 2006;71:310–321. doi: 10.1016/j.cardiores.2006.02.019. [DOI] [PubMed] [Google Scholar]