

Abstract
The development of mesenchymal stem cells (MSCs) as cell‐based drug delivery vectors for numerous clinical indications, including cancer, has significant promise. However, a considerable challenge for effective translation of these approaches is the limited tumor tropism and broad biodistribution observed using conventional MSCs, which raises concerns for toxicity to nontarget peripheral tissues (i.e., the bad). Consequently, there are a variety of synthetic engineering platforms in active development to improve tumor‐selective targeting via increased homing efficiency and/or specificity of drug activation, some of which are already being evaluated clinically (i.e., the good). Unfortunately, the lack of robust quantification and widespread adoption of standardized methodologies with high sensitivity and resolution has made accurate comparisons across studies difficult, which has significantly impeded progress (i.e., the ugly). Herein, we provide a concise review of active and passive MSC homing mechanisms and biodistribution postinfusion; in addition to in vivo cell tracking methodologies and strategies to enhance tumor targeting with a focus on MSC‐based drug delivery strategies for cancer therapy. Stem Cells Translational Medicine 2018;1–13
Keywords: Mesenchymal stem cell, Cell‐based therapy, Drug delivery, Homing, In vivo cell tracking, Cell size
Significance Statement.
As excitement for mesenchymal stem cell‐based therapies, and synthetic biology approaches in general, continues to build and as these therapies increasingly undergo evaluation in the clinic, this review represents a sobering reminder of the broad biodistribution and poor homing efficiency to most target tissues observed using current methodologies, thereby justifying the need for enhanced targeting strategies to potentiate efficient and effective clinical translation of these strategies.
Introduction
There is enormous enthusiasm regarding the potential for cell‐based therapies to treat a diverse array of pathological indications as the technology to engineer cells with specific attributes is maturing and entered clinical testing in some cases. This has been most visible with the emergence of chimeric antigen receptor (CAR) T‐cells, although multiple other cell types are also in active development as platforms for synthetic biology approaches. Among the most promising of these engineered cell platforms are mesenchymal stem cells (MSCs). MSCs are defined analytically and functionally based upon positive (CD73, CD90, and CD105) and negative (CD45, CD34, CD14/CD11b, CD19/CD20/CD79α, and HLA‐DR) cell surface markers, plastic adherence, and the ability to differentiate into osteoblasts, adipocytes, and chondrocytes. However, it should be noted this definition leaves room for significant phenotypic diversity, and these minimal criteria clearly define a heterogeneous population of cells with implications for clinical development 1.
Despite this heterogeneity, MSCs have numerous advantages that potentiate their clinical translation. These properties include their ease of isolation from multiple tissues, ex vivo expansion capacity, multipotent differentiation potential, immunomodulatory functions, ability to be manipulated or genetically modified, and immune‐evasive or ‐privileged status, which permits use in an allogeneic setting. Although initial trials were premised on the ability of MSCs to repair damaged tissue via cell replacement, more recent clinical development has focused on their potent paracrine and immune regulatory functions 2. Significant efforts have also been made to exploit the innate ability of MSCs to traffic to sites of inflammation, including those present in cancer, to deliver a variety of therapeutic interventions, including apoptosis‐inducing agents, cytotoxic chemotherapy, drug‐loaded nanoparticles/microparticles, tumor‐ or tissue‐specific prodrugs, immunomodulatory agents, oncolytic viruses, and anti‐angiogenic factors (Fig. 1; Table 1) 3, 4, 5.
Figure 1.
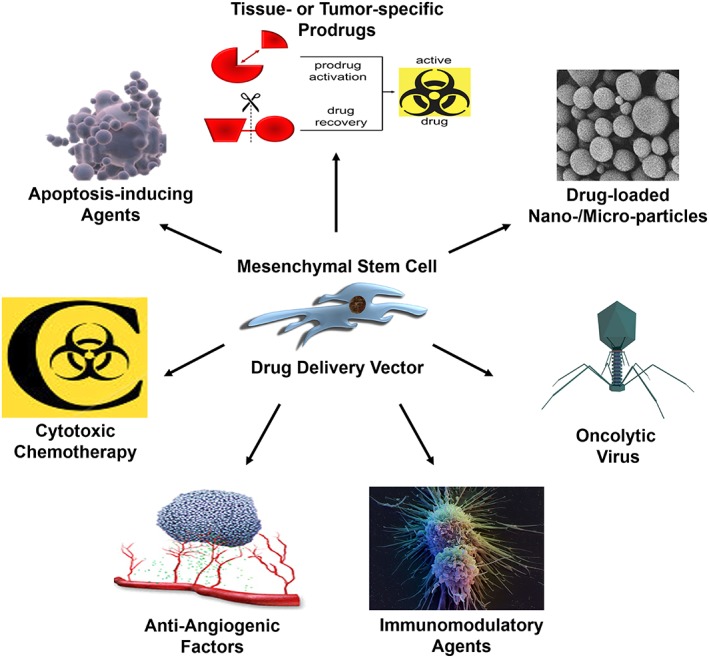
Mesenchymal stem cell (MSC)‐based drug delivery strategies. The tumor tropism of MSCs can be exploited to deliver a wide variety of therapeutic agents for the treatment of cancer, such as apoptosis‐inducing agents, cytotoxic chemotherapy, anti‐angiogenic factors, immunomodulatory agents, oncolytic viruses, drug‐loaded nanoparticles/microparticles, and tissue‐ or tumor‐specific prodrugs.
Table 1.
Classes and examples of MSC‐based anti‐cancer agent drug delivery strategies
| Anti‐cancer strategy | Common agents | Mechanism of action | Advantages | References |
|---|---|---|---|---|
| Oncolytic viruses | Adenovirus; Measles virus; Herpes simplex virus |
Viruses infect, replicate in, and lyse tumor cells | Amplification of anti‐tumor effect with multiple rounds of infection; Selective replication in tumor cells |
75, 76, 77, 78, 98 |
| Tumor‐ or tissue‐specific prodrugs | CD + 5‐5‐FU; Hsv‐tk + Ganciclovir; PSA‐activated thapsigargin peptide |
Cytotoxic drug metabolites induce cell death by inhibiting DNA synthesis (5‐FU, ganciclovir) or by inducing ER stress (thapsigargin) | Selective drug activation in tumor microenvironment | 79, 80, 81, 82, 83, 84 |
| Immunomodulatory agents | IL‐2; IL‐12; Interferon‐β; CX3CL1 |
Lymphocyte activation and induction of tumor‐specific T‐cell responses; Direct induction of tumor cell differentiation and growth arrest | Endogenous signaling molecules; Potential direct and indirect effects on tumor growth; Synergy with other immunotherapies |
73, 89, 90, 91, 92 |
| Apoptosis‐inducing agents | TRAIL | Direct induction of apoptosis via death receptors | Currently in clinical trials; Endogenous signaling molecule |
93, 94, 95, 96, 97 |
| Cytotoxic chemotherapy | Paclitaxel; Doxorubicin |
Induction of cell death via inhibition of microtubule depolymerization (paclitaxel) or topoisomerase II function (doxorubicin) | FDA‐approved chemotherapeutic drugs |
68 |
Abbreviations: CD, cytosine deaminase; 5‐FU, 5‐fluoruracil; Hsv‐tk, herpes simplex virus‐thymidine kinase; PSA, prostate specific antigen; TRAIL, TNF‐related apoptosis‐inducing ligand.
These efforts have culminated in more than 1,000 completed or ongoing clinical trials using MSCs across many disorders with varying degrees of success. The clinical benefits of repurposing MSCs for the treatment of diverse clinical indications are challenged by evolving techniques to improve cell function, localization, and tracking following systemic infusion. A significant limitation for many of these strategies has been the lack of robust MSC homing to target tissues 6. It has been posited that MSCs primarily ‘act at a distance’ via exosomes, polarization of phagocytic monocytes, and other paracrine effects 7, 8, 9, 10, 11, which may partially overcome poor targeting and broad biodistribution of systemically infused MSCs, particularly when coupled with exosome targeting strategies 9, 12. However, increased activity directly in target tissue would likely be of significant benefit for many applications, especially for those using MSCs as cell‐based drug delivery vectors; wherein limiting toxicity to peripheral nontarget tissues is of critical importance. Factors that influence MSC homing are multifactorial; these include culture conditions during ex vivo expansion, tissue source, population heterogeneity, cell size, and species‐specific differences in affinities for cognate receptor–ligand pairs when using xenogeneic models. The breadth of these factors necessitates that we focus this review on passive homing mechanisms related to cell size and mechanical entrapment, whereas others have recently been reviewed elsewhere 13. A central limitation in evaluating and improving MSC homing has been a lack of robust quantification and widespread adoption of standardized methodologies with high sensitivity and resolution across models and disease states. Herein, we summarize MSC homing mechanisms and biodistribution postinfusion, in vivo cell tracking methodologies, and strategies to enhance tumor targeting with a focus on MSC‐based drug delivery strategies for cancer therapy.
MSC Homing Mechanisms
The mechanisms for cellular trafficking via systemic circulation were first characterized for leukocyte homing to sites of inflammation, which involves a multistep adhesion and extravasation cascade. Given the role of MSCs in regulating the overall immune response 14, 15, 16, it is unsurprising that MSCs are thought to use similar mechanisms to migrate toward inflammatory cues emanating from sites of tissue damage including the tumor microenvironment 13, 17, 18, 19. Nitzsche et al. provide a thorough review of the important molecular determinants of MSC homing at each step of this migratory cascade 19.
‘Passive Homing’, Cell Size, and Mechanical Entrapment
Despite evidence that MSC homing is mediated by specific receptor–ligand pairs, passive entrapment of MSCs in the tumor or sites of injury occurs at least partially as a result of increased vascular permeability and mechanical entrapment in these microenvironments. An important distinction between MSCs and lymphocytes is their size (Fig. 2), with cell diameters ranging from 15–30 μm versus 4–12 μm, respectively 20, 21, 22. It is often unappreciated that relatively small increases in cell diameter translate into significant increases in cell volume, because this value increases as the cube of the radius. This larger cell size, particularly following ex vivo culture 23, leads to passive arrest of MSCs in small diameter vessels such as terminal arterioles, capillaries, and postcapillary venules as a result of mechanical entrapment. Indeed, the vast majority of MSCs infused intravenously (IV) are rapidly cleared from the blood and found within the capillary beds of the lungs within minutes of injection 24, 25, 26, 27, 28. In both humans and animal models, this rapid entrapment is followed by clearance from the lungs and accumulation in the liver and spleen over subsequent hours to days 24, 25, 26, 27, 28. Recent evidence suggests this “redistribution” may be a function of nonclassical phagocytic monocytes engulfing cellular debris (and tracking labels) from apoptotic MSCs entrapped in the lungs 10, 11.
Figure 2.
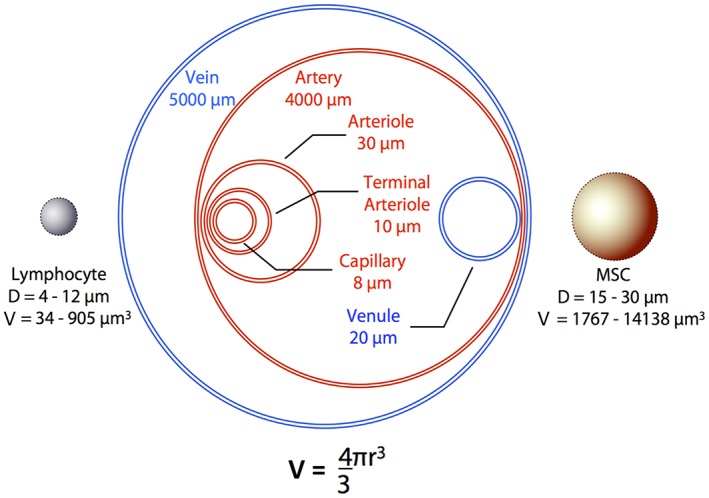
Mechanical barriers to MSC trafficking via systemic circulation. The large cell size of MSCs, particularly following ex vivo expansion, is a significant physical barrier that prevents efficient and complete dispersion through small vessels in the vascular network. This severely limits access of exogenously introduced MSCs to many target tissues, including tumors. Abbreviations: D, diameter; MSC, mesenchymal stem cell; V, volume.
Mechanical entrapment in the lungs results from the fact that pulmonary capillaries are ∼10–15 μm in diameter, a phenomenon termed the pulmonary ‘first‐pass effect’ 20, 21, 29, 30. Previous studies using microspheres have documented that objects ≥10 μm in diameter are highly susceptible to this effect 21, and particles <1 μm are necessary to ensure complete dispersion through the smallest capillaries 30. Importantly, endogenous MSCs in the bone marrow are smaller in size (∼10 μm) 23, which enable efficient trafficking via systemic circulation. Similar to lymphocytes, MSCs are thought to increase in size once activated (mimicked during ex vivo culture) within sites of inflammation and tissue damage.
Although cellular deformability can facilitate passage of larger cells through smaller vessels to some degree 23, 31, there is a physical limit to this property that is necessary to maintain cell viability and prevent vessel occlusion. Intra‐arterial (IA) infusions can circumvent the first‐pass effect to provide one pass through systemic circulation and exposure to peripheral tissues before entering the lungs. However, mechanical entrapment may still be a dominant driver of MSC biodistribution (Fig. 2). In one study, for example, >90% of MSCs injected into the iliac artery were found acutely arrested at the precapillary level in downstream microvessels (7 μm diameter) of the cremaster muscle 23. To date, there have been limited studies addressing the relative importance of active versus passive arrest in the lungs or other tissues; however, it is likely that both mechanisms are important and can be manipulated to increase homing efficiency to sites of interest.
In our own studies, we have identified MSCs in benign and malignant human prostate tissue 32. These observations led us to initiate studies characterizing MSC biodistribution, kinetics, and trafficking toward different prostate cancer xenografts postinfusion, in addition to assessing chemokine and cognate receptor profiles to identify key pathways mediating MSC tumor tropism in prostate cancer. For example, whole body PET imaging using zirconium‐89 (89Zr)‐labeled MSCs 33 (Fig. 3A), revealed that ∼0.2% of the injected cells trafficked to subcutaneous PC3 prostate cancer xenografts per gram of tissue at 7 days postinfusion (Fig. 3B). This uptake is consistent across MSC administration methods used (intra‐cardiac [IC]: 0.18 ±0.02 vs. IV: 0.23 ±0.06%ID/g). Broad biodistribution was observed at this time point with >1% of the injected dose detected in the heart, kidney, liver, lung, and spleen independent of the route of administration (Fig. 3B). Autoradiography confirmed tissue distribution of infiltrating MSCs with tumor localization restricted to the periphery as expected based on the vascular pattern of subcutaneous tumors (Fig. 3C).
Figure 3.
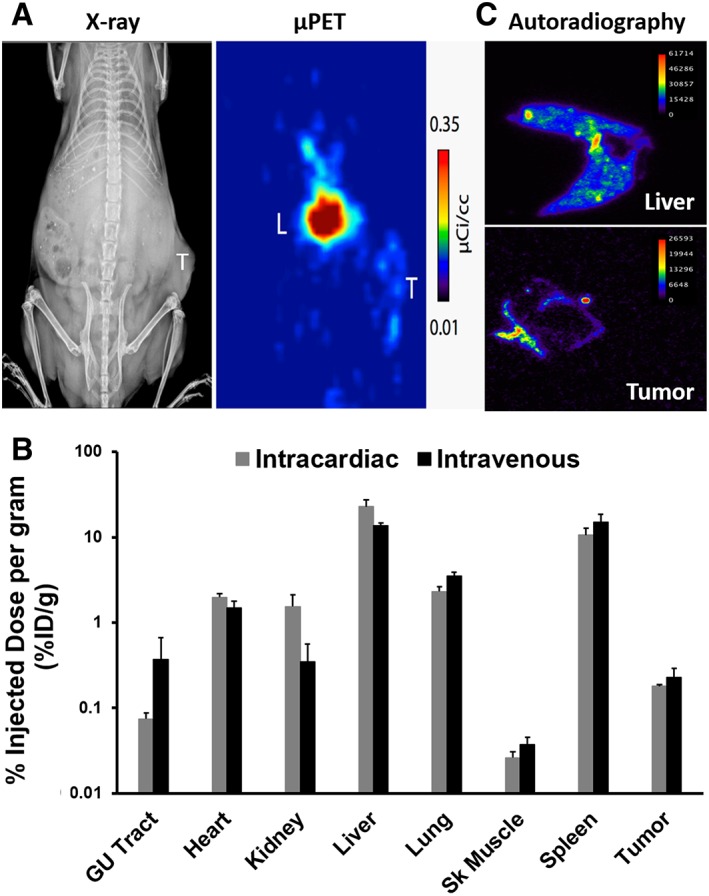
In vivo cell tracking of systemically‐infused 89Zr‐labeled human MSCs in a prostate cancer xenograft model. (A): X‐ray and μPET imaging documenting accumulation of the radiolabel in the liver and tumor (PC3) at 7 days post‐IV infusion. (B): Biodistribution of 89Zr‐labeled MSCs at 7 days post‐IV or ‐IC infusion determined by ex vivo scintigraphy. (C): Autoradiography detailing sub‐organ distribution of 89Zr‐labeled MSCs, confirming localization restricted to the tumor periphery. Abbreviation: μPET, microPET.
Strategies to Overcome Mechanical Perfusion Barriers
Collectively, these findings indicate the vast majority of exogenously introduced MSC‐based therapeutics have limited access to target tissue outside of the lung as a result of mechanical entrapment. To partially overcome this barrier and improve targeting, preadministration of vasodilators such as sodium nitroprusside have been used to reduce lung entrapment in mouse models 21, 29. In addition, multiple investigators have developed ex vivo expansion protocols reported to generate MSC cultures with smaller average cell diameters 34, 35, 36, 37.
Recently, Luo et al. generated “synthetic MSCs” (synMSCs) by packaging MSC conditioned media into PLGA microparticles coated with MSC membranes, which demonstrated efficacy in a model of myocardial infarction 38. In this study, the synMSCs were designed to be equal in size to culture‐expanded MSCs for comparison, but this raises the interesting possibility of engineering synMSCs (or microparticles coated with other cell membranes/targeting moieties) with engineered diameters designed to avoid first‐pass entrapment in pulmonary capillaries and deliver bioactive molecules based on active targeting mechanisms. A similar alternative approach is to incorporate anti‐cancer agents of interest (e.g. drugs, proteins, RNA, etc.) into targeted MSC‐derived extracellular vesicles or exosomes via active or passive loading mechanisms 9, 12. Despite these approaches, lung entrapment remains a critical challenge for MSC‐based drug delivery; although evidence suggests this is a surmountable engineering challenge.
Methods to Track MSC Homing In Vivo
Standardization of robust quantitative methods with high sensitivity and resolution along with standardized reporting metrics are essential for cross‐study comparisons and validation. Methods to track MSCs have largely been variations of the same core techniques, each with advantages and disadvantages (Table 2). Of these approaches, ex vivo histological analysis is the most common. This is typically performed using MSCs labeled with a fluorescent lipophilic vital dye (e.g. CM‐DiI or PKH26) pre‐infusion or stained immunohistochemically postinfusion for an exogenously introduced marker such as green fluorescent protein (GFP) followed by counting labeled cells in random fields after tissue processing. A variation of this approach uses in situ hybridization to label DNA sequences such as the Y‐chromosome or human Alu sequences in sex‐ or species‐mismatched samples, respectively. These ex vivo methods to quantify MSC homing are cost‐effective and do not require specialized equipment; however, tissue collection is highly invasive, not amenable to repeated measurements for kinetic analyses, and highly susceptible to sampling bias because only a small portion of the tissue of interest is typically analyzed, in addition to the potential for false positives as a result of label redistribution by phagocytic monocytes following uptake of cellular debris from dying MSCs.
Table 2.
Advantages and disadvantages of common in vivo and ex vivo cell tracking methodologies
| Technique | Common imaging agents | Limit of detection | Depth of penetration | Spatial resolution | Temporal resolution | Useful applications advantages | Limitations | References |
|---|---|---|---|---|---|---|---|---|
| In vivo techniques | ||||||||
| BLI | D‐luciferin/Firefly luciferase, Coelenterazine/Renilla luciferase | 10–10,000 cells (depending on tissue depth) | ∼3 cm (∼10‐fold loss of signal intensity per cm) | 50 μm | 1 min | Whole‐body imaging;Reporter enzyme expressed only inmetabolically activelive cells; Cheap; Ease of use | 2D image; Limited to small animal studies;Signal attenuation in deeper tissues; Signal quantitation not comparable across tissues; Susceptible to vascular disrupting agents | 21, 56, 65, 76, 95, 102, 106 |
| MRI | Ferumoxide;Ferumoxytol;Ferritin;Gd chelates | 1,000–10,000 cells (depending on imaging agent and instrumentation) | No limit | 100–200 μm | 1 min | Whole‐body imaging;Anatomical imaging;Ferritin reporter gene enables detection exclusively of live cells;Clinical integration | T2 hypointensity indistinguishablefrom tumor hemorrhage;False positives due to macrophage uptake;2D image; Label diluted with each cell division; Potential cytotoxicity and ROS generation; Sophisticated equipment and expertise needed | 26, 55, 56 |
| PET | 18F‐FDG;Hsv‐TK + 18F‐FDG;64Cu‐PTSM;89Zr;124I | 100–25,000 cells (depending on imaging agent and instrumentation) | No limit | 1–2 mm | 15 min | Whole‐body imaging; 3D imaging; Multiple radioisotopes and labeling chemistries available; Hsv‐TK reporter gene enables detection exclusively of live cells; Clinical integration | Use of radioisotopes;Hazardous transfection reagents; Potential false positives aftercell death/label efflux; Sophisticated equipment and expertise needed | 33, 48 |
| In vivo microscopy | GFP;Vital dyes (DID/DiL/DiD/DiR);CFDA/CFSE | Single cell | 150–250 μm | Single cell | Video‐rate | Imaging in context of natural microenvironment;Visualization of dynamic processes(migration, vascular adhesion, TEM) | Limited to preselected areas with low required path length; Tissue exteriorization may influence MSC recruitment/retention; Tissue auto‐fluorescence; Sophisticated equipment and expertise needed | 18, 23, 46, 58, 60, 61, 99, 100 |
| In vivo flow cytometry | GFP;Vital dyes (DID/DiL/DiD/DiR);CFDA/CFSE;PKH26 | 1–10 cells/mL | 150–250 μm | Single cell | Continuous | Enumeration of circulating cells; Tracking dynamic entry and exit from circulation; Detection of cell flow velocities;Analysis of large blood volume; No artifacts from cell isolation and processing | Quantification influenced by flow velocityand vessel size; Tissue auto‐fluorescence;Antibody labeling can lead to target cell depletion; Sophisticated equipment and expertise needed | 62, 63, 64 |
| Ex vivo techniques | ||||||||
| Histology | X‐gal/β‐gal;Prussian blue/Iron oxide | Single cell | N/A | Single cell | Single time point/sample | In situ visualization within contextual tissue structure | Loss of signal from gene silencing or cell division; Sampling bias | 26, 34, 47, 94 |
| IHC | GFP/anti‐GFP;CFDA/anti‐fluorescein; PKH26; Vital dyes (DiI/DiL/DiD/DiR); Quantum dots;Fluc/anti‐FLuc | Single cell | N/A | Single cell | Single time point/sample | In situ visualization within contextual tissue structure | Loss of signal from gene silencing or cell division; Sampling bias; Tissue auto‐fluorescence;Potential transfer of membrane dyes | 18, 43, 44, 45, 46, 56, 71, 72, 73, 77 |
| FISH | Y‐Chromosome;Alu sequences | Single cell | N/A | Single cell | Single time point/sample | In situ visualization within contextual tissue structure;No loss of signal | Sampling bias;Tissue auto‐fluorescence | 71 |
| Ex vivo flow cytometry | GFP | 1–10 cells/mL | N/A | Single cell | Single time point/sample | Enumeration of circulating cells | Potential artifacts from cell isolationand processing; Cell auto‐fluorescence;Sophisticated equipment and expertise needed | 26, 34, 63, 64 |
| qRT‐PCR | Y‐Chromosome;Alu sequences | 1/600,000 cells (∼0.0002%) | N/A | Single cell | Single time point/sample | Quantification from whole tissue | Rare transcripts difficult to detect due to stochasticity of PCR amplification | 25, 28, 40 |
| ddPCR/BEAMing | Y‐Chromosome;Alu sequences | 1/10,000 cells (0.01%) | N/A | Single cell | Single time point/sample | Quantification from whole tissue; Identification of rare transcripts/cell populations | Sophisticated equipment and expertise needed | 41 |
| Scintigraphy | 111In‐oxine; 18F‐FDG;64Cu‐PTSM; 51Cr; 125I;99mTc‐HMPAO | Single cell (depending on labeling efficiency) | N/A | Single cell | Single time point/sample | Quantification from whole tissue | Use of radioisotopes | 20, 26, 33, 47, 51, 52 |
Abbreviations: BLI, bioluminescence imaging; FISH, fluorescence in situ hybridization; Gd, gadolinium; Hsv‐tk, herpes simplex virus‐thymidine kinase; IHC, fluorescence imaging and immunohistochemistry; MRI, magnetic resonance imaging; PET, positron emission tomography; qRT‐PCR, quantitative real‐time PCR.
PCR‐based techniques are another commonly used method to quantify MSC homing. Frequently, these techniques take advantage of species‐mismatched donor/recipient pairs to detect species‐specific sequences (e.g. GAPDH or Alu sequences) 28. Conventional PCR‐based methods have an estimated threshold of detection of ∼50,000 cells 39, which does not achieve the sensitivity required to accurately quantify the level of MSC homing to most tissues. Although quantitative RT‐PCR (qRT‐PCR) has been estimated to have a threshold of detection as low as one human MSC in 600,000 murine cells 40, the detection of rare transcripts (i.e., injected MSCs) among a population (i.e., tissue) is still highly problematic for accurate quantification due to the stochastic nature of PCR‐based amplification and competition for reagents. More recently developed methodologies such as droplet digital PCR (ddPCR) and BEAMing address this problem by segregating individual transcripts into separate compartments (i.e., droplets/microemulsions) before PCR amplification. This methodology can accurately quantify rare transcripts in the range of 0.01% 41, and new advancements in next generation sequencing (NGS) have pushed the sensitivity even lower depending on the number of genome equivalents analyzed 42.
Whole Body Imaging Methods
Unlike the techniques described earlier, whole‐body imaging permits serial imaging over time to determine kinetics of biodistribution in multiple organs simultaneously in live animals. Several approaches have been used including: optical methods such as bioluminescent imaging (BLI) of MSCs expressing firefly luciferase (Fluc) or fluorescent dye‐labeled cells; magnetic resonance (MR) imaging using MSCs loaded with superparamagnetic particles; and nuclear imaging techniques (e.g., positron emission tomography [PET] and single photon emission computed tomography [SPECT]) with radiolabeled MSCs.
Of the whole‐body imaging techniques, optical methods require the least expensive equipment and have been widely used in proof‐of‐principle preclinical studies. However, BLI and fluorescence have limited translational application, and for BLI, the signal is dependent on vascular delivery of the luciferase substrate. Consequently, the signal is not necessarily proportional to MSC homing and is highly sensitive to vascular disrupting agents. In addition, both BLI and fluorescent signals have a limited depth of tissue penetration, meaning that signal quantification is not directly comparable for tissues at different depths. In contrast, both nuclear and MR imaging modalities are well‐integrated into clinical practice, potentially allowing quantitative in vivo tracking of MSCs for clinical trials.
Fluorescence imaging with cell labeling dyes has been widely used in the small animal imaging field to track the distribution of cells labeled ex vivo with membrane binding dyes. Light propagation through tissue is heavily dependent on wavelength, and redshifted or near infrared dyes enable visualization of labeled cells through several millimeters of tissue. To that end, membrane‐anchored dyes that contain a lipophilic anchor and a near infrared fluorescent component, such as PKH‐26 and the Dioctadecyl family of dyes (including DiI, DiD, and DiR), have been used to label MSCs for noninvasive tracking 43, 44, 45, 46. The limited depth of penetration, potential impact of the surface label on MSC behavior, the dilution of the label through cell division, and uptake by phagocytic cells following death are limitations to the use of this technique; however, the fluorescent label does enhance the ability to rapidly detect the presence of the cells in ex vivo analyses with the caveat of sampling bias and re‐distribution of the tracking label by phagocytic monocytes as described earlier.
In addition, there are a variety of SPECT and PET‐compatible radiolabels that have been used for quantitative in vivo cell tracking of infused MSCs, each with advantages and disadvantages 27, 33, 47, 48, 49, 50. DeGrado et al. recently developed a novel cell labeling methodology using 89Zr, which achieves high labeling efficiency and displays robust stability in vitro and in vivo with minimal impact on viability 33. The relatively long half‐life of 89Zr (t1/2 = 78.4 hours) enables in vivo cell tracking at high resolution over a 2–3 week period 33. Another intriguing approach has been to engineer MSCs to express the sodium iodide symporter (NIS), which can be exploited for imaging (124I) or therapeutic (131I) applications 48, 49. This strategy was recently translated into a phase I/II clinical trial using MSCs infected with an oncolytic measles virus encoding NIS to treat recurrent or chemotherapy‐resistant ovarian cancer [NCT02068794]. Ex vivo scintigraphy of radiolabeled MSCs is also frequently performed to complement in vivo nuclear methods to obtain semi‐quantitative counts in tissues of interest.
Although each of these whole‐body techniques enable serial imaging for kinetic analyses of MSC homing and biodistribution, it is important to consider the half‐life, stability, andcell retention of the particular tracking label selected depending on the specific application and length of study. For example, efflux of 111In‐oxine, a commonly used leukocyte labeling agent, from cells is reported to be as high as 70%–80% after just 24–96 hours post labeling 33. 99mTc‐hexamethylpropylene amine oxime (99mTc‐HMPAO) has a longer retention profile, but is similar to 111In‐oxine in that it is a lipophilic complex that can penetrate the cell membrane, which then becomes entrapped once internalized 51. 99mTc‐HMPAO labeled MSCs have enabled imaging and quantitation of uptake at sites of brain injury in mice 52 and cardiac damage in rats 47. Labeling with radioactive sodium chromate (Na2Cr51O4) involves intracellular reduction of the cell permeable hexavalent chromium (CrO4 2−) to the impermeable trivalent chromic (CR+++) ion following binding to macromolecules 53. Alternatively, the novel 89Zr‐labeling strategy mentioned earlier also labels macromolecules, but is restricted to primary amines on the cell surface due to poor membrane permeability of the reagents 33. The fate of such intracellular and membrane‐bound radiolabeled proteins following secretion or cell death (e.g., uptake by macrophages/monocytes) and the potential for subsequent redistribution is often not considered but can be monitored. It should be noted that many unbound radionuclides inherently accumulate in the bone and liver when released into systemic circulation. Therefore, significant efforts should be made to confirm that radioactivity or other detection labels observed in tissues of interest is associated with infiltrating MSCs by an independent method whenever possible.
In contrast, 125I‐5‐iodo‐2′‐deoxyuridine incorporates into DNA and is only released upon cell death. Furthermore, it is poorly re‐used and quickly eliminated following release, meaning that radioactivity is predominantly a direct readout of live cells 54. Unfortunately, however, 125I SPECT imaging requires a large activity dose, limiting its usefulness for applications with low rates of genome incorporation. The consideration of dose to the cell, which may affect cellular physiology and function, is very important. Ideally, labels are incorporated for tracking in a truly noninvasive setting. As an example, MR imaging provides high spatial and anatomic resolution but relative to nuclear methods a low sensitivity. Thus, MR approaches using superparamagnetic agents, which have recently been optimized for high labeling efficiency and stability (≥21 days), require large mass amounts of cell‐internalized particles 55, 56. Interestingly, Huang et al. found that iron oxide magnetic nanoparticles induced CXCR4 cell surface expression on MSCs in a HIF‐1α‐dependent manner and enhanced homing to traumatic brain injury 56. Collectively, these data suggest that significant care must be taken when selecting an appropriate cell tracking modality, as there may be unintended consequences and varying potential for false positives depending on the application and study design.
Localized Imaging Methods
Confocal, two‐photon, or intravital microscopy can be used to visualize fluorescently labeled MSCs in real‐time in specific in vivo settings. By introducing additional targeted fluorophores, MSCs can be visualized in the context of other labeled structures, such as blood vessels, with high spatial definition. This methodology allows for high resolution quantification down to the single‐cell level and can be used to interrogate various stages of the homing cascade, such as MSC rolling, entrapment, and extravasation. However, imaging is limited to preselected areas with penetration up to 250 μm. Due to this limitation, LPS‐induced inflammation in the murine ear (∼200 μm thick) is a commonly used model when employing this technique 57, 58. in vivo microscopy has been used for other applications such as measurement of tumor cell or MSC homing to the bone marrow of murine skulls 59, 60 or MSC passage through blood vessels of an exteriorized cremaster muscle 23. However, these procedures involve surgery to expose tissues, and such tissue injury may influence recruitment or retention of MSCs. Notably, video‐rate two‐photon imaging of T‐cell infiltration into subcutaneous tumors has been performed at depths of up to 150 μm from the tumor surface 61 and could similarly be used to quantify MSC infiltration.
More recent approaches to quantify MSC homing include ex vivo and in vivo flow cytometry. Although both techniques are based on the same principles, the former is the more traditional form to analyze cells labeled ex vivo with a fluorescent dye or antibody following sample collection, whereas the latter applies transillumination of a narrow slit along an artery with a focused laser to allow detection of circulating cells that are fluorescently labeled before infusion. Like intravital microscopy, arteries within the murine ear are typically used due to accessibility and the low required path length. This approach has been used to quantify circulating cells, including MSC clearance rates from peripheral blood of healthy and tumor‐bearing mice 59, 62, 63, 64. Although both ex vivo and in vivo flow cytometry are able to detect rare populations of cells, ∼1–10 cells per ml of blood, in vivo flow cytometry is approximately twice as sensitive but significantly more challenging to perform 63.
Each set of methods has advantages and limitations, including the inherent sensitivities of each method (Table 2), that may contribute to the variability often observed between MSC homing studies. In addition, homing efficiency is often reported as a relative measure between experimental and control groups rather than an absolute number. These facts, coupled with the lack of positive controls available for MSC homing studies, makes standardization and comparison of homing efficiencies across platforms extremely difficult.
Implications for Clinical Translation and Efficacy
Although clinical efficacy has been observed in several applications despite poor tissue‐targeting 2, 14, 15, 16, homing efficiency and selectivity are critical for developing MSCs as cell‐based delivery vehicles for anti‐cancer therapies. Although MSC homing to the tumor microenvironment has been observed 65, 66, 67, it is a very inefficient process as discussed earlier with the exception of strategies targeting the lung via entrapment or potentially the liver as a result of clearance. The typical broad biodistribution of systemically infused MSCs in nontarget tissue has significant consequences for potential toxicity to peripheral tissues in drug delivery applications. In addition, MSC infiltration must reach sufficient levels within the tumor microenvironment to deliver a complete tumoricidal dose of the cytotoxic agent, which is dependent upon drug potency, release kinetics, and the amount of drug that can be delivered per cell. Using in vitro co‐culture assays, Pessina et al. demonstrated that Paclitaxel‐primed MSCs needed to represent 2%–33% of the culture to reach 90% cytotoxicity against various cancer cell lines depending on the sensitivity of each line to the drug 68. In proof‐of‐principle in vivo studies, we have documented that MSCs loaded with microparticles encapsulating a PSA‐activated prodrug are required to reach levels of ∼1%–10% to achieve a significant anti‐tumor effect in a prostate cancer xenograft model 69.
In these preclinical studies, the homing endpoints necessary for efficacy are often not met. For example, only ∼0.2% of injected human bone marrow‐derived MSCs (i.e., 2,000 cells) per gram of tissue were found in subcutaneous PC3 prostate cancer xenografts at 7 days postinfusion (Fig. 2B). Importantly, our group recently completed a phase 0 pre‐prostatectomy clinical trial to quantify homing of IV‐infused allogeneic BM‐MSCs via BEAMing 41 and NGS haplotype counting 42 to sites of primary prostate cancer [NCT019833709], which documented that MSC homing to prostate tissue was below the level of detection under the conditions and time points tested. In addition, homing of MSCs to the bone marrow, a site of significant disease burden in men with lethal metastatic prostate cancer and other tumor types such as breast cancer, has been analyzed in many studies, often revealing that culture‐expanded MSCs have poor bone marrow tropism and are frequently undetectable 20, 70. Limited tumor tropism was also demonstrated in mouse models of glioma; no eGFP+ MSCs were detected in N29 or N32 glioma xenografts 2 or 7 days after IV injection 71. In contrast, MSCs were detected in U87, U251, and LN229 glioma xenografts 7 days after local IA injection via the internal carotid artery, and MSCs expressing IFNβ extended survival in treated animals, suggesting that >2.5 × 104 cells (i.e., 2.5% of injected dose) reached the tumor based on controls 72. The same group also demonstrated that IFNβ‐MSCs at fractions as low as 1% of the tumor mass suppressed growth of subcutaneous A375 melanoma xenografts, that MSCs were preferentially localized in the tumor periphery 8 days after IV injection, and that IV‐injected IFNβ‐MSCs significantly prolonged survival in a metastatic melanoma model 73. Collectively, these studies highlight the importance of quantifying MSC homing and determining endpoints necessary for efficacy in each application and disease model.
MSC‐Based Anti‐Cancer Strategies
Although the role of MSCs in cancer initiation and progression remains unclear 74, this has not dampened interest in exploiting their (albeit limited) tumor tropism for the delivery of anti‐cancer agents (Fig. 1 ; Table 1). Consequently, delivery of these anti‐cancer agents to nontarget tissues and the potential for toxicity are a critical concern for MSC‐based drug delivery strategies. Approaches to increase either tumor selective delivery or specificity of the targeted agent are of significant interest. One such strategy has been to infect MSCs with an oncolytic adeno or measles virus that selectively replicates in tumor cells, which has the added advantage of amplifying the anti‐tumor effect with subsequent rounds of infection and lysis 75, 76, 77, 78. Another promising approach is the selective activation of prodrugs within the tumor microenvironment. These include engineering MSCs to express cytosine deaminase (CD) or herpes simplex virus‐thymidine kinase (Hsv‐TK) to convert an inactive systemically injected substrate [5‐fluorouracil (5‐FU) and ganciclovir, respectively] into their respective active cytotoxic metabolites 79, 80. Preclinical development of TK‐expressing MSCs by Bruns and Nelson et al. led to the landmark TREAT‐ME1 study in gastrointestinal adenocarcinomas [NCT02008539], the first clinical trial to use genetically engineered autologous MSCs. Due to the small study size (n = 6), conclusions are limited, but it should be noted the treatment was safe and tolerable with no significant adverse events attributed to the MSC infusion product 5, 81.
Prodrug strategies such as these alter pharmacokinetics and limit systemic drug exposure; however, the biodistribution of MSCs in nontarget tissue remains a significant safety concern as this is still the primary determinant of drug activation in peripheral tissue. Furthermore, to enhance specificity, multiple investigators have placed TK under the control of conditionally expressed promoters (e.g. CCL5 or Tie2) to restrict activity to the tumor microenvironment 82, 83. An alternative approach being explored by our group is the use of prodrugs activated by tissue‐ or tumor‐specific proteases, such as prostate‐specific antigen (PSA) to target prostate cancer cells, prostate‐specific membrane antigen (PSMA) to target the tumor neovasculature, or fibroblast activation protein (FAP) to target MSCs and the tumor‐associated stroma 69, 84, 85, 86, 87. This strategy couples a highly potent nonselective drug or toxin to an activating peptide substrate selectively recognized by a proteolytic enzyme whose expression is restricted to the target site, thereby engineering a tissue‐ or tumor‐specific ‘molecular grenade’ 88.
Another common MSC‐based drug delivery approach is to exogenously introduce various immunomodulatory proteins (e.g. type I interferons, chemokines, interleukins, etc.) 57, 73, 89, 90, 91, 92. Similarly, engineering MSCs to express TNF‐related apoptosis‐inducing ligand (TRAIL) has shown particular promise in multiple preclinical cancer models 93, 94, 95. This preclinical data together with decades of clinical observations regarding MSC biodistribution patterns led to the recently initiated TACTICAL trial, a phase I/II study of MSC‐TRAIL in combination with cisplatin and pemetrexed in non‐small cell lung cancer (NSCLC) patients [NCT03298763] 96. Previous efforts using systemic delivery of TRAIL as an anti‐cancer strategy failed clinical testing due to toxicity and poor responses at the administered doses 97. This provides the rationale for the selective delivery of TRAIL by MSCs within the tumor microenvironment to limit systemic toxicity to non‐target peripheral tissues. The rapid entrapment of MSCs in lung capillaries following IV infusion suggests this platform may be uniquely suited to act as a ‘biological micro‐factory’ producing an anti‐cancer agent (i.e., TRAIL) directly at the target site, at least for this clinical scenario (i.e., NSCLC); although hepatotoxicity may still be of significant concern.
Another potential application for therapeutic MSCs may be in the adjuvant setting for localized treatment of residual disease following surgery or radiotherapy. This could be particularly useful when extensive surgical resection or large radiation fields are difficult or associated with significant risks (e.g. glioblastoma) 50, 95, 98. Local delivery directly to the site of action obviously circumvents limitations associated with homing efficiency, and the high degree of overall safety observed in clinical trials using MSCs to date in other disease settings make this a feasible strategy.
Synthetic Strategies to Enhance MSC Homing
Due to the low homing efficiency of MSCs to many tissues of interest, novel methods to synthetically modify MSCs for enhanced targeting are critically needed. Using bone tropism as an example, low homing has been attributed to the lack of chemokine and adhesion molecules such as PSGL‐1, CXCR4, and E‐selectin ligands, particularly following ex vivo expansion in tissue culture 60, 70. In a seminal paper, Sackstein et al. developed a strategy to mimic hematopoietic stem cell (HSC) selectin‐mediated homing to the bone via glycan engineering 60. Essentially, exogenously introduced fucosyltransferases are used to modify CD44 expressed by MSCs into HCELL (hematopoietic cell E−/L‐selectin ligand), a potent E‐selectin ligand critical for HSC homing to the bone marrow 60, 99. This approach is currently being evaluated in two ongoing clinical trials [NCT02566655, NCT03096782], the results of which are eagerly anticipated. Of note, it was recently shown that CD44 expression on MSCs can be transiently increased via culturing on hyaluronic acid (HA)‐coated plates 100, potentially providing a simple method to enhance HCELL levels on MSCs and subsequent homing to the bone when used in combination with the fucosylation strategies described earlier.
In addition, Karp et al. have attached the prototypical E‐selectin ligand, sialyl Lewis X (sLeX), to the MSC surface via biotin‐streptavidin bridges introduced via a series of techniques with stabilities ranging from 8 hrs to 7 days 101. The same group has recently used a transient multiplex cell engineering strategy (i.e., triple mRNA transfection) to combine homing modification (PSGL‐1 and sLeX) with delivery of an anti‐inflammatory agent (IL‐10) 57. This approach increased MSC homing to γ‐irradiation‐induced inflammation in the murine ear by 31% and reduced inflammation‐induced swelling by 50% compared to unmodified MSCs. Small molecules upregulating the expression of other important homing ligands, such as those for intracellular adhesion molecule 1 (ICAM‐1), have been identified using high‐throughput screens 58. Treating MSCs with the top hit from this screen resulted in 5‐fold increased expression of CD11a, an important ICAM‐1 ligand, which resulted in an ∼2‐fold increase in homing toward LPS‐induced inflammation in the ear 58.
The biotin‐streptavidin bridge concept is a versatile technique that could be adapted for modification of MSCs with many readily‐available biotinylated molecules. Similar techniques using palmitated protein A/G or bi‐specific antibodies have also been developed for decorating MSCs with a variety of ligands or receptors 102, 103. Recently, Won et al. optimized conjugation chemistry for attaching lipid‐PEG to CXCR4 for noninvasive and rapid insertion into the cell membrane 104. Another approach has been developed using a NHS‐PEG2‐maleimide linker to conjugate thiol‐containing molecules to amine residues in native MSC cell surface proteins 105. Other studies suggest that relatively simple preconditioning regimens can be used to enhance tumor tropism, including hypoxia, estrogen exposure, and incubation with conditioned media from irradiated cancer cells 106, 107, 108, 109. In aggregate, techniques such as these have the potential to optimize synthetically engineered MSCs for homing to specific targets of interest; however, the breadth and diversity of modification techniques being applied to MSC homing presents a significant challenge for standardization across studies. Furthermore, the usefulness of these modifications may be mitigated in the absence of strategies to overcome first‐pass entrapment, which would potentiate active homing mechanisms by providing a window of operation.
Conclusion
As excitement for the promise of MSC‐based therapies, and synthetic biology approaches in general, continues to build and as these therapies increasingly undergo evaluation in the clinic, this review represents a sobering reminder of the broad biodistribution and poor homing efficiency to most target tissues observed using current methodologies (i.e., the ugly). MSC‐based drug delivery strategies are particularly sensitive to this challenge and will require clever bioengineering strategies to enhance the therapeutic index between benign and malignant tissue. Furthermore, rational study design regarding the choice of MSC populations, culture conditions for ex vivo expansion, in vivo tracking methodologies, and cell modification strategies is crucial. Employing robust, quantitative methodologies with standardized reporting metrics will facilitate accurate comparisons across studies and enable more rapid and efficient translation of the platforms that are most likely to succeed in the clinic (i.e., the good). Fortunately, there are multiple synthetic strategies in active development that will hopefully enable innovative MSC‐based strategies to reach the full promise of their potential.
Author Contributions
T.E.G.K., D.L.J.T., S.R.D., J.T.I., and W.N.B.: conception and design; T.E.G.K., D.L.J.T., and W.N.B.: collection and/or assembly of data; T.E.G.K., D.L.J.T., S.R.D., J.T.I., and W.N.B.: data analysis and interpretation; T.E.G.K., D.L.J.T., S.R.D., J.T.I., and W.N.B.: manuscript writing; T.E.G.K., D.L.J.T., S.R.D., J.T.I., and W.N.B.: final approval of manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
Acknowledgments
We acknowledge the expert assistance of Dr. Kenneth Valkenburg and Marie‐France Penet for their help with IC and IV injections, respectively. This work was supported by the following sources: Prostate Cancer Foundation Young Investigator Award (W.N.B. and D.L.J.T.), Patrick C. Walsh Prostate Cancer Research Fund (W.N.B. and D.L.J.T.), SKCCC CCSG developmental funds [P30 CA006973, (W.N.B.)], NIH‐Prostate SPORE [P50 CA058236, (S.R.D. and J.T.I.)], Department of Defense Prostate Cancer Research Program [W81XWH‐16‐1‐0410, (J.T.I.) and W81XWH‐17‐1‐0528, (W.N.B.)], and NCI [R01CA201035, (D.L.J.T.)].
References
- 1. Phinney DG. Functional heterogeneity of mesenchymal stem cells: Implications for cell therapy. J Cell Biochem 2012;113:2806–2812. 10.1002/jcb.24166 [DOI] [PubMed] [Google Scholar]
- 2. Caplan AI. Adult mesenchymal stem cells: When, where, and how. Stem Cells Int 2015;2015:628767–628766. 10.1155/2015/628767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brennen WN, Denmeade SR, Isaacs JT. Mesenchymal stem cells as a vector for the inflammatory prostate microenvironment. Endocr Relat Cancer 2013;20:R269–R290. 10.1530/ERC-13-0151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. D'Souza N, Rossignoli F, Golinelli G et al. Mesenchymal stem/stromal cells as a delivery platform in cell and gene therapies. BMC Med 2015;13:186 10.1186/s12916-015-0426-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hagenhoff A, Bruns CJ, Zhao Y et al. Harnessing mesenchymal stem cell homing as an anticancer therapy. Expert Opin Biol Ther 2016;16:1079–1092. 10.1080/14712598.2016.1196179 [DOI] [PubMed] [Google Scholar]
- 6. Ankrum J, Karp JM. Mesenchymal stem cell therapy: Two steps forward, one step back. Trends Mol Med 2010;16:203–209. 10.1016/j.molmed.2010.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Prockop DJ, Oh JY. Mesenchymal stem/stromal cells (MSCs): Role as guardians of inflammation. Mol Ther 2012;20:14–20. 10.1038/mt.2011.211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Phinney DG, Pittenger MF. Concise review: MSC‐derived exosomes for cell‐free therapy. Stem Cells 2017;35:851–858. 10.1002/stem.2575 [DOI] [PubMed] [Google Scholar]
- 9. Crivelli B, Chlapanidas T, Perteghella S et al. Mesenchymal stem/stromal cell extracellular vesicles: From active principle to next generation drug delivery system. J Control Release 2017;262:104–117. 10.1016/j.jconrel.2017.07.023 [DOI] [PubMed] [Google Scholar]
- 10. Galleu A, Riffo‐Vasquez Y, Trento C et al. Apoptosis in mesenchymal stromal cells induces in vivo recipient‐mediated immunomodulation. Sci Transl Med 2017;9:eaarn7828 10.1126/scitranslmed.aam7828 [DOI] [PubMed] [Google Scholar]
- 11. de Witte SFH, Luk F, Sierra Parraga JM et al. Immunomodulation by therapeutic mesenchymal stromal cells (MSC) is triggered through phagocytosis of MSC by monocytic cells. Stem Cells 2018;36:602–615. 10.1002/stem.2779 [DOI] [PubMed] [Google Scholar]
- 12. Luan X, Sansanaphongpricha K, Myers I et al. Engineering exosomes as refined biological nanoplatforms for drug delivery. Acta Pharmacol Sin 2017;38:754–763. 10.1038/aps.2017.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. De Becker A, Riet IV. Homing and migration of mesenchymal stromal cells: How to improve the efficacy of cell therapy? World J Stem Cells 2016;8:73–87. 10.4252/wjsc.v8.i3.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. English K, Mahon BP. Allogeneic mesenchymal stem cells: Agents of immune modulation. J Cell Biochem 2011;112:1963–1968. 10.1002/jcb.23119 [DOI] [PubMed] [Google Scholar]
- 15. Prockop DJ. Concise review: Two negative feedback loops place mesenchymal stem/stromal cells at the center of early regulators of inflammation. Stem Cells 2013;31:2042–2046. 10.1002/stem.1400 [DOI] [PubMed] [Google Scholar]
- 16. Caplan AI, Sorrell JM. The MSC curtain that stops the immune system. Immunol Lett 2015;168:136–139. 10.1016/j.imlet.2015.06.005 [DOI] [PubMed] [Google Scholar]
- 17. Spaeth E, Klopp A, Dembinski J et al. Inflammation and tumor microenvironments: Defining the migratory itinerary of mesenchymal stem cells. Gene Ther 2008;15:730–738. 10.1038/gt.2008.39 [DOI] [PubMed] [Google Scholar]
- 18. Ruster B, Gottig S, Ludwig RJ et al. Mesenchymal stem cells display coordinated rolling and adhesion behavior on endothelial cells. Blood 2006;108:3938–3944. 10.1182/blood-2006-05-025098 [DOI] [PubMed] [Google Scholar]
- 19. Nitzsche F, Müller C, Lukomska B et al. Concise review: MSC adhesion cascade‐insights into homing and transendothelial migration. Stem Cells 2017;35:1446–1460. 10.1002/stem.2614 [DOI] [PubMed] [Google Scholar]
- 20. Gao J, Dennis JE, Muzic RF et al. The dynamic in vivo distribution of bone marrow‐derived mesenchymal stem cells after infusion. Cells Tissues Organs 2001;169:12–20. 10.1159/000047856 [DOI] [PubMed] [Google Scholar]
- 21. Schrepfer S, Deuse T, Reichenspurner H et al. Stem cell transplantation: The lung barrier. Transplant Proc 2007;39:573–576. 10.1016/j.transproceed.2006.12.019 [DOI] [PubMed] [Google Scholar]
- 22. Brennen WN, Kisteman LN, Isaacs JT. Rapid selection of mesenchymal stem and progenitor cells in primary prostate stromal cultures. Prostate 2016;76:552–564. 10.1002/pros.23145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Toma C, Wagner WR, Bowry S et al. Fate of culture‐expanded mesenchymal stem cells in the microvasculature: in vivo observations of cell kinetics. Circ Res 2009;104:398–402. 10.1161/CIRCRESAHA.108.187724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Koc ON, Gerson SL, Cooper BW et al. Rapid hematopoietic recovery after coinfusion of autologous‐blood stem cells and culture‐expanded marrow mesenchymal stem cells in advanced breast cancer patients receiving high‐dose chemotherapy. J Clin Oncol 2000;18:307–316. 10.1200/JCO.2000.18.2.307 [DOI] [PubMed] [Google Scholar]
- 25. Devine SM, Cobbs C, Jennings M et al. Mesenchymal stem cells distribute to a wide range of tissues following systemic infusion into nonhuman primates. Blood 2003;101:2999–3001. 10.1182/blood-2002-06-1830 [DOI] [PubMed] [Google Scholar]
- 26. Kraitchman DL, Tatsumi M, Gilson WD et al. Dynamic imaging of allogeneic mesenchymal stem cells trafficking to myocardial infarction. Circulation 2005;112:1451–1461. 10.1161/CIRCULATIONAHA.105.537480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gholamrezanezhad A, Mirpour S, Bagheri M et al. In vivo tracking of 111In‐oxine labeled mesenchymal stem cells following infusion in patients with advanced cirrhosis. Nucl Med Biol 2011;38:961–967. 10.1016/j.nucmedbio.2011.03.008 [DOI] [PubMed] [Google Scholar]
- 28. Lee RH, Pulin AA, Seo MJ et al. Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti‐inflammatory protein TSG‐6. Cell Stem Cell 2009;5:54–63. 10.1016/j.stem.2009.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fischer UM, Harting MT, Jimenez F et al. Pulmonary passage is a major obstacle for intravenous stem cell delivery: The pulmonary first‐pass effect. Stem Cells Dev 2009;18:683–692. 10.1089/scd.2008.0253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dutly AE, Kugathasan L, Trogadis JE et al. Fluorescent microangiography (FMA): An improved tool to visualize the pulmonary microvasculature. Lab Invest 2006;86:409–416. 10.1038/labinvest.3700399 [DOI] [PubMed] [Google Scholar]
- 31. Lipowsky HH, Bowers DT, Banik BL et al. Mesenchymal stem cell deformability and implications for microvascular sequestration. Ann Biomed Eng 2018;46:640–654. 10.1007/s10439-018-1985-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brennen WN, Zhang B, Kulac I et al. Mesenchymal stem cell infiltration during neoplastic transformation of the human prostate. Oncotarget 2017;8:46710–46727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bansal A, Pandey MK, Demirhan YE et al. Novel (89)Zr cell labeling approach for PET‐based cell trafficking studies. EJNMMI Res 2015;5:19 10.1186/s13550-015-0098-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zanetti A, Grata M, Etling EB et al. Suspension‐expansion of bone marrow results in small mesenchymal stem cells exhibiting increased transpulmonary passage following intravenous administration. Tissue Eng Part C Methods 2015;21:683–692. 10.1089/ten.TEC.2014.0344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sekiya I, Larson BL, Smith JR et al. Expansion of human adult stem cells from bone marrow stroma: Conditions that maximize the yields of early progenitors and evaluate their quality. Stem Cells 2002;20:530–541. 10.1634/stemcells.20-6-530 [DOI] [PubMed] [Google Scholar]
- 36. Alimperti S, Lei P, Wen Y et al. Serum‐free spheroid suspension culture maintains mesenchymal stem cell proliferation and differentiation potential. Biotechnol Prog 2014;30:974–983. 10.1002/btpr.1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baksh D, Zandstra PW, Davies JE. A non‐contact suspension culture approach to the culture of osteogenic cells derived from a CD49elow subpopulation of human bone marrow‐derived cells. Biotechnol Bioeng 2007;98:1195–1208. 10.1002/bit.21556 [DOI] [PubMed] [Google Scholar]
- 38. Luo L, Tang J, Nishi K et al. Fabrication of synthetic mesenchymal stem cells for the treatment of acute myocardial infarction in mice. Circ Res 2017;120:1768–1775. 10.1161/circresaha.116.310374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pittenger MF, Martin BJ. Mesenchymal stem cells and their potential as cardiac therapeutics. Circ Res 2004;95:9–20. 10.1161/01.RES.0000135902.99383.6f [DOI] [PubMed] [Google Scholar]
- 40. Iso Y, Spees JL, Serrano C et al. Multipotent human stromal cells improve cardiac function after myocardial infarction in mice without long‐term engraftment. Biochem Biophys Res Commun 2007;354:700–706. 10.1016/j.bbrc.2007.01.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Diehl F, Li M, Dressman D et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Nat Acad Sci USA 2005;102:16368–16373. 10.1073/pnas.0507904102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Debeljak M, Mocci E, Morrison MC et al. Haplotype counting for sensitive chimerism testing: Potential for early leukemia relapse detection. J Mol Diagn 2017;19:427–436. 10.1016/j.jmoldx.2017.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tatebe M, Nakamura R, Kagami H et al. Differentiation of transplanted mesenchymal stem cells in a large osteochondral defect in rabbit. Cytotherapy 2005;7:520–530. 10.1080/14653240500361350 [DOI] [PubMed] [Google Scholar]
- 44. Weir C, Morel‐Kopp MC, Gill A et al. Mesenchymal stem cells: Isolation, characterisation and in vivo fluorescent dye tracking. Heart Lung Circ 2008;17:395–403. 10.1016/j.hlc.2008.01.006 [DOI] [PubMed] [Google Scholar]
- 45. Polzer H, Volkmer E, Saller MM et al. Long‐term detection of fluorescently labeled human mesenchymal stem cell in vitro and in vivo by semi‐automated microscopy. Tissue Eng Part C Methods 2012;18:156–165. 10.1089/ten.TEC.2011.0275 [DOI] [PubMed] [Google Scholar]
- 46. Perez JR, Ybarra N, Chagnon F et al. Tracking of mesenchymal stem cells with fluorescence endomicroscopy imaging in radiotherapy‐induced lung injury. Sci Rep 2017;7:40748 10.1038/srep40748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Barbash IM, Chouraqui P, Baron J et al. Systemic delivery of bone marrow‐derived mesenchymal stem cells to the infarcted myocardium: Feasibility, cell migration, and body distribution. Circulation 2003;108:863–868. 10.1161/01.CIR.0000084828.50310.6A [DOI] [PubMed] [Google Scholar]
- 48. Knoop K, Kolokythas M, Klutz K et al. Image‐guided, tumor stroma‐targeted 131I therapy of hepatocellular cancer after systemic mesenchymal stem cell‐mediated NIS gene delivery. Mol Ther : J Am Soc Gene Ther 2011;19:1704–1713. 10.1038/mt.2011.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dwyer RM, Ryan J, Havelin RJ et al. Mesenchymal Stem Cell‐mediated delivery of the sodium iodide symporter supports radionuclide imaging and treatment of breast cancer. Stem Cells 2011;29:1149–1157. 10.1002/stem.665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Martinez‐Quintanilla J, Bhere D, Heidari P et al. Therapeutic efficacy and fate of bimodal engineered stem cells in malignant brain tumors. Stem Cells 2013;31:1706–1714. 10.1002/stem.1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ell PJ, Hocknell JM, Jarritt PH et al. A 99Tcm‐labelled radiotracer for the investigation of cerebral vascular disease. Nucl Med Commun 1985;6:437–441 [DOI] [PubMed] [Google Scholar]
- 52. Park BN, Shim W, Lee G et al. Early distribution of intravenously injected mesenchymal stem cells in rats with acute brain trauma evaluated by (99m)Tc‐HMPAO labeling. Nucl Med Biol 2011;38:1175–1182. 10.1016/j.nucmedbio.2011.05.009 [DOI] [PubMed] [Google Scholar]
- 53. Rajam PC, Jackson AL. Distribution and valence state of radiochromium in intracellularly labeled ehrlich mouse ascites carcinoma cells. Proc Soc Exp Biol Med 1958;99:210–213 [DOI] [PubMed] [Google Scholar]
- 54. Fidler IJ. Metastasis: Quantitative analysis of distribution and fate of tumor emboli labeled with 125 I‐5‐iodo‐2′‐deoxyuridine. J Natl Cancer Inst 1970;45:773–782 [PubMed] [Google Scholar]
- 55. Kim KS, Park W, Na K. Gadolinium‐chelate nanoparticle entrapped human mesenchymal stem cell via photochemical internalization for cancer diagnosis. Biomaterials 2015;36:90–97. 10.1016/j.biomaterials.2014.09.014 [DOI] [PubMed] [Google Scholar]
- 56. Huang X, Zhang F, Wang Y et al. Design considerations of iron‐based nanoclusters for noninvasive tracking of mesenchymal stem cell homing. ACS Nano 2014;8:4403–4414. 10.1021/nn4062726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Levy O, Zhao W, Mortensen LJ et al. mRNA‐engineered mesenchymal stem cells for targeted delivery of interleukin‐10 to sites of inflammation. Blood 2013;122:e23–e32. 10.1182/blood-2013-04-495119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Levy O, Mortensen LJ, Boquet G et al. A small‐molecule screen for enhanced homing of systemically infused cells. Cell Rep 2015;10:1261–1268. 10.1016/j.celrep.2015.01.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sipkins DA, Wei X, Wu JW et al. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature 2005;435:969–973. 10.1038/nature03703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sackstein R, Merzaban JS, Cain DW et al. Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat Med 2008;14:181–187. 10.1038/nm1703 [DOI] [PubMed] [Google Scholar]
- 61. Boissonnas A, Fetler L, Zeelenberg IS et al. In vivo imaging of cytotoxic T cell infiltration and elimination of a solid tumor. J Exp Med 2007;204:345–356. 10.1084/jem.20061890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Novak J, Georgakoudi I, Wei X et al. In vivo flow cytometer for real‐time detection and quantification of circulating cells. Optics Lett 2004;29:77–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fan ZC, Yan J, Liu GD et al. Real‐time monitoring of rare circulating hepatocellular carcinoma cells in an orthotopic model by in vivo flow cytometry assesses resection on metastasis. Cancer Res 2012;72:2683–2691. 10.1158/0008-5472.Can-11-3733 [DOI] [PubMed] [Google Scholar]
- 64. Xie C, Yang Z, Suo Y et al. Systemically infused mesenchymal stem cells show different homing profiles in healthy and tumor mouse models. Stem Cells Translational Medicine 2017;6:1120–1131. 10.1002/sctm.16-0204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kidd S, Spaeth E, Dembinski JL et al. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells 2009;27:2614–2623. 10.1002/stem.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Brennen WN, Chen S, Denmeade SR et al. Quantification of mesenchymal stem cells (MSCs) at sites of human prostate cancer. Oncotarget 2013;4:106–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pessina A, Leonetti C, Artuso S et al. Drug‐releasing mesenchymal cells strongly suppress B16 lung metastasis in a syngeneic murine model. J Exp Clin Cancer Res 2015;34:82 10.1186/s13046-015-0200-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pessina A, Bonomi A, Coccè V et al. Mesenchymal stromal cells primed with paclitaxel provide a new approach for cancer therapy. PLoS One 2011;6:e28321 10.1371/journal.pone.0028321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Levy O, Brennen WN, Han E et al. A prodrug‐doped cellular Trojan Horse for the potential treatment of prostate cancer. Biomaterials 2016;91:140–150. 10.1016/j.biomaterials.2016.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rombouts WJ, Ploemacher RE. Primary murine MSC show highly efficient homing to the bone marrow but lose homing ability following culture. Leukemia 2003;17:160–170. 10.1038/sj.leu.2402763 [DOI] [PubMed] [Google Scholar]
- 71. Bexell D, Gunnarsson S, Tormin A et al. Bone marrow multipotent mesenchymal stroma cells act as pericyte‐like migratory vehicles in experimental gliomas. Mol Ther : J Am Soc Gene Ther 2009;17:183–190. 10.1038/mt.2008.229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nakamizo A, Marini F, Amano T et al. Human bone marrow‐derived mesenchymal stem cells in the treatment of gliomas. Cancer Res 2005;65:3307–3318. 10.1158/0008-5472.Can-04-1874 [DOI] [PubMed] [Google Scholar]
- 73. Studeny M, Marini FC, Champlin RE et al. Bone marrow‐derived mesenchymal stem cells as vehicles for interferon‐beta delivery into tumors. Cancer Res 2002;62:3603–3608 [PubMed] [Google Scholar]
- 74. Klopp AH, Gupta A, Spaeth E et al. Concise review: Dissecting a discrepancy in the literature: Do mesenchymal stem cells support or suppress tumor growth? Stem Cells 2011;29:11–19. 10.1002/stem.559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Stoff‐Khalili MA, Rivera AA, Mathis JM et al. Mesenchymal stem cells as a vehicle for targeted delivery of CRAds to lung metastases of breast carcinoma. Breast Cancer Res Treat 2007;105:157–167. 10.1007/s10549-006-9449-8 [DOI] [PubMed] [Google Scholar]
- 76. Hakkarainen T, Särkioja M, Lehenkari P et al. Human mesenchymal stem cells lack tumor tropism but enhance the antitumor activity of oncolytic adenoviruses in orthotopic lung and breast tumors. Human Gene ther 2007;18:627–641. 10.1089/hum.2007.034 [DOI] [PubMed] [Google Scholar]
- 77. Dembinski JL, Spaeth EL, Fueyo J et al. Reduction of nontarget infection and systemic toxicity by targeted delivery of conditionally replicating viruses transported in mesenchymal stem cells. Cancer Gene Ther 2010;17:289–297. 10.1038/cgt.2009.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Castleton A, Dey A, Beaton B et al. Human mesenchymal stromal cells deliver systemic oncolytic measles virus to treat acute lymphoblastic leukemia in the presence of humoral immunity. Blood 2014;123:1327–1335. 10.1182/blood-2013-09-528851 [DOI] [PubMed] [Google Scholar]
- 79. Kucerova L, Altanerova V, Matuskova M et al. Adipose tissue‐derived human mesenchymal stem cells mediated prodrug cancer gene therapy. Cancer Res 2007;67:6304–6313. 10.1158/0008-5472.Can-06-4024 [DOI] [PubMed] [Google Scholar]
- 80. Cavarretta IT, Altanerova V, Matuskova M et al. Adipose tissue‐derived mesenchymal stem cells expressing prodrug‐converting enzyme inhibit human prostate tumor growth. Mol Ther : J Am Soc Gene Ther 2010;18:223–231. 10.1038/mt.2009.237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. von Einem JC, Sylvia P, Christine G et al. Treatment of advanced gastrointestinal cancer with genetically modified autologous mesenchymal stem cells ‐ TREAT‐ME‐1 – A phase I, first in human, first in class trial. Oncotarget 2017;8:80156–80166. https://doi.org/10.18632/oncotarget.20964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zischek C, Niess H, Ischenko I et al. Targeting tumor stroma using engineered mesenchymal stem cells reduces the growth of pancreatic carcinoma. Annals Surg 2009;250:747–753. 10.1097/SLA.0b013e3181bd62d0 [DOI] [PubMed] [Google Scholar]
- 83. Conrad C, Hüsemann Y, Niess H et al. Linking transgene expression of engineered mesenchymal stem cells and angiopoietin‐1‐induced differentiation to target cancer angiogenesis. Annals Surg 2011;253:566–571. 10.1097/SLA.0b013e3181fcb5d8 [DOI] [PubMed] [Google Scholar]
- 84. Denmeade SR, Jakobsen CM, Janssen S et al. Prostate‐specific antigen‐activated thapsigargin prodrug as targeted therapy for prostate cancer. J Natl Cancer Inst 2003;95:990–1000 [DOI] [PubMed] [Google Scholar]
- 85. Williams SA, Merchant RF, Garrett‐Mayer E et al. A prostate‐specific antigen‐activated channel‐forming toxin as therapy for prostatic disease. J Natl Cancer Inst 2007;99:376–385. 10.1093/jnci/djk065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Brennen WN, Rosen DM, Wang H et al. Targeting carcinoma‐associated fibroblasts within the tumor stroma with a fibroblast activation protein‐activated prodrug. J Natl Cancer Inst 2012;104:1320–1334. 10.1093/jnci/djs336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Denmeade SR, Mhaka AM, Rosen DM et al. Engineering a prostate‐specific membrane antigen‐activated tumor endothelial cell prodrug for cancer therapy. Sci Transl Med 2012;4:140ra186 10.1126/scitranslmed.3003886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Denmeade SR, Isaacs JT. Engineering enzymatically activated “molecular grenades” for cancer. Oncotarget 2012;3:666–667. https://doi.org/10.18632/oncotarget.562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Stagg J, Lejeune L, Paquin A et al. Marrow stromal cells for interleukin‐2 delivery in cancer immunotherapy. Human Gene ther 2004;15:597–608. 10.1089/104303404323142042 [DOI] [PubMed] [Google Scholar]
- 90. Xin H, Kanehira M, Mizuguchi H et al. Targeted delivery of CX3CL1 to multiple lung tumors by mesenchymal stem cells. Stem Cells 2007;25:1618–1626. 10.1634/stemcells.2006-0461 [DOI] [PubMed] [Google Scholar]
- 91. Ren C, Kumar S, Chanda D et al. Cancer gene therapy using mesenchymal stem cells expressing interferon‐beta in a mouse prostate cancer lung metastasis model. Gene Ther 2008;15:1446–1453. 10.1038/gt.2008.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Seo SH, Kim KS, Park SH et al. The effects of mesenchymal stem cells injected via different routes on modified IL‐12‐mediated antitumor activity. Gene Ther 2011;18:488–495. 10.1038/gt.2010.170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Luetzkendorf J, Mueller LP, Mueller T et al. Growth inhibition of colorectal carcinoma by lentiviral TRAIL‐transgenic human mesenchymal stem cells requires their substantial intratumoral presence. J Cell Mol Med 2010;14:2292–2304. 10.1111/j.1582-4934.2009.00794.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Loebinger MR, Eddaoudi A, Davies D et al. Mesenchymal stem cell delivery of TRAIL can eliminate metastatic cancer. Cancer Res 2009;69:4134–4142. 10.1158/0008-5472.Can-08-4698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Sasportas LS, Kasmieh R, Wakimoto H et al. Assessment of therapeutic efficacy and fate of engineered human mesenchymal stem cells for cancer therapy. Proc Nat Acad Sci USA 2009;106:4822–4827. 10.1073/pnas.0806647106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Sage EK, Thakrar RM, Janes SM. Genetically modified mesenchymal stromal cells in cancer therapy. Cytotherapy 2016;18:1435–1445. 10.1016/j.jcyt.2016.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. von Karstedt S, Montinaro A, Walczak H. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat Rev Cancer 2017;17:352–366. 10.1038/nrc.2017.28 [DOI] [PubMed] [Google Scholar]
- 98. Duebgen M, Martinez‐Quintanilla J, Tamura K et al. Stem cells loaded with multimechanistic oncolytic herpes simplex virus variants for brain tumor therapy. J Natl Cancer Inst 2014;106:dju090 10.1093/jnci/dju090 [DOI] [PubMed] [Google Scholar]
- 99. Dykstra B, Lee J, Mortensen LJ et al. Glycoengineering of E‐selectin ligands by intracellular versus extracellular fucosylation differentially affects osteotropism of human mesenchymal stem cells. Stem Cells 2016;34:2501–2511. 10.1002/stem.2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Corradetti B, Taraballi F, Martinez JO et al. Hyaluronic acid coatings as a simple and efficient approach to improve MSC homing toward the site of inflammation. Sc Rep 2017;7:7991 10.1038/s41598-017-08687-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Sarkar D, Spencer JA, Phillips JA et al. Eng. Cell Homing. Blood 2011;118:e184–e191. 10.1182/blood-2010-10-311464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Ko IK, Kim BG, Awadallah A et al. Targeting improves MSC treatment of inflammatory bowel disease. Mol Ther : J Am Soc Gene Therapy 2010;18:1365–1372. 10.1038/mt.2010.54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Lee RJ, Fang Q, Davol PA et al. Antibody targeting of stem cells to infarcted myocardium. Stem Cells 2007;25:712–717. 10.1634/stemcells.2005-0602 [DOI] [PubMed] [Google Scholar]
- 104. Won YW, Patel AN, Bull DA. Cell surface engineering to enhance mesenchymal stem cell migration toward an SDF‐1 gradient. Biomaterials 2014;35:5627–5635. 10.1016/j.biomaterials.2014.03.070 [DOI] [PubMed] [Google Scholar]
- 105. Cheng H, Byrska‐Bishop M, Zhang CT et al. Stem cell membrane engineering for cell rolling using peptide conjugation and tuning of cell‐selectin interaction kinetics. Biomaterials 2012;33:5004–5012. 10.1016/j.biomaterials.2012.03.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Klopp AH, Spaeth EL, Dembinski JL et al. Tumor irradiation increases the recruitment of circulating mesenchymal stem cells into the tumor microenvironment. Cancer Res 2007;67:11687–11695. 10.1158/0008-5472.CAN-07-1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Das R, Jahr H, van Osch GJ et al. The role of hypoxia in bone marrow‐derived mesenchymal stem cells: Considerations for regenerative medicine approaches. Tissue Eng Part B Rev 2010;16:159–168. 10.1089/ten.TEB.2009.0296 [DOI] [PubMed] [Google Scholar]
- 108. Mirzamohammadi S, Aali E, Najafi R et al. Effect of 17beta‐estradiol on mediators involved in mesenchymal stromal cell trafficking in cell therapy of diabetes. Cytotherapy 2015;17:46–57. 10.1016/j.jcyt.2014.06.009 [DOI] [PubMed] [Google Scholar]
- 109. Smith CL, Chaichana KL, Lee YM et al. Pre‐exposure of human adipose mesenchymal stem cells to soluble factors enhances their homing to brain cancer. Stem Cells Translational Medicine 2015;4:239–251. 10.5966/sctm.2014-0149 [DOI] [PMC free article] [PubMed] [Google Scholar]