

Abstract
Background
In subcutaneous adipose tissue (SAT), higher stearoyl-CoA desaturase-1 (SCD1) expression has been related to improved insulin sensitivity in thiazolidinedione-treated type 2 diabetes mellitus patients. In animal models, deficiency of the free fatty acid receptor (FFAR) 2 associated with higher and FFAR4-deficiency with lower insulin sensitivity. We hypothesized that increased FFAR2 expression and reductions in FFAR4 and SCD1 expression in SAT of type 2 diabetes mellitus patients associate positively with insulin resistance and impaired beta cell function.
Methods
Twenty-five type 2 diabetes mellitus patients and 25 glucose-tolerant humans (CON) matched for sex, age, and BMI underwent mixed-meal tests to assess insulin sensitivity (OGIS) and beta cell function (ΔAUC(C-peptide)0–180 min/ΔAUC(glucose)0–180 min) in a cross-sectional study. Gene and protein expression of SCD1 and FFAR2/4 were quantified in SAT biopsies.
Results
Insulin sensitivity was 14% and beta cell function 71% (both p < 0.001) lower in type 2 diabetes mellitus patients. In type 2 diabetes mellitus, SCD1 mRNA was fivefold (p < 0.001) and protein expression twofold (p < 0.01) lower. While FFAR2/4 mRNA and protein expression did not differ between groups, FFAR2 protein levels correlated negatively with beta cell function only in CON (r = −0.74, p < 0.01). However, neither SCD1 nor FFAR2/4 protein expression correlated with insulin sensitivity in both groups.
Conclusions
Type 2 diabetes patients have lower SCD1, which does not associate with insulin resistance. Only in non-diabetic humans, FFAR2 associated with impaired beta cell function.
Introduction
Type 2 diabetes is characterized by insufficient insulin secretion to compensate for peripheral insulin resistance1. Studies in animal models and humans implicate free fatty acid receptors (FFAR), also known as G-protein coupled receptors (GPR), as receptors for non-esterified fatty acids (NEFA) in the pathogenesis of beta cell dysfunction and progression to insulin resistance and type 2 diabetes mellitus2,3.
FFAR2 and FFAR4 (also known as GPR43 and GPR120) serve as receptors for acetate and long-chain fatty acids (FA)4,5, respectively, and are supposed to contribute to the regulation of glucose homeostasis through FA signaling pathways6. Recent studies provided evidence that FFAR2-deficient mice on high-fat diet are protected from the increase in body fat mass and dyslipidemia, accompanied by increased insulin sensitivity (IS)7. In addition, in mouse islets FFAR2 gene expression was increased during the insulin-resistant phase of pregnancy8,9. One translational study further identified gene expression of FFAR2 in mouse and human islets, and suggested FFAR2 to mediate inhibition of insulin secretion by coupling to Gi-type G proteins3. Beta cell-specific deletion of FFAR2 in another mouse model led to increased insulin secretion and improved glucose tolerance3. These findings in beta cells from mouse models and human in vitro studies point to an involvement of FFAR2 in maintaining glucose homeostasis. Adipose tissue (AT) expansion in obesity associates with insulin resistance and progressive immune cell infiltration in AT10. Pro-inflammatory cytokines activate lipolysis11 causing dyslipidemia12, lipid-induced insulin resistance in peripheral tissues13, and impairment of beta cell function14. In contrast, FFAR2 knock out mice were protected from high-fat diet-induced AT inflammation and obesity7. Thus, FFAR2 may serve as a potential target for diabetes prevention strategies via inhibition of lipid-induced insulin resistance.
A previous study showed that FFAR4 activation by omega-3 fatty acid protected human islets from palmitate-induced apoptosis, whereas FFAR4 knock out attenuated omega-3 fatty acid-related anti-apoptotic effects15. Compared to wild-type mice, high-fat fed FFAR4-deficient mice developed more severe obesity, liver fat accumulation, and insulin resistance16,17. However, these findings were accompanied by lower stearoyl-CoA desaturase-1 (SCD1) gene expression in AT18. In murine models, four isoforms (SCD1, SCD2, SCD3, and SCD4) have been identified, whereas humans express only two Δ9 desaturases (SCD1 and SCD5). Our study focused on SCD1 as the most highly expressed SCD isoform in AT. SCD1 in AT facilitates the protective conversion of lipotoxic lipid species (saturated into monounsaturated FA). Circulating palmitoleate, an AT derived product of SCD1, increased insulin signaling in both skeletal muscle and the liver, increased insulin secretion from beta cells, and improved whole-body glucose uptake in mice19. Furthermore, palmitoleate treatment reduced cytokine expression in cultured adipocytes19. SCD1 in AT facilitates the last step of de novo lipogenesis and induces incorporation of FA into triglycerides (TG), both associating positively with whole-body insulin sensitivity. Accordingly, thiazolidinedione treatment promoted TG esterification in cultured adipocytes20 and increased SCD1 gene expression in subcutaneous adipose tissue (SAT) with subsequent improvement of IS in patients with type 2 diabetes mellitus, suggesting a potential role of SCD1 in AT on systemic glucose homeostasis21.
Although FFAR2/4 and SCD1 seem to be involved in maintaining glucose homeostasis in mice3,16,17,22, the relevance of their expression in human SAT for glucose homeostasis has not yet been elucidated. FFARs and SCD1 are expressed in various tissues, but might be especially important in AT due to its prominent role in lipid turnover. Here, we hypothesized that increased FFAR2 expression and reduced FFAR4 and SCD1 expression in SAT of patients with type 2 diabetes mellitus in the fasted state associate positively with insulin resistance and inversely with beta cell function. Furthermore, we hypothesized that increased FFAR2 and reduced FFAR4 expression in AT of type 2 diabetes patients associate with parameters of dyslipidemia. Finally, we hypothesized that higher SCD1 expression in AT of type 2 diabetes patients associates negatively with high-sensitivity C-reactive protein (hsCRP) in plasma. To this end, we analyzed FFAR2 as well as FFAR4 and SCD1 mRNA and protein expression in SAT of 25 metabolically well-characterized patients with newly diagnosed type 2 diabetes mellitus and 25 age-matched, sex-matched, and BMI-matched glucose-tolerant humans (CON).
Materials and methods
Study participants
The study population comprised 25 patients with recently diagnosed type 2 diabetes mellitus and 25 age-matched, sex-matched, and BMI-matched CON. All participants gave their written informed consent before inclusion into the study (ClinicalTrial.gov registration no: NCT01055093), which was performed according to the Declaration of Helsinki and approved by the ethics board of Heinrich Heine University, Düsseldorf, Germany. Participants were recruited via general practitioners, internet, or advertisements in newspapers. For three days prior to each visit, participants refrained from physical activity and alcohol ingestion and fasted for 10 h on the day before the metabolic studies. Exclusion criteria comprised medical history of acute or chronic diseases including cancer, insulin or thiazolidinedione treatment, medication affecting the immune system and/or a HbA1c > 9.0% (75 mmol mol−1), diabetes other than type 2 diabetes mellitus. Patients with type 2 diabetes mellitus were treated with metformin only (n = 15), sulfonylurea only (n = 2), metformin and sulfonylurea (n = 2), glucagon-like peptide-1 receptor agonist and metformin (n = 1), or diet only (n = 5). They withdrew their oral glucose-lowering medication for at least 3 days before all measurement to exclude acute effects on glucose metabolism23. All patients with type 2 diabetes mellitus also participated in the baseline cohort of the ongoing German Diabetes Study (GDS), a prospective observational study investigating the natural course of recently diagnosed diabetes and the development of diabetes-associated complications. The study design and cohort profile of the GDS are described in detail elsewhere23. Age-matched, sex-matched, and BMI-matched glucose-tolerant participants were recruited as control group (CON).
Mixed-meal test (MMT)
All participants underwent a standardized MMT to assess whole-body IS and beta cell function. For the MMT, each participant consumed 360 ml of Boost High Protein (Nestlé Nutrition, Vervey, Switzerland) containing 41 g of glucose, 9 g of fat, and 23 g protein before 10 am within 5 min followed by defined blood sampling for specific parameters described elsewhere23,24. Dynamic IS was assessed by the oral glucose insulin sensitivity index (OGIS), which allows calculating whole-body IS during both oral glucose tolerance test (OGTT) and MMT, provided that the dose of glucose administered during the test is taken into account25,26. Beta cell function was assessed from incremental AUC of plasma glucose, insulin, and C-peptide concentrations during MMT. The insulinogenic index from ΔAUC(C-peptide)0–180 min/ΔAUC(glucose)0–180 min, and ΔAUC(insulin)0–180 min/ΔAUC(glucose)0–180 min was used to describe insulin secretion in relationship to glucose as a measure of beta cell function27.
Oral glucose tolerance test (OGTT)
All CON underwent a 75 g-OGTT (Accu-Chek Dextro O.G-T., Roche, Basel, Switzerland) after at least 10 h overnight fasting to assess glucose tolerance and exclude participants with (pre-) diabetes mellitus. Blood samples were taken at time points −5, 30, 60, 120, and 180 min and glucose tolerance was categorized according to internationally accepted criteria28.
Laboratory analyses
Plasma glucose, HbA1c, NEFA, high-density lipoprotein (HDL) cholesterol, low-density lipoprotein (LDL) cholesterol, TG and hsCRP, insulin, and C-peptide were measured as previously described23. Plasma glucagon was measured using radioimmunoassay (Millipore, St. Charles, MO, USA).
Adipose tissue analyses
A biopsy was obtained from abdominal SAT at the level of umbilicus by needle suction technique after administration of local anesthesia (5–10 ml of 1% lidocaine) under fasting conditions. Fat tissue specimen were immediately frozen in liquid nitrogen, and stored −80 °C until analysis. For analyses of mRNA expression, total RNA was isolated (miRNeasy Mini Kit, Qiagen, Hilden, Germany) including on-column DNase digestion. For gene expression analyses, RNA quantity and quality were determined by Nanodrop (Peqlab, Erlangen, Germany) and RNA 6000 Nano Kit (Agilent Technologies, Böblingen, Germany). The complementary DNA equivalent of 20 ng RNA was analyzed by RT-qPCR using predesigned assays with gene-specific hydrolysis probes (FFAR2: Hs00271142_s1; FFAR4: Hs00699184_m1; SCD1: Hs01682761_m1; peptidylprolyl isomerase A (PPIA): Hs04194521_s1; Thermo Fisher Scientific, Darmstadt, Germany). Data were analyzed for relative expression differences using PPIA as reference gene with standard Ct method as previously described29. For protein expression analyses, reagents for SDS-PAGE were supplied by GE Healthcare (Freiburg, Germany). Complete protease inhibitor cocktail and PhosStop phosphatase inhibitor cocktail were provided by Roche (Mannheim, Germany). All other chemicals were of the highest analytic grade commercially available and purchased from Sigma-Aldrich. The following antibodies were used: anti-G-protein coupled receptor (GPR) 43 (FFAR2) (sc-293202) from Santa Cruz (Dallas, TX, USA), anti-GPR120 (FFAR4) (NBP1-00858) from Novus Biologicals (Abingdon, UK), anti-SCD1 (ab39969), and anti-actin (ab6276) from Abcam (Cambridge, UK). Horseradish peroxidase (HRP)-conjugated goat anti-rabbit and goat anti-mouse IgG antibodies were supplied by Promega (Mannheim, Germany). AT biopsies were lysed in a buffer containing 50 mmol l−1 HEPES, pH 7.4, 1% Triton X-100, complete protease inhibitor, and PhosStop phosphatase inhibitor cocktail. After incubation for 2 h at 4 °C, the suspension was centrifuged at 10,000×g for 15 min. Thereafter, 10 µg of the lysates were separated by SDS-PAGE using gradient horizontal gels and transferred to polyvinylidene fluoride filters in a semidry blotting apparatus. Filters were blocked with Tris-buffered saline containing 0.1% Tween and 5% nonfat dry milk and subsequently incubated overnight with a 1:1000 dilution (1:40,000 for anti-actin) of the appropriate antibodies. After washing, filters were incubated with secondary HRP-coupled antibody and processed for enhanced chemiluminescence detection using Immobilon HRP substrate (Millipore, Billerica, MA, USA). Signals were visualized and evaluated on a ChemiDoc workstation (Bio-Rad Laboratories, Munich, Germany).
Statistical analyses
Results are given as median [first and third quartiles] or mean ± SEM. Data were compared using Mann–Whitney U test for unpaired samples to determine differences between groups. Relations between variables were investigated using Spearman rank correlation analyses. The total AUC for a specific variable was calculated as the integral of the time course of such variable during the test, while the incremental AUC (ΔAUC) was calculated by subtracting the basal area from the respective total AUC. All statistical tests were two-sided and a p-value ≤ 5% was accepted to indicate significant differences. All statistical analyses were performed using SPSS for Windows 23.0 (SPSS Inc., Chicago, IL, USA). All graphs were generated using GraphPad Prism, Version 7.01 (GraphPad Software, Inc., La Jolla, CA, USA).
Results
Participants’ characteristics
CON and patients with type 2 diabetes mellitus had similar age, BMI, waist circumference, waist-to-hip ratio, and relative body fat mass (Table 1). Patients with type 2 diabetes mellitus had 34% higher fasting plasma glucose, 28% higher fasting insulin, and 29% higher fasting C-peptide. Additionally, serum hsCRP levels were 56% higher in patients with type 2 diabetes mellitus compared to CON. Furthermore, HDL cholesterol was 25% lower and systolic blood pressure 8% higher in patients with type 2 diabetes mellitus (Table 1). Of note, one female patient with type 2 diabetes was in the luteal phase. However, exclusion of this patient did not affect the results. All other females investigated were either postmenopausal or studied in the follicular phase (day 1–14) of their menstrual cycle. Furthermore, one woman in the follicular phase was controlled by an oral contraceptive.
Table 1.
Characteristics of study population
| Variable | CON | T2D | p |
|---|---|---|---|
| Female/male [n] | 6/19 | 6/19 | |
| Age [years] | 51 [42; 60] | 49 [43; 58] | 0.84 |
| Weight [kg] | 98 [91; 98] | 99 [88; 112] | 0.61 |
| BMI [kg* m−2] | 33 [27; 41] | 34 [27; 37] | 0.81 |
| HbA1c [%] | 5.3 [5.1; 5.5] | 6.1 [5.5; 7.0] | <0.001 |
| HbA1c [mmol* mol−1] | 34 [32; 37] | 43 [36; 52] | <0.001 |
| Waist circumference [cm] | 106 [99; 116] | 107 [102; 118] | 0.54 |
| Hip circumference [cm] | 111 [97; 127] | 110 [102; 118] | 0.97 |
| WHR | 0.98 [0.90; 1.04] | 0.99 [0.93; 1.04] | 0.54 |
| Body fat [%] | 34 [27; 42] | 33 [28; 39] | 0.87 |
| Lean body weight [kg] | 66 [57; 74] | 65 [58; 72] | 0.87 |
| Systolic blood pressure [mmHg] | 124 [114; 143] | 144 [126; 159] | <0.05 |
| Diastolic blood pressure [mmHg] | 80 [69; 86] | 80 [73; 94] | 0.40 |
| Fasting blood glucose [mmol* l−1] | 4.6 [4.4; 4.7] | 6.7 [5.7; 8.0] | <0.001 |
| Fasting insulin [µU* ml−1] | 11 [6; 17] | 17 [11; 27] | <0.05 |
| Fasting C-peptide [ng* ml−1] | 2.3 [1.5; 3.1] | 3.4 [2.0; 4.7] | <0.05 |
| Fasting glucagon [pg* ml−1] | 118 [98; 137] | 110 [76; 138] | 0.34 |
| NEFA [µmol* l−1] | 603 [431; 655] | 481 [333; 652] | 0.21 |
| TG [mg* dl−1] | 119 [82; 144] | 117 [102; 252] | 0.17 |
| HDL cholesterol [mg* dl−1] | 53 [43; 66] | 38 [33; 46] | <0.001 |
| LDL cholesterol [mg* dl−1] | 135 [114; 156] | 127 [111; 139] | 0.21 |
| hsCRP [mg* dl−1] | 0.13 [0.07; 0.33] | 0.34 [0.14; 0.72] | <0.05 |
| OGIS0–180 min [ml* min−1* m−2] | 362 [349; 385] | 311 [293; 343] | <0.001 |
Data are median [first; third quartile], two-tailed Mann–Whitney U test (n = 22–25 for CON; n = 16–25 for type 2 diabetes patients). Significant differences between groups (p-value ≤ 5%) are indicated by bold values. Glucose-tolerant humans (CON), patients with type 2 diabetes mellitus (T2D), waist/hip ratio (WHR), diastolic and systolic blood pressure (BP), oral glucose insulin sensitivity index (OGIS). Triglycerides (TG), high-density lipoprotein (HDL) cholesterol, low-density lipoprotein (LDL) cholesterol, non-esterified fatty acids (NEFA), and high-sensitivity C-reactive protein (hsCRP) were analyzed in fasted state
Mixed-meal test (MMT)
In patients with type 2 diabetes mellitus, the glucose concentrations were higher at all time points during the MMT and AUC(glucose)0–180 min was also 31% higher (Table 2, Supplementary Figure 1a). Insulin at 30 min and C-peptide at 90, 120, and 180 min were higher in patients with type 2 diabetes mellitus vs CON, while AUC(insulin)0–180 min and AUC(C-peptide)0–180 min did not differ between groups (Table 2, Supplementary Figure 1b, c). AUC(NEFA)0–120 min, AUC(TG)0–180 min, and AUC(glucagon)0–180 min also did not differ between groups (data not shown). In patients with type 2 diabetes mellitus, OGIS was 14% lower and beta cell function assessed from ΔAUC(C-peptide)0–180 min/ΔAUC(glucose)0–180 min as well as from ΔAUC(insulin)0–180 min/ΔAUC(glucose)0–180 min was 71% and 78% lower than CON, respectively.
Table 2.
Characteristics of beta cell function in study population
| Variable | CON | T2D | p |
|---|---|---|---|
| AUC 0–180 min | |||
| AUC(glucose)0–180 min [mmol* l−1 min−1] | 842 [772; 901] | 1 188 [1 080; 1 407] | <0.001 |
| AUC(insulin)0–180 min [nmol* l−1 min−1] | 48 [25; 68] | 49 [31;65] | 0.77 |
| AUC(C-peptide)0–180 min [nmol* l−1 min−1] | 350 [238; 405] | 373 [290; 506] | 0.17 |
| Beta cell function | |||
| ΔAUC(insulin)0–180 min/ΔAUC(glucose)0–180 min [pmol* mmol−1] | 680 [408; 1367] | 174 [92; 228] | <0.001 |
| ΔAUC(C-peptide)0–180 min/ΔAUC(glucose)0–180 min [nmol* mmol−1] | 46 [20; 78] | 13 [8; 21] | <0.001 |
Data are median [first; third quartile], two-tailed Mann–Whitney U test (n = 13–25 for CON; n = 18–25 for patients with type 2 diabetes mellitus). Significant differences between groups (p-value ≤ 5%) are indicated by bold values. Glucose-tolerant humans (CON), patients with type 2 diabetes mellitus (T2D). Marker for beta cell function are ΔAUC(insulin)0–180 min/ΔAUC(glucose)0–180 min in pmol* mmol−1 and ΔAUC(C-peptide)0–180 min/ΔAUC(glucose)0–180 min in nmol* mmol−1
Gene and protein expression levels
FFAR2 mRNA and protein expression were similar in type 2 diabetes mellitus patients compared to CON (Fig. 1a, b, Supplementary Figure 2a). FFAR4 mRNA expression did not differ, while FFAR4 protein expression tended to be lower in patients with type 2 diabetes mellitus (Fig. 1c, d, Supplementary Figure 2b). Patients with type 2 diabetes mellitus had fivefold lower mRNA expression and twofold lower protein expression of SCD1 compared to CON (Fig. 1e, f, Supplementary Figure 2c).
Fig. 1. mRNA and protein expression analyses for genes involved in fatty acid metabolism.

Data are mean ± SEM (n = 23–25 for CON; n = 22–25 for patients with type 2 diabetes mellitus), two-tailed Mann–Whitney U test. **p < 0.01, ***p < 0.001. Data were analyzed for relative expression differences using peptidylprolyl isomerase A (PPIA) as reference gene with standard Ct method. Glucose-tolerant humans (CON), patients with type 2 diabetes mellitus (T2D). a mRNA expression levels of free fatty acid receptor 2 (FFAR2), b protein expression levels of FFAR2, c mRNA expression levels of free fatty acid receptor 4 (FFAR4), d protein expression levels of FFAR4, e mRNA expression levels of stearoyl-CoA desaturase-1 (SCD1), f protein expression levels of SCD1
Correlation analyses
FFAR2 protein expression neither correlated with OGIS in type 2 diabetes mellitus patients nor in CON. Only in CON, protein expression levels of FFAR2 associated negatively with beta cell function, as assessed from ΔAUC(insulin)0–180 min/ΔAUC(glucose)0–180 min (r = −0.78, p < 0.01) and ΔAUC(C-peptide)0–180 min/ΔAUC(glucose)0–180 min (Fig. 2). Of note, fasted NEFA levels and AUC(NEFA)0–120 min did not correlate with beta cell function. Only in patients with type 2 diabetes mellitus, protein levels of FFAR2 correlated positively with AUC(TG)0–180 min (r = 0.48, p < 0.05) and negatively with HDL cholesterol (r = −0.43, p < 0.05). FFAR4 protein expression did not correlate with OGIS or beta cell function in both groups. Protein expression levels of FFAR4 associated negatively with fasting TG and AUC(TG)0–180 min in patients with type 2 diabetes mellitus (r = −0.62 and r = −0.59, respectively, both p < 0.01), but not in CON (r = 0.15, p = 0.48 and r = 0.25, p = 0.26, respectively). FFAR2/4 did not associate with body weight, BMI, or relative body fat content in both groups.
Fig. 2. Association of protein expression levels of FFAR2 with beta cell function.
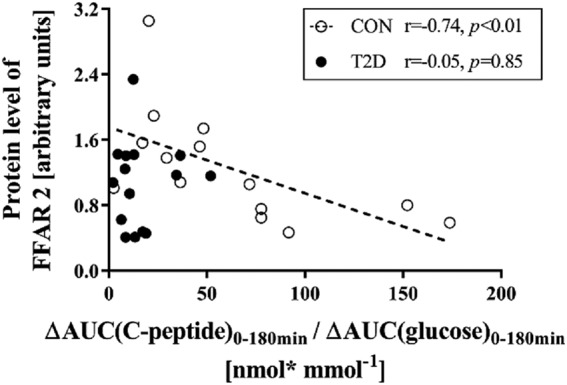
r-Spearman’s rank correlation coefficient. Glucose-tolerant humans (CON), patients with type 2 diabetes mellitus (T2D), free fatty acid receptor 2 (FFAR2), marker for beta cell function from ΔAUC(C-peptide)0–180 min/ΔAUC(glucose)0–180 min in nmol* mmol−1
SCD1 protein expression did not correlate with OGIS or beta cell function in both groups. However, SCD1 mRNA and protein levels associated positively with insulin sensitivity across the whole cohort (r = 0.38, p < 0.01 and r = 0.31, p < 0.05, respectively). SCD1 protein expression correlated positively with mRNA levels (r = 0.60, p < 0.001). In CON, protein levels of SCD1 associated negatively with fasting TG (r = −0.57, p < 0.01) and AUC(TG)0–180 min (r = −0.52, p < 0.05). In patients with type 2 diabetes mellitus, these associations were absent (TG: r = −0.17, p = 0.44; AUC(TG)0–180 min: r = 0.05, p = 0.85). Only in patients with type 2 diabetes mellitus, SCD1 protein levels correlated negatively with hsCRP (r = −0.45, p < 0.05).
Discussion
This study found no differences in FFAR2/4 mRNA and protein expression between patients with type 2 diabetes mellitus vs CON of similar body weight. Our findings are in contrast to results in mice, which might be biased by differences in body weight explaining a large part of their phenotypes30. We found markedly lower gene and protein expression of SCD1 in subcutaneous AT of patients with type 2 diabetes mellitus when compared with sex-matched, age-matched, and BMI-matched glucose-tolerant humans. Furthermore, FFAR2 protein expression correlated negatively with beta cell function in glucose-tolerant humans. However, this association was not found in patients with type 2 diabetes mellitus.
In mice, a recent study of whole-body or beta cell selective deletion of FFAR2 also provided evidence that insulin secretion is increased, accompanied by improved glucose tolerance3. In the present study, FFAR2 expression did not differ between patients with type 2 diabetes mellitus and CON, while its protein levels negatively associated with beta cell function during MMT in CON. This points to a potential impact of the receptor on glucose homeostasis after food intake rather than in the fasted state. In addition, the association between FFAR2-deficiency and IS in mice7 was not confirmed in humans with or without diabetes in the present study, indicating that reported findings in FFAR2-deficient mice cannot directly be translated to humans. Although previous studies indicated an assumed role in AT lipolysis11 causing impaired beta cell function14, we did not find a significant correlation of NEFA levels either during fasting or after the MMT with beta cell function. However, suppression of NEFA concentrations after the MMT might not be an optimal method to assess AT lipolysis, because of the exogenous oral intake of lipids and the individual variability of lipid absorption. Thus, the absence of a significant correlation between suppression of NEFA levels after the MMT and beta cell function does not exclude effects of insulin-mediated adipose tissue lipolysis.
In humans, a deleterious non-synonymous mutation (p.R270H) that inhibits FFAR4 signaling was found to associate with increased risk of obesity in a European population16. Of note, development of severe obesity, liver fat accumulation, and insulin resistance in FFAR4-deficient mice under high-fat diet16,17 were accompanied by lower SCD1 gene expression18. We found no association between FFAR4 and body weight or differences in human FFAR4 expression between patients with type 2 diabetes mellitus and CON, but FFAR4 protein expression tended to be lower in type 2 diabetes mellitus. One possible explanation for the lack of differences between groups in FFAR4 expression might be that these receptors are dependent on acute increase in NEFAs triggering FFAR4 expression in AT. In the present study, fasted NEFA and TG levels did not differ between groups, mainly because our study only included patients with recently diagnosed and well controlled type 2 diabetes mellitus.
Interestingly, our study revealed markedly lower gene and protein expression of SCD1 in SAT of patients with type 2 diabetes mellitus. These findings are in line with the previously reported decrease in SCD1 gene expression in FFAR4-deficient mice18 and support a role of SCD1 (Δ9 desaturase) in glucose homeostasis. Desaturases are key enzymes in converting saturated to unsaturated FAs by introducing a double-bond in the growing FA chain. Of note, the large prospective population-based Kuopio Ischaemic Heart Disease (KIHD) Risk Factor Study indicated that higher serum Δ5 desaturase activity associate with a lower risk for type 2 diabetes mellitus and increased Δ6 desaturase activity with a higher risk among middle-aged and older Finish men31. A putative mechanism behind the negative association of higher Δ5 desaturase activity with lower risk of type 2 diabetes mellitus may be the simultaneous improvement in IS32. In the European Prospective Investigation into Cancer (EPIC) and Nutrition-Potsdam Study, SCD1 activity (assessed from product-to-precursor ratios) of erythrocyte membrane was positively associated with risk of type 2 diabetes mellitus33. In agreement with our findings and previously reported beneficial effects of SCD1 in type 2 diabetes mellitus21,34, thiazolidinedione treatment led to increased SCD1 expression in AT of patients with type 2 diabetes mellitus together with improved TG esterification and IS20. The mechanisms underlying insulin resistance are still not fully understood, but increased NEFA release from AT is generally recognized as an important factor for the development of insulin resistance35,36. Thus, dysfunctional AT as indicated by decreased SCD1 expression in type 2 diabetes mellitus compared to CON may contribute to the development of type 2 diabetes mellitus. Previously, we showed that an intravenous lipid infusion as well as a single oral fat load rich in long-chain polyunsaturated FAs can induce insulin resistance29,37. Especially, saturated FAs are thought to induce inflammation and insulin resistance38–40. Accordingly, SCD1-deficient mice exhibited increased inflammation41. In agreement with our results, AT-specific SCD1 deletion in a mouse model induced glucose transporter 1 upregulation in AT, which was associated with increased tumor necrosis factor-alpha production42. The possible protective effect of SCD1 in CON is underlined by our observation that protein levels of SCD1 correlated negatively with hsCRP in patients with type 2 diabetes mellitus. Moreover, SCD1 protein levels in CON associated negatively with fasting TG and postprandial AUC(TG)0–180 min, respectively. The decrease in plasma TG levels with increasing SCD1 protein expression is in accordance with previous studies, where patients with type 2 diabetes mellitus exhibited enhanced TG esterification in SAT under thiazolidinedione treatment with subsequently increased SCD1 gene expression20.
The strength of our study lies in the deep phenotyping of patients with type 2 diabetes mellitus and well-matched glucose-tolerant controls. However, the conclusions from our study are limited by the small sample size and the lack of muscle samples. Due to the nature of a cross-sectional design, this study also does not allow to draw conclusions as to causality or temporal relationships.
In conclusion, patients with recent onset type 2 diabetes mellitus have lower SCD1, but not FFAR2 or 4 expression in SAT compared to CON. Our findings suggest that SCD1 expression may be important in early development of type 2 diabetes mellitus, but is not as effective in modulating beta cell function as FFAR2. Our study implies that FFAR2 could negatively influence glucose homeostasis by decreasing beta cell function in CON. Thus, both FFAR2 and SCD1 may be potential treatment targets in diabetes prevention strategies.
Data availability
To ensure data privacy of the study participants, the generated datasets of the currently still running GDS are not publicly available. Especially, since they are subject to national data protection laws and restrictions by the ethics committee. However, access to the data can be requested through an individual project agreement within the GDS.
Disclaimer
The funding sources had neither influence on design and conduct of this study, collection, analysis, and interpretation of the data; nor on the preparation, review, or approval of this article.
Electronic supplementary material
Acknowledgements
We would like to thank the staff of the Clinical Research Center, Institute for Clinical Diabetology at the German Diabetes Center, Düsseldorf, Germany for excellent help with the experiments. We thank the participants for their invaluable contributions to the study. Some of the data were presented as an abstract at the 52nd EASD Annual Meeting in 2016. This work was supported by the Ministry of Culture and Science of the State of North Rhine-Westphalia (MIWF NRW) and the German Federal Ministry of Health (BMG). This study was supported in part by a grant of the Federal Ministry for Research (BMBF) to the German Center for Diabetes Research (DZD e.V.).
Members of GDS Group
A.E. Buyken, B. Belgardt, G. Geerling, H. Al-Hasani, C. Herder, A. Icks, J. Kotzka, O. Kuss, E. Lammert, D. Markgraf, K. Müssig, W. Rathmann, J. Szendroedi, D. Ziegler, and M. Roden (speaker).
Authors’ contributions
M.R. designed the study. K.B. wrote the article and researched the data. S.K., M.S., and M.R. researched the data, contributed to the discussion, and reviewed and edited the article. D.M. performed laboratory analyses. J.S., K.S., V.B., K.M., Z.Z., B.K., H.S. and A.T. researched data, contributed to the discussion, and reviewed and edited the article. M.R. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. All authors gave final approval of this version to be published.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Kálmán Bódis, Sabine Kahl, Marie-Christine Simon
Members of the GDS Study Group are listed below Acknowledgements.
Contributor Information
Michael Roden, Phone: +49-211-3382-201, Email: michael.roden@ddz.uni-duesseldorf.de.
for the GDS Study Group:
A. E. Buyken, B. Belgardt, G. Geerling, H. Al-Hasani, C. Herder, A. Icks, J. Kotzka, O. Kuss, E. Lammert, D. Markgraf, K. Müssig, W. Rathmann, J. Szendroedi, D. Ziegler, and M. Roden
Electronic supplementary material
Supplementary Information accompanies this paper at (10.1038/s41387-018-0054-9).
References
- 1.Prentki M, Nolan CJ. Islet beta cell failure in type 2 diabetes. J. Clin. Invest. 2006;116:1802–1812. doi: 10.1172/JCI29103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miyamoto J, et al. Nutritional signaling via free fatty acid receptors. Int. J. Mol. Sci. 2016;17:450. doi: 10.3390/ijms17040450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tang C, et al. Loss of FFA2 and FFA3 increases insulin secretion and improves glucose tolerance in type 2 diabetes. Nat. Med. 2015;21:173–177. doi: 10.1038/nm.3779. [DOI] [PubMed] [Google Scholar]
- 4.Tazoe H, et al. Roles of short-chain fatty acids receptors, GPR41 and GPR43 on colonic functions. J. Physiol. Pharmacol. 2008;59(Suppl 2):251–262. [PubMed] [Google Scholar]
- 5.Hirasawa A, et al. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat. Med. 2005;11:90–94. doi: 10.1038/nm1168. [DOI] [PubMed] [Google Scholar]
- 6.Vangaveti V, Shashidhar V, Jarrod G, Baune BT, Kennedy RL. Free fatty acid receptors: emerging targets for treatment of diabetes and its complications. Ther. Adv. Endocrinol. Metab. 2010;1:165–175. doi: 10.1177/2042018810375656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bjursell M, et al. Improved glucose control and reduced body fat mass in free fatty acid receptor 2-deficient mice fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2011;300:E211–E220. doi: 10.1152/ajpendo.00229.2010. [DOI] [PubMed] [Google Scholar]
- 8.Layden BT, et al. Regulation of pancreatic islet gene expression in mouse islets by pregnancy. J. Endocrinol. 2010;207:265–279. doi: 10.1677/JOE-10-0298. [DOI] [PubMed] [Google Scholar]
- 9.Rieck S, et al. The transcriptional response of the islet to pregnancy in mice. Mol Endocrinol. 2009;23:1702–1712. doi: 10.1210/me.2009-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muoio DM, Newgard CB. Obesity-related derangements in metabolic regulation. Annu. Rev. Biochem. 2006;75:367–401. doi: 10.1146/annurev.biochem.75.103004.142512. [DOI] [PubMed] [Google Scholar]
- 11.Glass CK, Olefsky JM. Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab. 2012;15:635–645. doi: 10.1016/j.cmet.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanaka N, et al. Adipocyte-specific disruption of fat-specific protein 27 causes hepatosteatosis and insulin resistance in high-fat diet-fed mice. J. Biol. Chem. 2015;290:3092–3105. doi: 10.1074/jbc.M114.605980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gancheva S, Jelenik T, Alvarez-Hernandez E, Roden M. Interorgan metabolic crosstalk in human insulin resistance. Physiol. Rev. 2018;98:1371–1415. doi: 10.1152/physrev.00015.2017. [DOI] [PubMed] [Google Scholar]
- 14.Giacca A, Xiao C, Oprescu AI, Carpentier AC, Lewis GF. Lipid-induced pancreatic beta-cell dysfunction: focus on in vivo studies. Am. J. Physiol. Endocrinol. Metab. 2011;300:E255–E262. doi: 10.1152/ajpendo.00416.2010. [DOI] [PubMed] [Google Scholar]
- 15.Taneera J, et al. A systems genetics approach identifies genes and pathways for type 2 diabetes in human islets. Cell Metab. 2012;16:122–134. doi: 10.1016/j.cmet.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 16.Ichimura A, et al. Dysfunction of lipid sensor GPR120 leads to obesity in both mouse and human. Nature. 2012;483:350–354. doi: 10.1038/nature10798. [DOI] [PubMed] [Google Scholar]
- 17.Waguri T, Goda T, Kasezawa N, Yamakawa-Kobayashi K. The combined effects of genetic variations in the GPR120 gene and dietary fat intake on obesity risk. Biomed. Res. 2013;34:69–74. doi: 10.2220/biomedres.34.69. [DOI] [PubMed] [Google Scholar]
- 18.Ichimura A, Hara T, Hirasawa A. Regulation of energy homeostasis via GPR120. Front. Endocrinol. 2014;5:111. doi: 10.3389/fendo.2014.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao H, et al. Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell. 2008;134:933–944. doi: 10.1016/j.cell.2008.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tordjman J, et al. Thiazolidinediones block fatty acid release by inducing glyceroneogenesis in fat cells. J. Biol. Chem. 2003;278:18785–18790. doi: 10.1074/jbc.M206999200. [DOI] [PubMed] [Google Scholar]
- 21.Riserus U, et al. Rosiglitazone increases indexes of stearoyl-CoA desaturase activity in humans: link to insulin sensitization and the role of dominant-negative mutation in peroxisome proliferator-activated receptor-gamma. Diabetes. 2005;54:1379–1384. doi: 10.2337/diabetes.54.5.1379. [DOI] [PubMed] [Google Scholar]
- 22.Pinnamaneni SK, Southgate RJ, Febbraio MA, Watt MJ. Stearoyl CoA desaturase 1 is elevated in obesity but protects against fatty acid-induced skeletal muscle insulin resistance in vitro. Diabetologia. 2006;49:3027–3037. doi: 10.1007/s00125-006-0427-9. [DOI] [PubMed] [Google Scholar]
- 23.Szendroedi J, et al. Cohort profile: the German Diabetes Study (GDS) Cardiovasc. Diabetol. 2016;15:59. doi: 10.1186/s12933-016-0374-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greenbaum CJ, et al. Mixed-meal tolerance test versus glucagon stimulation test for the assessment of beta-cell function in therapeutic trials in type 1 diabetes. Diabetes Care. 2008;31:1966–1971. doi: 10.2337/dc07-2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mari A, Pacini G, Murphy E, Ludvik B, Nolan JJ. A model-based method for assessing insulin sensitivity from the oral glucose tolerance test. Diabetes Care. 2001;24:539–548. doi: 10.2337/diacare.24.3.539. [DOI] [PubMed] [Google Scholar]
- 26.Pacini G, Mari A. Methods for clinical assessment of insulin sensitivity and beta-cell function. Best. Pract. Res. Clin. Endocrinol. Metab. 2003;17:305–322. doi: 10.1016/S1521-690X(03)00042-3. [DOI] [PubMed] [Google Scholar]
- 27.Alsalim W, et al. Mixed meal ingestion diminishes glucose excursion in comparison with glucose ingestion via several adaptive mechanisms in people with and without type 2 diabetes. Diabetes Obes. Metab. 2016;18:24–33. doi: 10.1111/dom.12570. [DOI] [PubMed] [Google Scholar]
- 28.American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2006;29(Suppl 1):S43–S48. [PubMed] [Google Scholar]
- 29.Nowotny B, et al. Mechanisms underlying the onset of oral lipid-induced skeletal muscle insulin resistance in humans. Diabetes. 2013;62:2240–2248. doi: 10.2337/db12-1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oh DY, et al. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell. 2010;142:687–698. doi: 10.1016/j.cell.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yary T, et al. Serum n-6 polyunsaturated fatty acids, Delta5- and Delta6-desaturase activities, and risk of incident type 2 diabetes in men: the Kuopio Ischaemic Heart Disease Risk Factor Study. Am. J. Clin. Nutr. 2016;103:1337–1343. doi: 10.3945/ajcn.115.128629. [DOI] [PubMed] [Google Scholar]
- 32.Takkunen MJ, et al. Longitudinal associations of serum fatty acid composition with type 2 diabetes risk and markers of insulin secretion and sensitivity in the Finnish Diabetes Prevention Study. Eur. J. Nutr. 2016;55:967–979. doi: 10.1007/s00394-015-0911-4. [DOI] [PubMed] [Google Scholar]
- 33.Jacobs S, et al. Evaluation of various biomarkers as potential mediators of the association between Delta5 desaturase, Delta6 desaturase, and stearoyl-CoA desaturase activity and incident type 2 diabetes in the European Prospective Investigation into Cancer and Nutrition-Potsdam Study. Am. J. Clin. Nutr. 2015;102:155–164. doi: 10.3945/ajcn.114.102707. [DOI] [PubMed] [Google Scholar]
- 34.Flowers JB, et al. Loss of stearoyl-CoA desaturase-1 improves insulin sensitivity in lean mice but worsens diabetes in leptin-deficient obese mice. Diabetes. 2007;56:1228–1239. doi: 10.2337/db06-1142. [DOI] [PubMed] [Google Scholar]
- 35.Krssak M, et al. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1H NMR spectroscopy study. Diabetologia. 1999;42:113–116. doi: 10.1007/s001250051123. [DOI] [PubMed] [Google Scholar]
- 36.Petersen KF, Shulman GI. Pathogenesis of skeletal muscle insulin resistance in type 2 diabetes mellitus. Am. J. Cardiol. 2002;90(5A):11G–18GG. doi: 10.1016/S0002-9149(02)02554-7. [DOI] [PubMed] [Google Scholar]
- 37.Roden M, et al. Mechanism of free fatty acid-induced insulin resistance in humans. J. Clin. Invest. 1996;97:2859–2865. doi: 10.1172/JCI118742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kausch C, et al. Skeletal muscle cells from insulin-resistant (non-diabetic) individuals are susceptible to insulin desensitization by palmitate. Horm. Metab. Res. 2003;35:570–576. doi: 10.1055/s-2003-43501. [DOI] [PubMed] [Google Scholar]
- 39.Staiger K, et al. Saturated, but not unsaturated, fatty acids induce apoptosis of human coronary artery endothelial cells via nuclear factor-kappaB activation. Diabetes. 2006;55:3121–3126. doi: 10.2337/db06-0188. [DOI] [PubMed] [Google Scholar]
- 40.Staiger H, et al. Palmitate-induced interleukin-6 expression in human coronary artery endothelial cells. Diabetes. 2004;53:3209–3216. doi: 10.2337/diabetes.53.12.3209. [DOI] [PubMed] [Google Scholar]
- 41.MacDonald ML, et al. Despite antiatherogenic metabolic characteristics, SCD1-deficient mice have increased inflammation and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2009;29:341–347. doi: 10.1161/ATVBAHA.108.181099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hyun CK, et al. Adipose-specific deletion of stearoyl-CoA desaturase 1 up-regulates the glucose transporter GLUT1 in adipose tissue. Biochem. Biophys. Res. Commun. 2010;399:480–486. doi: 10.1016/j.bbrc.2010.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
To ensure data privacy of the study participants, the generated datasets of the currently still running GDS are not publicly available. Especially, since they are subject to national data protection laws and restrictions by the ethics committee. However, access to the data can be requested through an individual project agreement within the GDS.