

Abstract
Bacteria belonging to the genera Dickeya and Pectobacterium are responsible for significant economic losses in a wide variety of crops and ornamentals. During last years, increasing losses in potato production have been attributed to the appearance of Dickeya solani. The D. solani strains investigated so far share genetic homogeneity, although different virulence levels were observed among strains of various origins. The purpose of this study was to investigate the genetic traits possibly related to the diverse virulence levels by means of comparative genomics. First, we developed a new genome assembly pipeline which allowed us to complete the D. solani genomes. Four de novo sequenced and ten publicly available genomes were used to identify the structure of the D. solani pangenome, in which 74.8 and 25.2% of genes were grouped into the core and dispensable genome, respectively. For D. solani panregulon analysis, we performed a binding site prediction for four transcription factors, namely CRP, KdgR, PecS and Fur, to detect the regulons of these virulence regulators. Most of the D. solani potential virulence factors were predicted to belong to the accessory regulons of CRP, KdgR, and PecS. Thus, some differences in gene expression could exist between D. solani strains. The comparison between a highly and a low virulent strain, IFB0099 and IFB0223, respectively, disclosed only small differences between their genomes but significant differences in the production of virulence factors like pectinases, cellulases and proteases, and in their mobility. The D. solani strains also diverge in the number and size of prophages present in their genomes. Another relevant difference is the disruption of the adhesin gene fhaB2 in the highly virulent strain. Strain IFB0223, which has a complete adhesin gene, is less mobile and less aggressive than IFB0099. This suggests that in this case, mobility rather than adherence is needed in order to trigger disease symptoms. This study highlights the utility of comparative genomics in predicting D. solani traits involved in the aggressiveness of this emerging plant pathogen.
Keywords: adhesin, genome comparison, Pectobacteriaceae, prophages, regulon comparison
Introduction
Bacteria belonging to the genera Dickeya and Pectobacterium (formerly genus Erwinia, soft-rot Enterobacteriaceae), recently reclassified into the new family Pectobacteriaceae (Adeolu et al., 2016), have been responsible for significant economic losses in European crop production (Perombelon, 2002; Toth et al., 2011; Mansfield et al., 2012). These bacterial phytopathogens cause blackleg symptoms on potato and soft rot disease on many other host plants. Despite the fact that D. solani strains were described as one of the most aggressive blackleg and soft rot-causing bacteria, their mechanism of virulence has not been fully elucidated yet. The competitive advantage of D. solani relies on a wider range of temperatures favorable to disease development than in the case of other Dickeya spp., as well as on the fact that lower inoculum levels are sufficient for an effective infection spread (Czajkowski et al., 2013). Outside symptomatic plants, D. solani can also be found as saprophytic bacterium in rhizosphere soil (Heuer et al., 2010; Potrykus et al., 2014). However, different virulence levels were observed among D. solani isolates of various origins (Potrykus et al., 2014; Golanowska et al., 2017).
All of the D. solani strains investigated so far share genetic homogeneity. In more detail, dnaX, dnaN, fusA, gapA, gyrA, purA, rplB, rpoS, recA, and 16S rDNA sequences of D. solani showed 100% identity in Multilocus Sequence Analysis (MLSA) (Slawiak et al., 2009; van der Wolf et al., 2014; Potrykus et al., 2016). Likewise, no genomic differences were shown by classical DNA fingerprinting methods, i.e., REP PCR and PFGE, even though the analyzed strains originated from various European countries (Slawiak et al., 2009; Degefu et al., 2013; van der Wolf et al., 2014; Potrykus et al., 2016; Golanowska et al., 2017). Similar outcomes resulted from the variable number of tandem repeats (VNTR) method differentiating only 3 patterns for 54 D. solani isolates (Parkinson et al., 2014). Such a low genetic variation was attributed to a limited number of introductions and/or a recent emergence of D. solani in Europe.
The species D. solani is closely related to the well described species Dickeya dadantii. The virulence of D. dadantii relies on the coordinated production of high levels of multiple secreted enzymes, including pectinases, cellulases and proteases, which breakdown the plant cell wall and release nutrients used for bacterial growth (Barras et al., 1994; Hugouvieux-Cotte-Pattat et al., 1996; Py et al., 1998; Thomson et al., 1999; Perombelon, 2002; Hugouvieux-Cotte-Pattat, 2016). Analyses of D. solani genomes revealed the existence of additional genes potentially involved in pathogenicity and production of toxins, including clusters that encode polyketide synthases (PKS), non-ribosomal peptide synthetases (NRPS), amino acid adenylation domain, and proteins transported via T5SS/T6SS (Garlant et al., 2013; Pédron et al., 2014).
Efficiency to attach to the plant organs and cell motility can also influence virulence (Khayi et al., 2015; Weller-Stuart et al., 2017). It is usually considered that the ability of bacteria to attach to plant tissue increases its aggressiveness (Nair et al., 2003; Liao et al., 2014; Chen et al., 2017). However, some studies demonstrated inverse effects. For instance, a highly virulent mutant of Xylella fastidiosa has lower attachment to the xylem vessels than the wild type strain (Gottig et al., 2009; Ionescu et al., 2014). The authors concluded that strong attachment of bacteria to plant surfaces could restrict their movement, thus capacity to colonize the plants and finally limit disease severity.
The involvement of prophages in disease symptoms was demonstrated in different plant pathogens, such as Pectobacterium, Pseudomonas, Ralstonia and Streptomyces (Varani et al., 2013). Phages and prophages can affect the bacterial genome in many different ways for instance by gene disruption or shuffling and affect expression of adjacent genes. By introducing new fitness factors, including pathogenicity determinants, phages can even cause an avirulent strain to become virulent (Varani et al., 2013).
The appropriate regulation of genes encoding virulence factors is essential for setting off pathogenesis. It was first shown in D. dadantii 3937 that a set of transcriptional factors (TF), acting as global or specific regulators, enables bacterial adaptation during the infection process (Hugouvieux-Cotte-Pattat, 2016; Reverchon et al., 2016). The combined action of the regulators KdgR, CRP, Fur, PecS, and PecT, plays a decisive role in the proper synchronization of virulence factors production. The KdgR regulator triggers induction of pectinases after the pathogen have sensed the presence of pectin, a major component of middle lamellae, which is a part of plant cell wall. CRP (catabolite activator protein) is a highly conserved global regulator, whose function is to direct bacteria toward the utilization of preferential carbon sources, depending on the nutrient availability (Hugouvieux-Cotte-Pattat, 2016). The iron-dependent repressor Fur (ferric uptake regulator) is necessary to control iron uptake and iron homeostasis (Franza et al., 1999, 2002). The global regulators PecS and PecT play an essential role in the switch from epiphytic to pathogenic lifestyle, by preventing the premature expression of genes encoding virulence factors (Reverchon et al., 2016). Potrykus et al. (2014) showed that the regulators KdgR, PecS, and PecT have similar functions in D. solani as in D. dadantii.
Apart from genomic homogeneity stated for D. solani strains, our former studies (Potrykus et al., 2014, 2016, 2018) showed significant differences between various isolates in their ability to cause disease symptoms on diverse plant hosts, such as potato or chicory. Most of all, the highest virulence level was attributed to the strains originating from diseased plants in comparison to the strains isolated from the rhizosphere of healthy potatoes. The goal of the present study was to extensively describe the variability of the genomes of D. solani strains isolated from various origins, in search for genetic signatures possibly related to diverse virulence of these strains. To our knowledge, this is the first work providing an insight into pangenome and panregulon of the economically important plant pathogen D. solani.
Materials and methods
Dickeya solani strains and genomic sequences
D. solani strains exhibiting different levels of virulence were included in this work (Table 1). Two strains were isolated in Poland from rotten potato plants and showed a high level of virulence on potato tubers and chicory leaves (IFB0099, IFB0158), while 2 other isolates originated from healthy potato rhizosphere in Germany (IFB0221, IFB0223), showing either intermediate or low virulence on potato tubers and chicory leaves. The genomes of these 4 strains were assembled and annotated in our laboratory. The draft genomic sequence of IFB0099 had been published previously, with the GenBank accession no. JXRS00000000 (Golanowska et al., 2015). The version discussed here has an accession no. CP024711. Three other genome sequences were deposited in GenBank with the following accession numbers: PENA00000000, PEMZ00000000 and CP024710, for IFB0158, IFB0221 and IFB0223, respectively. In addition, 10 D. solani strains isolated in different countries, whose genomes were sequenced and deposited in GenBank, were enclosed for pangenome and panregulon analyses, including the D. solani Type strain - IPO 2222 (NCPPB4479T, LMG25993T) and the strains GBBC 2040, MK16, MK10, D s0432-1, RNS 08.23.3.1A, RNS 05.1.2A, RNS 07.7.3B, PPO 9019, and PPO 9134 (origin and references in Table 1).
Table 1.
Bacterial strains used in this study and their genomic features.
| D. solani strain | Country, year of isolation, source | No. of scaffolds | No. of N bases | Genome size (bp) | % GC | GenBank accession (Reference) |
|---|---|---|---|---|---|---|
| IFB0099 | Poland, 2005, potato stem | 1 | 0 | 4,932,920 | 56.24 | CP024711 (this work) |
| IFB0158 | Poland, 2009, potato | 37 | 395 | 4,879,070 | 56.24 | PENA00000000 (this work) |
| IFB0221 | Germany, 2005, potato rhizosphere | 38 | 394 | 4,878,255 | 56.24 | PEMZ00000000 (this work) |
| IFB0223 | Germany, 2005, potato rhizosphere | 1 | 0 | 4,937,554 | 56.24 | CP024710 (this work) |
| NCPPB4479T IPO2222 | The Netherlands, 2007, potato | 1 | 9,200 | 4,867,258 | 56.22 | AONU01000000 (Pritchard et al., 2013) |
| GBBC 2040 | Belgium, 2007, potato | 1 | 27,548 | 4,860,047 | 56.34 | AONX01000000 (Pritchard et al., 2013) |
| MK10 | Israel, potato | 3 | 3,800 | 4,935,237 | 56.21 | AOOP01000000 (Pritchard et al., 2013) |
| MK16 | Scotland, river water | 3 | 2,100 | 4,870,382 | 56.23 | AOOQ01000000 (Pritchard et al., 2013) |
| D s0432-1 | Finland, 2004, potato stem | 4 | 0 | 4,904,518 | 56.20 | AMWE01000000 (Garlant et al., 2013) |
| RNS 08.23.3.1A Dsl 3337 | France, 2008, potato | 1 | 12,124 | 4,923,743 | 56.25 | AMYI01000000 (Khayi et al., 2014) |
| PPO 9019 | The Netherlands, 2006, grape hyacinth | 24 | 30 | 4,866,823 | 56.25 | JWLS01000000 (Khayi et al., 2015) |
| PPO 9134 | The Netherlands, 2008, hyacinth | 22 | 187 | 4,870,830 | 56.24 | JWLT01000000 (Khayi et al., 2015) |
| RNS 05.1.2A | France, 2005, potato | 37 | 0 | 4,985,571 | 56.13 | JWMJ01000000 (Khayi et al., 2015) |
| RNS 07.7.3B | France, 2007, potato | 24 | 325 | 4,871,815 | 56.24 | JWLR01000000 (Khayi et al., 2015) |
For the phenotypic analysis, we used 9 strains of D. solani that were available at Intercollegiate Faculty of Biotechnology University of Gdansk and Medical University of Gdansk at the time of performing experiments: IFB0099, IFB0158, IFB0221, IFB0223, IPO 2222, GBBC 2040, MK16, D s0432-1 and RNS 08.23.3.1A. All the strains were grown at 30°C on crystal violet pectate medium (CVP) (Helias et al., 2012), Luria broth agar (LA), or in Luria broth (LB) (Lennox, 1955) for 24–48 h, unless otherwise stated. Liquid cultures were agitated at 200 rpm.
Potato tuber maceration assay
Potato tubers (cv. Caesar) were inoculated with the tested D. solani strains as described previously (Hugouvieux-Cotte-Pattat, 2004). Briefly, potato tubers were washed in tap water and dried. Ten potato tubers, each containing a small hole made with a sterile pipette tip, were inoculated with 10 μl of the bacterial suspension (~5 × 108 CFU ml−1). Inoculated holes were then covered with mineral oil to provide anaerobic conditions. After 48 h of incubation at 30°C under high (99%) humidity, the rotten potato tissue was removed and weighted. The experiment was performed three times.
Quantitative determination of pectate lyase activity
The pectate lyase activity was measured in liquid bacterial cultures grown for 24 h at 30°C with agitation 200 rpm in M63 minimal medium (Miller, 1992), supplemented with 2 g l−1 glycerol for noninduced conditions, and with both 2 g l−1 glycerol and 2 g l−1 polygalacturonic acid (PGA, Sigma) for induced conditions. The pectate lyase activity was determined spectrophotometrically by monitoring (Specord, Analytic Jena) the formation of unsaturated products from polygalacturonate at 235 nm (Tardy et al., 1997). The pectate lyase specific activity is expressed as micromoles of unsaturated products liberated per minute per milligram of bacterial dry weight (μmol min−1 mg−1). The experiment was performed three times with three replicates.
Plate assays for cellulase and protease activities
The ability to produce cellulases was analyzed on M63 agar plates supplemented with 2 g l−1 glycerol and 10 g l−1 carboxymethylcellulose (CMC). Two microliter of bacterial suspension containing 108 CFU ml−1 were spotted onto this medium and incubated for 24 h at 30°C. Afterwards, the plates were flooded with 10 mg ml−1 of Congo Red solution for 10 min and subsequently washed for 5 min with 1 M NaCl (Wood, 1980). Protease production was detected on LA medium containing skim milk (12.5 g l−1) after incubation for 24 h at 30°C (Ji et al., 1987). For both tests, diameters of the clear halo zones appearing around the colonies were measured. All experiments were performed three times with three replicates.
Plate assay for swimming ability
The bacterial ability to swim was tested on LA medium solidified by a low amount of agar (3 g l−1). The plates were stabbed with 0.2 μl inoculation loop. After incubation for 24 h at 30°C, the diameters of the bacteria spreading zones were measured. The experiment was performed once with three replicates.
Statistical analysis
In order to analyze the significance of the phenotypic results, we utilized statistical computing and graphics R 3.3.1 (GNU project) programming environment. To check whether the data follow normal distribution, the Shapiro–Wilk test was used. Levene's test was implemented to verify whether the data variances are equal. As the requirements of ANOVA were not fulfilled, Kruskal–Wallis test was applied for multiple comparisons (R agricolae package) followed by post-hoc test using the Fisher's least significant difference criterion. All statistical hypotheses were tested at p < 0.05.
Whole-genome shotgun sequencing and establishment of the genome assembly pipeline
Reads for the genomic sequences of strains IFB0099, IFB0158, IFB0221 and IFB0223 were acquired from BaseClear (The Netherlands). Different sequencing technologies and assembly methods have been tested in order to propose an optimized genome assembly pipeline leading to closing the genomes of D. solani. Regarding the first approach applied to the strains IFB0158 and IFB0221, respectively, 5,654,985 and 3,330,639 reads generated by MiSeq paired-end Illumina have been cleared out from adapters and low quality bases with the use of Trimmomatic (Bolger et al., 2014) (parameters: -phred33 LEADING:20 TRAILING:20 SLIDINGWINDOW:5:20 MINLEN:100). The application of SPAdes run together with MismatchCorrector (Bankevich et al., 2012) resulted in assembling IFB0158 and IFB0221 genomes into accordingly 37 or 38 scaffolds with ~395 N bases (Table 1). The IFB0099 and IFB0223 genomes were sequenced with PacBio platform (BaseClear, The Netherlands). 118,344 and 102,248 PacBio reads of IFB0099 (the previously reported draft genome of IFB0099 consisted of 97 scaffolds; Golanowska et al., 2015) and IFB0223 have been corrected, trimmed and assembled by using Canu (Berlin et al., 2015) (parameters: -pacbio-raw minOverlapLength = 500). The final genome polishing including getting consensus and variant calling was achieved by utilizing Quiver with the default settings (Chin et al., 2013). The above-proposed genome assembly pipeline (Figure 1) resulted in closing the genomes of IFB0099 and IFB0223 (with no N bases) by utilizing the reads derived solely from PacBio SMRT technology. All 14 genomes of D. solani strains used in this study were annotated by Prokka v1.11 (Seemann, 2014) for assuring uniformity of subsequent predictions. All the annotations of non-coding or regulatory RNAs yielded by Prokka's dependencies have been manually edited to meet the Genbank submission requirements. The genome of D. dadantii 3937 (Glasner et al., 2011) was utilized throughout as a reference.
Figure 1.
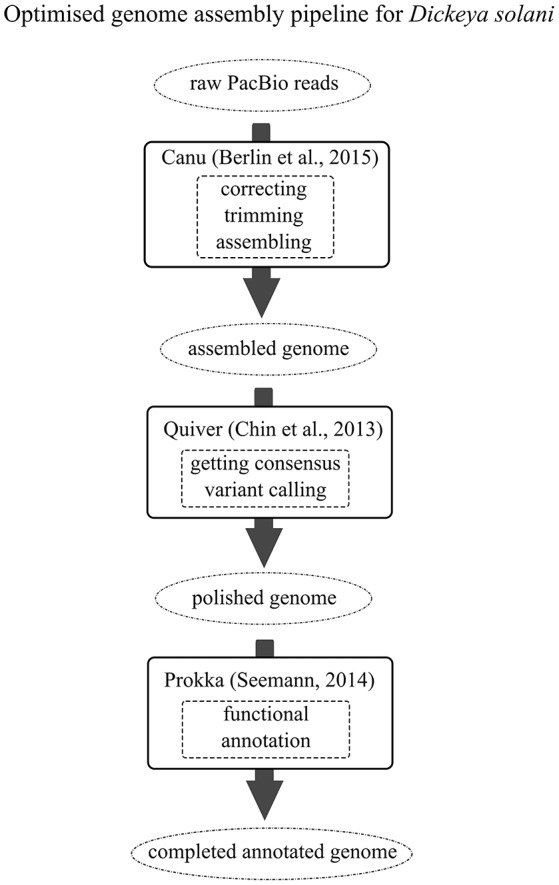
The optimized genome assembly pipeline for D. solani. Black arrows indicate the genomic assembly pipeline to follow. Different completeness stages of the constructed genome are presented within the dotted circles. Black boxes include data on the open source software and the corresponding literature references. Functions provided by certain software are enclosed within the dashed boxes.
Bioinformatics analyses
General genome analyses
Average Nucleotide Identity (ANI) values were calculated for the available D. solani genomes by applying ChunLab's online Average Nucleotide Identity Calculator (EzBioCloud) (Yoon et al., 2017). This tool is based on OrthoANI algorithm, improved by using USEARCH instead of BLAST (Lee et al., 2016). The D. dadantii 3937 genome was used as an interspecies reference.
Synteny analysis was performed on genomes successfully closed to only one scaffold (IFB0099, IFB0223, IPO 2222, GBBC 2040, and RNS 08.23.3.1A) with the use of progressive Mauve available in Mauve 2.4.0 (development snapshot Mauve_2015_02_26) and the default settings (Darling et al., 2010). In this analysis, the IFB0099 genome was used as a reference.
The orthology assessment was performed by Mauve (Darling et al., 2004). Annotated genomes of 14 strains were aligned by Mauve and the orthological relationships between genes were extracted using the default parameters (min. 60% sequence identity, min. 70% alignment coverage). Each annotated gene was classified within the pangenome structure comprising the core (present in all analyzed genomes), accessory (present in more than a single genome, but not all) or unique (present in only one genome) pangenome fraction.
For protein motif prediction, the protein structures and the presence of signal peptides were analyzed at http://www.ebi.ac.uk/Tools/hmmer/, by utilizing hmmscan against Pfam database (HmmerWeb version 2.15.0; Finn et al., 2015).
The presence of prophages within the D. solani genomes was predicted by PHAST on-line web server (http://phast.wishartlab.com/). The completeness of the prophages and their classification as intact, questionable, or incomplete, which is linked with their potential viability, was assigned basing on the score calculations as described previously (Zhou et al., 2011).
Regulon and panregulon analyses
Regulon predictions were conducted on the basis of experimentally established and previously described sequences of the binding sites for four TFs: KdgR, Fur, CRP, and PecS (de Lorenzo et al., 1987; Sauvage et al., 1996; Rodionov et al., 2004; Rouanet et al., 2004; Franza et al., 2005). Regulon predictions have been performed with the use of MAST (version 4.10.1.,) (Bailey and Elkan, 1994) and Bio.motifs package from Biopython library (version 1.62b) (Cock et al., 2009) as reported previously (Galardini et al., 2015). In-house built Python (version 1.65, Cock et al., 2009) scripts were used for this purpose and are available here: https://github.com/combogenomics/regtools. Basically, the MAST suite was used for computing the k-mer frequencies for each genome (fasta-get-markov command) with a maximum k-mer length of 3 bp.
Regulatory motifs were searched in all the genomes by using the two algorithms reported above and retaining only concordant prediction obtained with both of them. Each regulatory motif correctly predicted has been parsed by grouping all the hits present in the upstream region of the putative target gene (up to 400 bp from the first codon). The panregulon was defined as the sum of all putative targets for a given TF in all the genomes considered (Galardini et al., 2011). The panregulon was subsequently divided into three parts—the core regulon (the set of targets for a given TF shared by all the tested genomes), accessory regulon (the set of targets found in not all but more than one genome) and unique regulon (targets found in only one genome). Similarity between strains that was based on panregulon analyses was represented by the UPGMA methods on Jaccard distance.
Results
Phenotypic comparison of the D. solani strains
Several phenotypic features were determined for 9 D. solani strains available in our collection and whose genomes have been sequenced (IFB0099, IFB0158, IFB0221, IFB0223, IPO 2222, GBBC 2040, MK16, RNS 08.23.3.1A, and D s0432-1). The virulence of these D. solani strains, measured as the ability to macerate potato tissue, was compared. We distinguished three groups of strains on the basis of their virulence: high, intermediate and low virulent strains. High virulence was exhibited by IFB0099, IFB0158, GBBC 2040, MK16, RNS 08.23.3.1A, and D s0432-1. Strains IFB0221 and IPO 2222 showed intermediate virulence with maceration decreased by 53 and 46%, respectively, in comparison to IFB0099. The lowest virulence level was observed for the strain IFB0223, with a 96% reduction in comparison to IFB0099 (Figure 2A). The differences between the high, intermediate and low virulent strains were statistically significant (Figure 2A).
Figure 2.

Potato tuber tissue maceration ability and pectinolytic activity of the D. solani strains. (A) Maceration ability in grams of macerated tissue, 48 h post inoculation. (B) Total pectate lyase specific activity (μmol min−1 mg−1) in the presence of polygalacturonic acid. Means ± standard errors are depicted, n = 10 for (A) and n = 3 for (B). Within each panel mean values marked with different letters are significantly different by Kruskal–Wallis test followed by post hoc analysis using Fisher's least significant difference criterion (R agricolae package). All statistical hypotheses were tested at p < 0.05.
The activity of pectate lyase, being one of the main virulence factors, was analyzed. In noninduced conditions, all the D. solani strains showed a low pectate lyase activity (lower than 0.5 μmol min−1 mg−1) (data not shown). In induced conditions, IFB0099, GBBC 2040, MK16, D s0432-1, and RNS 08.23.3.1A strains showed the highest pectate lyase activity, from 7.7 to 8.7 μmol min−1 mg−1. Strains IFB0158, IFB0221, and IPO 2222 exhibited an intermediate pectate lyase activity, from 3.9 to 4.8 μmol min−1 mg−1. The lowest pectate lyase activity of ~1.3 μmol min−1 mg−1 was observed for the strain IFB0223 (Figure 2B). For the cellulase activity, two groups were differentiated (Figure 3A). Six strains (IFB0099, IPO 2222, GBBC 2040, MK16, D s0432-1, RNS 08.23.3.1A) exhibited high cellulase activity while compared to three other strains (IFB0158, IFB0221, IFB0223). All the tested strains, except for IFB0223, were able to produce proteases (Figure 3B). The strains RNS 08.23.3.1A, IFB0099, IFB0221, MK16, and D s0432-1 showed the highest protease activity.
Figure 3.
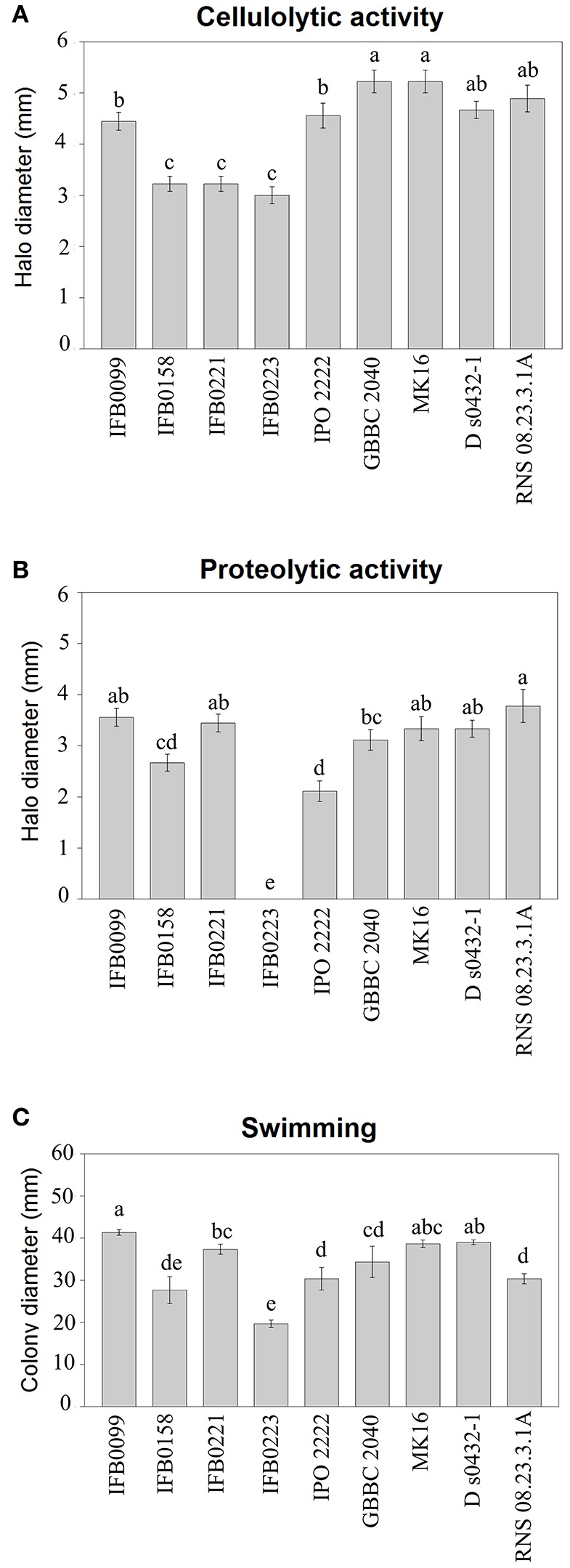
Phenotypic characterization of D. solani strains. (A) Cellulase production was estimated by the diameter (mm) of the haloes observed on the detection plates. (B) Protease production was estimated by the diameter (mm) of the haloes observed on the detection plates. (C) Motility was estimated by the diameter (mm) of the spread of the colonies in the low amount of agar (3 g l−1) medium. All the experiments were performed three times. Means ± standard errors are depicted, n = 3. Within each panel, mean values marked with different letters are significantly different by Kruskal–Wallis test followed by post hoc analysis using Fisher's least significant difference criterion (R agricolae package). All statistical hypotheses were tested at p < 0.05.
The swimming ability of the strains was also analyzed in low concentration agar medium (Figure 3C). The mobility of the D. solani strains was variable and the relationship between motility and virulence was not evident. However, the reduced swimming capacity of strain IFB0223 matched with its low virulence. In contrast, strains IFB0099 with the best swimming capacity in the agar medium showed the greatest ability to macerate potato tuber tissue (Figures 2, 3C).
In our further analyzes of the relationship between virulence, phenotypic traits and genomic signatures, we focused on the comparison of two strains IFB0099 and IFB0223 exhibiting extremely different levels of virulence, with IFB0099 still showing the highest virulence and IFB0223 the lowest virulence, regardless of the experimental conditions.
The D. solani genome structure
For 14 analyzed genomes, the number of scaffolds, the number of N bases added to the genome sequence during the assembly process, the genome size, and the GC content are reported in Table 1. The number of scaffolds ranged from 1 (5 genomes, namely IFB0099, IFB0223, IPO 2222, GBBC 2040, RNS 08.23.3.1A) to 38 (IFB0221). The length of these D. solani genomes varied from 4,860,047 to 4,985,571 bp for the strains GBBC 2040 and RNS 05.1.2A, respectively. Their GC content ranged from 56.34 to 56.13% for the strains GBBC 2040 and RNS 05.1.2A, respectively (Table 1).
Synteny analysis was performed on 5 genomes successfully closed to one scaffold and showed no strong rearrangements among these D. solani strains (Figure 4). There are only 3 syntenic blocks, with two inversions in the case of the strains IFB0099, IFB0223, and RNS 08.23.3.1A, in comparison to GBBC 2040 and IPO 2222. With the Prokka annotation, the number of total predicted genes varied from 4,273 for the strain GBBC 2040 to 4,536 for RNS 08.23.3.1A (Table 2). The number of protein encoding genes varie between 4,138 for the strain RNS 07.7.3B and 4,303 for RNS 05.1.2A (Table 2). The overall ANI analysis of the 14 D. solani strains showed low genomic variability between them, with ANI values ranging from 98.60 to 99.99% (Supplementary Table 1).
Figure 4.

Synteny of the D. solani genomes. Pairwise alignments of genomes were generated by Mauve 2.4.0 (development snapshot Mauve_2015_02_26). Inside each block a similarity profile of the genomic sequence is presented. Height of the similarity profile corresponds to the average level of genomic conservation in that region. Areas that are completely white were not aligned and probably contain sequence elements specific to a particular genome. Height of the similarity profile is inversely proportional to the average alignment column entropy over a region of alignment.
Table 2.
Genome and pangenome contents of D. solani strains.
| Total number of genes | Genome | Pangenomea | ||||||
|---|---|---|---|---|---|---|---|---|
| D. solani genome | Predicted number of genes encoding | Core genes | Accessory genes | Unique genes | ||||
| Proteins | rRNAs | tRNAs | tmRNA | |||||
| IFB0099 | 4,326 | 4164 | 22 | 75 | 1 | 3,756 | 387 | 21 |
| IFB0158 | 4,289 | 4149 | 4 | 64 | 1 | 3,756 | 384 | 9 |
| IFB0221 | 4,283 | 4148 | 4 | 60 | 1 | 3,756 | 383 | 9 |
| IFB0223 | 4,328 | 4167 | 22 | 75 | 1 | 3,756 | 382 | 29 |
| NCPPB4479T IPO 2222 | 4,327 | 4199 | 3 | 62 | 1 | 3,756 | 429 | 14 |
| GBBC 2040 | 4,273 | 4145 | 3 | 62 | 1 | 3,756 | 368 | 21 |
| MK10 | 4,376 | 4245 | 3 | 64 | 1 | 3,756 | 356 | 133 |
| MK16 | 4,288 | 4157 | 3 | 64 | 1 | 3,756 | 380 | 21 |
| D s0432-1 | 4,331 | 4167 | 22 | 74 | 1 | 3,756 | 385 | 26 |
| RNS 08.23.3.1A | 4,536 | 4151 | 18 | 72 | 1 | 3,756 | 380 | 15 |
| PPO 9019 | 4,280 | 4146 | 1 | 64 | 1 | 3,756 | 380 | 10 |
| PPO 9134 | 4,282 | 4144 | 3 | 65 | 1 | 3,756 | 384 | 4 |
| RNS 05.1.2A | 4,436 | 4303 | 1 | 64 | 1 | 3,756 | 175 | 372 |
| RNS 07.7.3B | 4,274 | 4138 | 1 | 64 | 1 | 3,756 | 376 | 6 |
Solely genes encoding proteins were included in the analysis.
The D. solani pangenome
The pangenome of the selected 14 strains contains 5,020 protein encoding genes with a core genome of 3,756 genes (74.8%) and an accessory genome of 574 genes (11.5%) (Table 2, Supplementary Table 2). The number of unique genes, as calculated by Mauve, was 690 (13.7%) (Figure 5A, Table 2) and 281 of them were annotated as encoding hypothetical proteins. The accessory genome fraction contains from 356 to 429 genes of the strains MK10 and IPO 2222, respectively, with a notable exception of the strain RNS 05.1.2A which has only 175 genes in this fraction (Table 2). The unique genome fraction varies from only 4 genes for the strain PPO 9134 to 372 genes for RNS 05.1.2A. Twelve strains possess from 4 to 29 unique genes. Interestingly, MK10 and RNS 05.1.2A were the most diverse strains with 133 and 372 unique genes, respectively (Table 2). If the most diverse strain RNS 05.1.2A is excluded from the pangenome analysis, the pangenome is reduced to 4,651 genes, the core genome increases up to 3,901 genes, the number of accessory genes decreases to 432 and the number of unique genes goes down to only 318 (Figure 5B).
Figure 5.

The D. solani pangenome shape. (A) Pangenome shape based on the number of genes in all 14 strains. (B) Pangenome shape after exclusion of strain RNS 05.1.2A. Blue—represents the core genome. Orange—represents the accessory genome. Gray—represents the unique genome. Lighter shades of each color represent the fractions of genes annotated as hypothetical proteins.
Conservation of virulence and regulatory genes and repertoire of prophages in the D. solani genomes
Because of their involvement in the soft-rot symptoms, we verified the presence of genes encoding PCWDEs and their regulators in 14 D. solani genomes. All the genes encoding PCWDEs previously described in D. dadantii (pectate lyases, pectin methylesterases, cellulases, or proteases) are found in the D. solani core genome. Each gene has an identical sequence in all the 14 D. solani strains. In the case of pectate lyases, protein sequence identities between D. solani and D. dadantii 3937 reache 98% for PelB, 97% for PelC and PelZ, 96% for PelA, PelE, PelW and PelX, 95% for PelL and PelN, 94% for PelD and 84% for PelI. For the proteases and cellulase, the identities level between D. solani and D. dadantii 3937 reache 98% for PrtA and PrtB, 95% for PrtG and 93% for CelZ.
The genes encoding transcriptional regulators of virulence genes, previously identified in D. dadantii 3937 (such as KdgR, PecS, PecT, CRP, Fur, Fis, H-NS), are also well conserved and found in the D. solani core genome. These regulators are very similar to the corresponding proteins of D. dadantii 3937, with protein identity of 100% for CRP, Fis and H-NS, 99% for KdgR and Fur, 98% for PecS, and 92% for PecT. As for PCWDEs, their protein sequences are identical among the D. solani strains.
Due to their importance in genome evolution, the presence of prophages was analyzed in the 14 D. solani genomes. Of 35 predicted prophages, 23 could be gathered into three main groups, I, II and III, according to their sequence similarity (Figure 6, Table 3). The sequence of the 12 other prophages greatly varied and they were assigned as ungrouped or “n” (Supplementary Figure 1, Table 3). In addition, each prophage was predicted as intact, questionable or incomplete. All group I prophages were predicted as intact, group II prophages were described as incomplete, and group III prophages as questionable (Figure 6). Interestingly, each genome contains at least one member of each defined group I, II, and III.
Figure 6.

Prophages present in the D. solani genomes. Prophages are divided into three groups: intact (IA and IB), incomplete (II) and questionable (III) on the basis of structural analysis based on data provided by PHAST on-line web server (http://phast.wishartlab.com/; Zhou et al., 2011).
Table 3.
Prophages present in the analyzed D. solani genomes.
| D. solani strain | No. of the prophage sequence | Length (kb) | Sequence completeness | Positions in the D. solani genome | Groupa |
|---|---|---|---|---|---|
| IFB0099 | PR1 PR2 |
27 14.1 |
intact questionable |
2748142-2775153 4672375-4686494 |
I A III |
| IFB0158 | PR1 PR2 |
27.4 14.1 |
intact questionable |
637761-665201 3565525-3579644 |
I A III |
| IFB0221 | PR1 PR2 |
27.4 14.1 |
intact questionable |
637761-665201 4859798-4874569 |
I A N |
| IFB0223 | PR2 PR1 PR3 |
48.3 44.9 14.1 |
intact incomplete questionable |
2744993-2793313 432810-477786 4677009-4691128 |
I B II III |
| NCPPB4479T IPO 2222 | PR1 PR2 |
16 14.1 |
incomplete questionable |
2913332-2929416 4611857-4625976 |
n III |
| GBBC 2040 | PR1 PR2 |
26.9 14.1 |
incomplete questionable |
2905276-2932269 4604558-4618677 |
n III |
| MK10 | PR2 PR1 |
27.2 14.1 |
intact questionable | 2756228-2783473 159129-173256 |
I A III |
| MK16 | PR2 PR1 |
27.2 14.1 |
intact questionable | 2687323-2714567 159136-173255 |
I A III |
| D s0432-1 | PR2 PR1 PR3 |
27.4 44.9 14.1 |
intact incomplete questionable |
2732844-2760284 435970-480946 4643974-4658093 |
I A II III |
| RNS 08.23.3.1A | PR1 PR3 PR2 |
28.3 44.9 14.5 |
intact incomplete questionable |
649811-678182 3258604-3303580 2561220-2575779 |
I A II III |
| PPO 9019 | PR1 | 14.1 | questionable | 1622153-1636272 | III |
| PPO 9134 | PR1 PR2 |
12.3 14.1 |
questionable questionable |
965401-977759 4823112-4837231 |
n III |
| RNS 05.1.2A | PR4 PR5 PR6 PR1 PR2 PR7 PR3 |
23.5 34 31.5 11.4 15.2 19.4 20.8 |
intact intact intact incomplete incomplete incomplete questionable |
2062173-2085685 2397356-2431394 4377687-4409244 522314-533731 871014-886304 4482742-4502177 1782074-1802942 |
n n n n n n n |
| RNS 07.7.3B | PR1 PR2 |
14.1 13.1 |
questionable questionable |
1644970-1659089 3091451-3104564 |
III n |
Unique prophage sequences were marked with ‘n'.
The prophage composition of each strain varies quantitatively and qualitatively among the 14 D. solani strains (Table 3). The prophage number varies from a single prophage for the strain PPO 9019 (group III) to seven prophages for RNS 05.1.2A, all attributed to “n” because of their diverse structures (three intact, three incomplete and one questionable prophage). For majority of D. solani genomes, i.e. nine, two prophages are predicted. Only four bacterial strains (IFB0099, IFB0158, MK10 and MK16) share the same prophage repertoire (group I and III). Three strains (IFB0223, RNS 08.23.3.1A and D s0432-1) have three predicted prophages in their genomes and a similar prophage repertoire (group I, II, and III).
Based on our classification, group I contains eight intact prophages, found in seven virulent strains (sub-classified in group IA). The gene structure is very similar, with a prophage length varying from 27 kb (strain IFB0099) to 28.3 kb (strain RNS 08.23.3.1A). The eighth group I prophage (sub-classified in group IB) is found in the weakly virulent strain IFB0223. Despite its size of 48.3 kb, it has a similar basal structure. Indeed, prophages of groups IA and IB differ by a deletion/insertion of about 20 kb in the middle of their sequence (Figure 6). The three prophages of group II, found in three genomes, are 44.9 kb long. Group III comprises 12 questionable prophages of 14.1 kb, found in 12 genomes. Prophages not classified in these three groups (“n”) are predicted to be intact (3 prophages), or questionable (4 prophages) or incomplete (5 prophages) (Supplementary Figure 1). It should be noted that many phage-related genes are classified in the unique genome fraction, demonstrating their genetic diversity.
The D. solani panregulon
In order to provide additional genomic information that can help to interpret the differences in virulence observed between D. solani strains, a prediction of the regulons of TFs known to be involved in Dickeya pathogenesis was performed. Large putative regulons were found for the major regulators: CRP, Fur, KdgR, and PecS. All strains contain a set of shared target genes, constituting the core regulon (binding sites present in all the genomes), but also a dispensable regulon fraction (Figure 7A). CRP has the largest predicted panregulon with 807 potential targets, of which 121 are in the core regulon, 554 in the accessory regulon and 132 in the unique regulon (Figure 7A). The predicted PecS panregulon includes 784 targets of which 84 are in the core regulon, 540 in the accessory regulon and 160 in the unique regulon. The Fur predicted panregulon has 174 targets, of which 24 are present in the core regulon, 133 in the accessory regulon and 17 in the unique regulon. KdgR has the smallest predicted panregulon with 130 targets, of which 16 are in the core regulon, 100 in the accessory regulon and 14 in the unique regulon (Figure 7A).
Figure 7.

Regulon analysis based on binding sites predictions. Predictions of the transcription regulators (CRP, Fur, KdgR, and PecS) binding sites were made with the use of MAST version 4.10.1. (Bailey and Elkan, 1994) and the version 1.62b of the Bio.motifs package from the Biopython library (Cock et al., 2009). (A) The regulon representation for the 4 transcription regulators (TFs): in orange for core, green for accessory, violet for unique TF targets. (B) Phylogenetic tree based on the total regulons predicted for the 4 TFs. The UPGMA methods on Jaccard distance were utilized for phylogenetic analysis. (C) Heatmaps for CRP, Fur, KdgR and PecS TFs, which represent all predicted genes that are putatively regulated by the selected TF. A red box indicates the presence whereas a black one indicates the absence of a TF binding site in the upstream region of the gene.
The D. solani strains grouped into three main clusters by considering the differential occurrence of all four TF targets (Figure 7B). The strain RNS 05.1.2A groups separately from other 13 D. solani strains, which form two clusters, one of 6 strains (MK10, MK16, IFB0099, IFB0223, D s0432-1 and RNS 08.23.3.1A) and the other of 7 strains (GBBC 2040, IPO 2222, IFB0158, IFB0221, PPO 9019, PPO 9134, and RNS 07.7.3B). The heatmaps show phylogenetic relationships of the strains based on TF binding sites present/absent in the regulatory region of the target genes (Figure 7C). Clustering was similar when considering each single TF or the phylogenetic model based on all four TFs. Small rearrangements of the tree contents are visible only in case of CRP and KdgR.
In more detail, the predicted CRP core and accessory regulon contains several genes involved in cellulose and pectin degradation such as celZ, pehN, pelB, pelC, pelD, pelE, pelI, pelW, pelX, kdgF, kdgM, kdgN, kdgT, kdsD, kduD, ogl, togM, and togT (Figure 8). The core or accessory regulon predicted for CRP also includes genes related to flagella (flgB, flgJ, flhC, flhD, fliD, fliJ), T3SS (hrpN and hrpS), polyketide synthesis (pksG, pksI, and pksJ), iron metabolism (bfd, bfr, cbsH, dps, fct, fhuC, ftnA, sfuA and sufA) and resistance to oxidative stress (ahpC). Interestingly, the predicted CRP regulon contains some genes constituting the two quorum sensing systems described for D. dadantii 3937, namely expR, vfmB and vfmD. CRP was also predicted to regulate other TFs, such as argP, argR, cpxR, crl, and gntR (Figure 8).
Figure 8.

Selected regulons of D. solani based on binding sites predictions. Predictions of the CRP, Fur, KdgR, and PecS (TFs) binding sites were conducted with the use of MAST version 4.10.1. (Bailey and Elkan, 1994) and Bio.motifs package from Biopython library (version 1.62b) (Cock et al., 2009) for the virulence factors: proteins involved in pectin and oligogalacturonide degradation, chemotaxis, and motility, iron metabolism, polyketide synthesis, resistance to oxidative stress, transcription factors, quorum sensing-related proteins and T3SS components. Gene names are marked in black when the genes belong to the core genome and in red when the genes belong to the accessory genome fractions. The TFs name is given in black to indicate the core regulon, and in red to indicate the accessory regulon.
As expected, the predicted Fur regulon contains mostly genes related to iron assimilation, namely cbuD, fhuC, ftnA, and sufA (Figure 8). It also includes a protease gene (prtA), a gene involved in oligogalacturonide transport (kdgN), a flagellar gene (fliT), a gene involved in resistance to oxidative stress (sodA) and TF genes, such as cdaR and fhlA (Figure 8).
The predicted KdgR core or accessory regulon includes several genes involved in cell wall component degradation and catabolism: kdgF, kdgK, kdgN, kdgT, kduI, ogl, togT, celZ, pehN, pehV, pelA, pelB, pelC, pelE, pelI, pelW, pelX, togT, (Figure 8) and other genes previously shown to be regulated by KdgR in D. dadantii (ppsA, pykF, sotA, spiX, tpgX, ydiA, ygjV, chmX) (Rodionov et al., 2004). More surprisingly, it contains several genes involved in regulation, which were not previously identified in the D. dadantii KdgR regulon (cyaY, expI, hns, asnC, betI, pecS) (Figure 8).
The predicted PecS core and accessory regulon contains genes encoding PCWDEs (pehV, pelA, pelD, pelE, pelW, prtA, prtB, and prtG), genes related to chemotaxis and motility (cheB, cheY, chmX, flgB, flgJ, flhD, fliD, fliE, fliF, fliJ, fliK, and fliL), T3SS (hrpA, hrpD, hrpF and hrpY) (Figure 8), polyketide synthesis (pksL, pksH, and pksI), iron metabolism (acsC, cbuB, cbuC, cbuD, cyaY, and feoB) and quorum sensing (expI). The predicted PecS regulon also includes other genes encoding TFs, such as argP, betI, cdaR, cpxR, cytR, and oxyR.
In summary, genes coding for PCWDEs and important for oligogalacturonide degradation are predicted to be regulated mainly by KdgR, PecS and CRP. Chemotaxis and motility related genes as well as genes encoding T3SS proteins and polyketide synthases are predicted to be regulated by CRP and PecS. Genes significant for iron-metabolism are predicted to be regulated by Fur, but also by PecS and CRP. Quorum sensing related genes seem to be regulated by CRP, KdgR and PecS. However, it should be underlined that for all regulon predictions the acquired hits are putative, and consequently, they provide only starting points for later investigations.
Genomic background of the differences in virulence between D. solani IFB0099 and IFB0223
This study performed on 14 D. solani strains showed that they have homogenous genomes despite different phenotypic features, not only in their ability to macerate plant tissue but also in pectinolytic, cellulolytic, and proteolytic activities and motility. In order to elucidate the genomic background of these differences, we concentrated our analysis on two D. solani strains: IFB0099 and IFB0223, which differ significantly in virulence level on potato (Figure 2A) and chicory (Potrykus et al., 2014).
All genes encoding PCWDEs and their regulators are present in both genomes and their sequences indicated 100% identity. D. solani IFB0099 exhibited a significantly higher activity of pectinolytic and cellulolytic enzymes than IFB0223 (Figures 2B, 3A). The strain IFB0223, which showed no protease production, possesses genes coding for three proteases: PrtA, PrtB, and PrtG and their T1SS secretion system, and all of these genes show 100% identity with those of D. solani IFB0099.
At the genome level, IFB0099 and IFB0223 are highly similar to each other. There are only 38 genes in IFB0099 genome that are not present in IFB0223. Inversely, 41 genes are found in the IFB0223 genome but absent in the IFB0099 genome. In both cases, most of these genes are prophage related. D. solani IFB0099 genome contains one intact prophage of group IA (27 kb) and one questionable prophage (about 14 kb) (Figure 6). IFB0223 contains one intact prophage of group IB (48.3 kb), one incomplete prophage (about 45 kb) and one questionable prophage (about 14 kb) (Figure 6). The long intact prophage of IFB0223 contains two copies of several genes: smrA (endonuclease), pcaK (4-hydroxybenzoate transporter), hin (invertase), cas1 (clavaminate synthase 1) and 16 other phage related genes. Moreover, the long intact prophage of IFB0223 (group IB, 48.3 kb) is homologous to the group IA prophage of strain FB0099, localized in the same genomic region but 27 kb long.
Apart from differences in the prophage related genes, IFB0223 lacks the genes encoding a peptidoglycan transglycosylase (mtgA), an isoprenoid biosynthesis protein with amidotransferase-like domain (elbB), a T3SS component (hrpQ) and a miscRNA called ArcZ, while these genes were present in all other D. solani genomes.
When looking into the nucleotide sequence similarity of the protein coding genes present in the genomes of both IFB0099 and IFB0223, only 10 SNPs were found and 5 of them determine non-synonymous variations. For instance, a SNP in rpoB caused a change from Ser in IFB0099 to Phe in IFB0223, and the SNP in dmlR_13 replaced Ser in IFB0099 with a Pro in IFB0223. More surprisingly, we observed 3 non-synonymous substitutions (2 × Ala → Thr and Ser → Ala) in the fhaB1 gene (IFB0099_02260) between the two strains. The fha genes encode large filamentous hemagglutinins secreted outside the cells. These FHA proteins may facilitate the attachment of the bacterial cells to their host. According to Prokka annotation, two fhaB genes are found in the genome of IFB0223 (IFB0223_02255 and IFB0223_02334) and three fhaB genes are present in the genome of IFB0099 (IFB0099_02260, IFB0099_02339 and IFB0099_02340). Both genomes contain fhaB1 (IFB0099_02260, IFB0223_2255), localized closely to the gene dadA coding for the d-amino acid dehydrogenase small subunit. Despite the 3 non-synonymous substitutions observed in their genes, the FhaB1 proteins of IFB0099 and IFB0223 possess a signal peptide of the same length (4165 amino acids) and a similar domain structure, as predicted by hmmscan against Pfam database (Finn et al., 2015). Thus, both FhaB1 proteins are supposed to be functional, even if they might show different efficiencies.
A second gene, fhaB2 (IFB0223_02334 and IFB0099_02339) is predicted in both genomes, closely to the region coding for the harpin HrpN. The fhaB2 gene present in the IFB0223 genome (IFB0223_02334, 13,791 bp) is homologous to the gene hecA described in the D. chrysanthemi strain EC16 (Rojas et al., 2002; Finn et al., 2015). Both proteins are similar in length and structure (predicted by hmmscan) and both possess a signal peptide, which may suggest that the IFB0223 FhaB2 protein is functional. In contrast, two successive shorter genes (IFB0099_02339, 6,525 bp and IFB0099_02340, 7,242 bp) are predicted in the corresponding position in the genome of IFB0099, and only one of them encodes a protein with a signal peptide. Comparison of the nucleotide sequences of the genomic regions coding for fhaB2 in IFB0223 and IFB0099 showed high similarity between them with only 2 differences in the 13,791 nucleotide long alignment. One of these differences leads to a synonymous substitution at position 6,504 of the alignment (C → T). More interestingly, at position 7,235 of the alignment, the deletion of a G nucleotide leads to a frameshift, giving rise to an Opal stop codon (TGA) at position 7,240 in the IFB0099 sequence. Because of this deletion, the translated protein is shorter in IFB0099 (IFB0099_02340) than in IFB0223, with 2,413 amino acids instead of 4,596. A third fhaB is then predicted in IFB0099 (IFB0099_02339); however this truncated protein has no signal peptide.
Discussion
In the present study, we combined the results obtained from the pangenome and the predicted panregulon to explore the background of diverse virulence levels of D. solani strains. First, we developed a genome assembly pipeline for D. solani which allowed us to close the genomes of a highly virulent strain, IFB0099, and a low virulent strain, IFB0223 (Figure 1). Reads originating from only one sequencing technology (PacBio) are sufficient for closing the genome. Instead of a set of contigs, we obtained a fully annotated chromosome without N-bases. Importantly, the software used is freely available and no home-made scripts were utilized during the assembly and annotation, making the pipeline easy to follow by other researchers.
High homogeneity of D. solani genomes and its effect on the pangenome structure
Genomes of 14 D. solani strains were explored in order to disclose some explanation for their diverse ability to macerate plant tissue (Tsror et al., 2013; Potrykus et al., 2014, 2016, 2018; Golanowska et al., 2017). Our comparative study, performed on 10 available and 4 newly sequenced D. solani genomes, confirmed the high genomic homogeneity in this species, even among strains isolated from soft-rotting plants or rhizosphere of healthy plants. The homogeneity of D. solani genomes is reflected by ANI values of about 99.9, with the exception of strain RNS 05.1.2A which gives values around 98.7. As noticed by Khayi et al. (2015), the strain RNS 05.1.2A forms a specific sub-group among D. solani strains. The D. solani genomes have a similar structure, revealed by their high synteny presented for five successfully closed genomes. The genes encoding major virulence determinants (pectinases, cellulases, and proteases) and their regulators (KdgR, PecS, PecT, Fis, H-NS, CRP, and Fur) shared 100% identity.
For the newly sequenced genomes, the number of predicted protein encoding genes varied from 4,148 (IFB0221) to 4,167 (IFB0223), with 60 to 75 genes encoding tRNAs. These results are comparable to those obtained for other D. solani strains (Garlant et al., 2013; Khayi et al., 2014, 2015, 2016; Pédron et al., 2014). Considering the 14 D. solani genomes, their mean size is 4,892,047 bp and their mean GC content is 56.23%. These 14 genomes were used for description of the species pangenome, with the following repartition of the genes: 74.8% into the core genome and 25.2% into the dispensable (accessory and unique) genome. If strain RNS 05.1.2A is excluded, the core genome increases by 4%, the accessory genome decreases by 25% and the number of unique genes decreases by about 50%. The pangenome of D. solani appears open and, after adding another genome to the pool of analyzed genomes, the unique genome fraction increases on average by 50 genes (by 25 genes if strain RNS 05.1.2A is excluded).
Pangenome analysis is a good way to describe bacterial lifestyle and to explain differences in the strain pathogenicity. The genes found in the dispensable genome fraction can be responsible for the strains ability to survive in rare or specific conditions. The dispensable genome of D. solani comprised only 25.2% of the genes and it mainly consisted of prophage-related genes. In contrast, recent data using Pectobacterium parmentieri strains showed that the dispensable genome of this more heterologous species comprises about 49% of the genes (Zoledowska et al. unpublished results).
Variability in prophage sequences present in D. solani genomes
A notable variability in the prophage sequences present in D. solani genomes is for the first time reported in this study. The D. solani strains generally contain two to three predicted prophages, among them: intact, incomplete and questionable ones. Most of these prophages belong to 3 groups but different repertoires are observed depending on the strain (Table 3). The presence of the prophage sets found in individual strains may be related to their origin, for example two strains isolated in Poland (IFB0099 and IFB0158) have the same prophage repertoire (Figure 6 and Table 3). The presence of similar prophages in the strains isolated in Scotland and Israel (MK10 and MK16) can be explained by the fact that majority of potato seeds imported by Israel are produced in Scotland (Figure 6 and Table 3). Strain RNS 05.1.2A contains the highest number of predicted prophages and all of them are found only in this strain. This confirms the report of Khayi et al. (2015) indicating that the strain RNS 05.1.2A forms a separate clade in the D. solani phylogenetic tree (Khayi et al., 2015). It could be speculated that strain RNS 05.1.2A does not possess strong tools to protect itself from phage infections.
The role of prophages in bacteria can be variable. Temperate bacteriophages are particularly important agents of horizontal gene transfer. They could be vectors of new virulence factors or/and toxins that may change a non-virulent strain into a pathogenic one (Varani et al., 2013). Prophage insertion could also have structural genomic impacts (Varani et al., 2013). Since chromosome organization is known to influence transcriptional networks in D. dadantii (Jiang et al., 2015), the expression of some genes, in D. dadantii and in D. solani, could be affected by modification of genom structure due to prophage insertion.
Analysis of D. solani panregulon, effect of regulators on virulence genes
Panregulon analysis is a powerful tool, complementary to comparative genomics, that helps to identify new targets for known TFs and to elucidate roles they play in microorganisms. We used TF binding site prediction tools to identify putative targets of four TFs, namely CRP, KdgR, PecS, and Fur in D. solani genomes. Each of them plays a crucial role in Dickeya virulence. However, until now, these regulators have been analyzed almost exclusively in the D. dadantii model strain 3937. Thus, it is not known whether the data obtained in strain 3937 are relevant in other Dickeya species and, even, in other D. dadantii strains. Originality of our approach relies on analysis the corresponding regulons in a set of D. solani strains. The binding site predictions allowed us to identify the D. solani panregulon for KdgR, PecS, CRP and Fur, and to differentiate the core and accessory regulons. For each TF, the core regulon contains from 10.7 to 15% of the predicted regulated genes. Thus, majority of the predicted targets, 85–89.7%, are in the accessory regulon. Many genes encoding virulence factors, such as PCWDEs and proteins involved in pectin degradation are found in the predicted accessory regulons of KdgR, PecS, and CRP. For instance, the pectate lyase gene pelE seems to be regulated by KdgR and CRP in all the tested D. solani strains, but only some of them are regulated by PecS (Figure 7). Thus, some virulence genes could have differential expression among D. solani strains.
KdgR is primarily a repressor of genes encoding pectinases and pectin catabolism. In D. dadantii 3937, more than 50 genes are induced in the presence of pectin and KdgR directly controls at least 13 operons (Hugouvieux-Cotte-Pattat and Robert-Baudouy, 1989; Hugouvieux-Cotte-Pattat et al., 1996). Comparative genomics of different Enterobacteriaceae predicted that KdgR could control as many as 32 operons, including several novel targets such as chmX, dhfX, gntB, ppsA pykF, spiX, sotA, tpfX, yeeO, and yjgK (Rodionov et al., 2004). In our study, we also found in the D. solani KdgR panregulon all the KdgR targets reported in D. dadantii 3937. In addition, we found novel putative members of the KdgR regulon, such as pir, gacA, uvrY, ybbH, nagC, expI, hns, nadR, asnC, betI, pecS, pecM. Function of these KdgR targets are not limited to pectin degradation and some of them are involved in regulation. For instance, GacA regulates T3SS at a post transcriptional level in D. dadantii 3937 (Yang et al., 2008). The expI gene encodes a homoserine lactone synthase involved in quorum sensing. Interactions between these two regulators could have an impact on the virulence of D. solani.
In D. dadantii, PecS acts as a repressor of genes encoding PCWDEs. The strongest repression is exerted on the genes encoding secreted proteins (pelC, pelE, pelL, pelN, celZ, prtABC, nipE, virK, avrL) and on the genes involved in the biosynthesis of excreted compounds, such as the blue pigment indigoidine (indABC), a surfactant (rhlA) and the quorum sensing signal VFM (vfmZ, vfmE, vfmAB) (Hommais et al., 2008). In addition to these genes, we also found protease genes in the PecS accessory regulon of D. solani. We previously noticed that a pecS mutant of IFB0223 has a de-repressed protease activity in comparison to the wild-type strain (Potrykus et al., 2014). In D. dadantii 3937, PecS also regulates genes involved in the flagellum biogenesis (fliE and fliFGHIJKLMNOPQR) (Rouanet et al., 2004). In our study, putative binding sites for PecS were predicted in the regulatory regions of several D. solani flagellum genes (fliE, fliD, fliF, fliJ, fliK, fliL). It was previously observed that pecS inactivation in D. solani resulted in variable phenotypes regarding swimming motility; the pecS mutants of 2 strains, among the 4 tested, were hypermotile (Potrykus et al., 2014). Transcriptomic analysis of D. dadantii pecS mutants showed that PecS directly or indirectly controls from 400 to more than 600 genes (Hommais et al., 2008; Pédron et al., 2018), placing PecS at the top of a major regulatory cascade. In D. solani, the predicted PecS panregulon contains approximately 780 genes, including other TFs like CRP, KdgR and Fur. Considering either KdgR or PecS, our regulon prediction seems credible and can be used with confidence to investigate bacterial regulatory networks.
The main role of CRP is to control assimilation of carbohydrates but it is also involved in various other cellular processes. As expected, the CRP regulon of D. solani includes several pectate lyase genes and genes involved in pectin catabolism. It also contains genes involved in mobility, T3SS, toxin production, iron metabolism regulation (Figure 8). Among them, we observed two genes vfmB and vfmD which are involved in the biosynthesis of a Dickeya specific communication signal of yet unknown structure (Nasser et al., 2013; Potrykus et al., 2018). This specific quorum sensing system influences the PCWDE production in D. solani, as well as in D. dadantii (Nasser et al., 2013; Potrykus et al., 2018).
PecT is an interesting case of a TF sensitive to DNA conformation changes (Hérault et al., 2014) and it has an important role in regulation of the PCWDE genes in D. solani (Potrykus et al., 2014). Unfortunately, prediction of PecT regulon on the basis of in silico analysis was not possible since the PecT binding site has no defined DNA sequence.
Genomic background related to differences in virulence between D. solani IFB0099 and IFB0223
In order to find information on virulence differences in the genomic background, we concentrated our analysis on strains IFB0099 and IFB0223 which differ in their virulence level on potato (Figure 2A) and chicory (Potrykus et al., 2014) and in addition their activity of PCWDE (Figures 2B, 3A,B). However, sequences encoding proteins involved in cell wall degrading indicated 100% homology.
When comparing these two strains, most strain-specific genes are prophage related. The genomes of IFB0099 and IFB0223 contain only one intact prophage of 27 kb and 48.3 kb, respectively, localized in the same genome region (Figure 6). The IFB0223 intact prophage contains two copies of 20 genes (smrA, pcaK, hin, cas1 and prophage related genes), suggesting a duplication event. The presence of a longer prophage could indirectly contribute to the expression of virulence genes in IFB0223 through changing the spatial organization of this DNA region. Some TFs involved in Dickeya virulence modulate their activity in function of the DNA conformation, as shown for PecT (Hérault et al., 2014) or even KdgR (Bouyioukos et al., 2016).
Concerning the phage elements encoded in the D. solani genomes, only weakly virulent IFB0223 possesses an additional phage genetic material (intact phage from group IB) which is twice as large as the phage occurring in the corresponding regions of IFB0099 and other virulent D. solani strains. The potential role of prophages in the variability of D. solani virulence will need further investigation.
In comparison to IFB0099 and all other virulent strains, IFB0223 lacks the genes mtgA, elbB, hrpQ and a gene encoding a miscRNA called ArcZ. ArcZ belongs to a family of small regulatory RNAs (sRNAs) which are important post-transcriptional regulatory components in bacteria. In Erwinia amylovora ArcZ participates in the positive control of T3SS, amylovoran exopolysaccharide production, biofilm formation, and motility (Zeng and Sundin, 2014). ArcZ is also responsible for the translational activation of RpoS in response to oxidative stress in E. coli (Mandin and Gottesman, 2010). These indications suggest that ArcZ could play a role in D. solani pathogenicity and its absence might be responsible for low virulence of IFB0223.
Following Prokka annotation 30 genes coding for filamentous hemagglutinins (adhesin, CdiA, FhaB) were found in the 14 D. solani genomes. Each D. solani strain possesses at least one (IPO 2222, GBBC 2040, MK10), two (IFB0158, IFB0221, IFB0223, PPO 9134, RNS 05.1.2A, RNS 07.7.3B) or even three predicted adhesin genes (IFB0099, MK16, D s0432-1, RNS 08.23.3.1A, PPO 9019). The fhaB genes annotated in D. solani genomes vary in length and code for proteins of as little as 246 (MK16) to as many as 4,596 (IFB0223) amino acids. The FhaB proteins have similar domain structure. The most variable region found in CdiA/FhaB is the cytotoxic domain after a common pre-toxin VENN motif. Since these proteins are highly variable, the phylogenetic relationship of the D. solani 21 cytotoxin domains extracted from adhesin proteins was examined (Supplementary Figure 2, Supplementary Table 3). Two FhaB proteins described for E. coli EC93 (CdiA) and E. chrysanthemi EC16 (HecA) were included in the analysis. Interestingly, majority of the cytotoxic domains extracted from D. solani adhesins form two distinct clades A and B (Supplementary Figure 2). Sequences from clade B are similar to HecA from E. chrysanthemi EC16, which exhibit a Dnase activity. Moreover, both types of cytotoxic domains are encoded by most of the D. solani genomes (except for IPO 2222, GBBC 2040, MK10, which have only one fhaB gene, Supplementary Table 2).
Genomic data suggested that strain IFB0223 produces two functional secreted adhesins, FhaB1 and FhaB2. These two adhesins could be responsible for its efficient adherence ability, but also might limit its mobility. In contrast, disruption of the gene fhaB2 in IFB0099 may cause a loss of function for the adhesin FhaB2 and less efficient attachment of IFB0099 to plant surfaces. Poor attachment may result in an increased cell mobility that, finally, could contribute to higher virulence of this strain. Such hypothesis resembles observations of Ionescu et al. (2014) who showed that a highly virulent mutant of X. fastidiosa has lower attachment to the xylem vessels than the wild type strain. Therefore, strong attachment of bacteria to plant surfaces could restrict their movement and their capacity to colonize the plants and to systemically spread within the plant host. Strain IFB0223 appeared significantly less mobile than strain IFB0099 (Figure 3). Khayi et al. (2015) underlined that non-synonymous variations in the flagellar genes fliC and fliN correlate with the absence of motility and weak virulence of the D. solani strain 3296. In the case of low and highly virulent strains, IFB0223 and IFB0099, flagellar genes and flagella motor genes (motA and motB) have identical DNA sequences. Thus, low mobility of IFB0223 could rather be related to a high attachment capacity.
Conclusions
The genome-wide comparison based on pangenome and panregulon analysis of 14 D. solani genomes, showed only few differences between the highly virulent potato isolate D. solani IFB0099 and the low virulent rhizosphere isolate D. solani IFB0223. In contrast, these two strains differ significantly in the production of virulence factors like pectinases, cellulases and proteases, and in their mobility. Their genomes diverge in the number and size of prophages. Only IFB0223 lacks the genes encoding peptidoglycan transglycosylase (mtgA), a T3SS component (hrpQ) and a miscRNA called ArcZ, a small regulatory RNA which can provide resistance to oxidative stress. Another relevant difference is the disruption of the gene fhaB2, encoding adhesin, in strain IFB0099 and other virulent D. solani strains. Inactivation of this gene may result in less efficient attachment of bacteria to plant surfaces and, concurrently, to increased mobility bacteria within plant intercellular spaces. The more adhesive but less motile strain IFB0223 showed weaker aggressiveness than the spreading faster and causing more severe disease symptoms strain IFB0099.
Author contributions
MalG conducted the majority of the experiments, analyzed, and interpreted the pangenome and panregulon data and prepared the first draft of the manuscript. MP and AM-P contributed to complete genomes, prepared figures and tables, discussed results, and contributed to the writing of the final version of the manuscript. MK made the pangenome bioinformatics analysis. GB made the panregulon bioinformatics analysis. MarG performed preliminary pangenome analysis and contributed to the writing of the manuscript. MB, IM, and KS critically revised the final version of the manuscript. EL, NH-C-P, and AM conceived the idea, designed research and critically revised the final version of the manuscript. All the authors read and approved the manuscript.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This work was financed by the National Science Centre in Poland via grants 2013/08/M/NZ9/00974 and 2014/14/M/NZ8/00501 awarded to EL, the Polish–French collaboration program Polonium 2012 awarded to EL and NH-C-P, and the Polish–Italian collaboration program Canaletto 2013 awarded to EL, MB, and AM.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01940/full#supplementary-material
{kind=link}
{kind=link}
References
- Adeolu M., Alnajar S., Naushad S., Gupta R. (2016). Genome-based phylogeny and taxonomy of the “Enterobacteriales”: proposal for Enterobacterales ord. nov. divided into the families Enterobacteriaceae, Erwiniaceae fam. nov., Pectobacteriaceae fam. nov., Yersiniaceae fam. nov., Hafniaceae fam. nov., Morganellaceae fam. nov., and Budviciaceae fam. nov. Int. J. Syst. Evol. Microbiol. 66, 5575–5599. 10.1099/ijsem.0.001485 [DOI] [PubMed] [Google Scholar]
- Bailey T. L., Elkan C. (1994). Fitting a Mixture Model by Expectation Maximization to Discover Motifs in Biopolymers. UCSD Technical Report CS94-351. Toronto, ON: University of California San Diego (UCSD). [PubMed] [Google Scholar]
- Bankevich A., Nurk S., Antipov D., Gurevich A. A., Dvorkin M., Kulikov A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. 10.1089/cmb.2012.0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barras F., van Gijsegem F., Chatterjee A. K. (1994). Extracellular enzymes and pathogenesis of soft-rot Erwinia. Annu. Rev. Phytopathol. 32, 201–234. 10.1146/annurev.py.32.090194.001221 [DOI] [Google Scholar]
- Berlin K., Koren S., Chin C. S., Drake J., Landolin J. M., Phillippy A. M. (2015). Assembling large genomes with single-molecule sequencing and locality sensitive hashing. Nat. Biotechnol. 33, 623–630. 10.1038/nbt.3238 [DOI] [PubMed] [Google Scholar]
- Bolger A. M., Lohse M., Usadel B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouyioukos C., Reverchon S., Képès F. (2016). From multiple pathogenicity islands to a unique organized pathogenicity archipelago. Sci. Rep. 6:27978. 10.1038/srep27978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Kandel P. P., Cruz L. F., Cobine P. A., De La Fuente L. (2017). The major outer membrane protein MopB is required for twitching movement and affects biofilm formation and virulence in two Xylella fastidiosa strains. Mol. Plant-Microbe Interact. 30, 896–905. 10.1094/MPMI-07-17-0161-R [DOI] [PubMed] [Google Scholar]
- Chin C. S., Alexander D. H., Marks P., Klammer A. A., Drake J., Heiner C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569. 10.1038/nmeth.2474 [DOI] [PubMed] [Google Scholar]
- Cock P. J., Antao T., Chang J. T., Chapman B. A., Cox C. J., Dalke A., et al. (2009). Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423. 10.1093/bioinformatics/btp163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czajkowski R., De Boer W. J., Van der Zouwen P. S., Kastelein P., Jafra S., De Haan E. G., et al. (2013). Virulence of “Dickeya solani” and Dickeya dianthicola biovar-1 and−7 strains on potato (Solanum tuberosum). Plant Pathol. 62, 597–610. 10.1111/j.1365-3059.2012.02664.x24843434 [DOI] [Google Scholar]
- Darling A. C. E., Mau B., Blattner F. R., Perna N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. 10.1101/gr.2289704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling A. E., Mau B., Perna N. T. (2010). Progressivemauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 5:e11147. 10.1371/journal.pone.0011147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lorenzo V., Wee S., Herrero M., Neilands J. B. (1987). Operator sequences of the aerobactin operon of plasmid colV-K30 binding the ferric uptake regulation (fur) repressor. J. Bacteriol. 169, 2624–2630. 10.1128/jb.169.6.2624-2630.1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degefu Y., Potrykus M., Golanowska M., Virtanen E., Lojkowska E. (2013). A new clade of Dickeya spp. plays a major role in potato blackleg outbreaks in North Finland. Ann. Appl. Biol. 162, 231–241. 10.1111/aab.1202024843434 [DOI] [Google Scholar]
- Finn R. D., Clements J., Arndt W., Miller B. L., Wheeler T. J., Schreiber F., et al. (2015). HMMER web server: 2015 update. Nucleic Acids Res. 43, W30–W38. 10.1093/nar/gkv397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franza T., Mahé B., Expert D. (2005). Erwinia chrysanthemi requires a second iron transport route dependent of the siderophore achromobactin for extracellular growth and plant infection. Mol. Microbiol. 55, 261–275. 10.1111/j.1365-2958.2004.04383.x [DOI] [PubMed] [Google Scholar]
- Franza T., Michaud-Soret I., Piquerel P., Expert D. (2002). Coupling of iron assimilation and pectinolysis in Erwinia chrysanthemi 3937. Mol. Plant-Microbe Interact. 15, 1181–1191. 10.1094/MPMI.2002.15.11.1181 [DOI] [PubMed] [Google Scholar]
- Franza T., Sauvage C., Expert D. (1999). Iron regulation and pathogenicity in Erwinia chrysanthemi 3937: role of the Fur repressor protein. Mol. Plant-Microbe Interact. 12, 119–128. 10.1094/MPMI.1999.12.2.119 [DOI] [PubMed] [Google Scholar]
- Galardini M., Brilli M., Spini G., Rossi M., Roncaglia B., Bani A., et al. (2015). Evolution of intra-specific regulatory networks in a multipartite bacterial genome. PLoS Comput. Biol. 11:e1004478. 10.1371/journal.pcbi.1004478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galardini M., Mengoni A., Brilli M., Pini F., Fioravanti A., Lucas S., et al. (2011). Exploring the symbiotic pangenome of the nitrogen-fixing bacterium Sinorhizobium meliloti. BMC Genomics 12, 235–250. 10.1186/1471-2164-12-235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlant L., Koskinen P., Rouhiainen L., Laine P., Paulin L., Auvinen P., et al. (2013). Genome sequence of Dickeya solani, a new soft rot pathogen of potato, suggests its emergence may be related to a novel combination of non-ribosomal peptide/polyketide synthetase clusters. Diversity 5, 824–842. 10.3390/d5040824 [DOI] [Google Scholar]
- Glasner J. D., Yang C. H., Reverchon S., Hugouvieux-Cotte-Pattat N., Condemine G., Bohin J. P., et al. (2011). Genome sequence of the plant-pathogenic bacterium Dickeya dadantii 3937. J. Bacteriol. 193, 2076–2077. 10.1128/JB.01513-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golanowska M., Galardini M., Bazzicalupo M., Hugouvieux-Cotte-Pattat N., Mengoni A., Potrykus M., et al. (2015). Draft genome sequence of a highly virulent strain of the plant pathogen Dickeya solani, IFB0099. Genome Announc. 3, e00109–e00115. 10.1128/genomeA.00109-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golanowska M., Kielar J., Lojkowska E. (2017). The effect of temperature on the phenotypic features and the maceration ability of Dickeya solani strains isolated in Finland, Israel and Poland. Eur. J. Plant Pathol. 147, 803–817. 10.1007/s10658-016-1044-126811110 [DOI] [Google Scholar]
- Gottig N., Garavaglia B. S., Garofalo C. G., Orellano E. G., Ottado J. (2009). A filamentous hemagglutinin-like protein of Xanthomonas axonopodis pv. citri, the phytopathogen responsible for citrus canker, is involved in bacterial virulence. PLoS ONE 4:e4358. 10.1371/journal.pone.0004358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helias V., Hamon P., Huchet E., Wolf J. V. D., Andrivon D. (2012). Two new effective semiselective crystal violet pectate media for isolation of Pectobacterium and Dickeya. Plant Pathol. 61, 339–345. 10.1111/j.1365-3059.2011.02508.x24843434 [DOI] [Google Scholar]
- Hérault E., Reverchon S., Nasser W. (2014). Role of the LysR-type transcriptional regulator PecT and DNA supercoiling in the thermoregulation of pel genes, the major virulence factors in Dickeya dadantii. Environ. Microbiol. 16, 734–745. 10.1111/1462-2920.12198 [DOI] [PubMed] [Google Scholar]
- Heuer H., Ebers J., Weinert N., Smalla K. (2010). Variation in permissiveness for broad-host-range plasmids among genetically indistinguishable isolates of Dickeya sp. from a small field plot. FEMS Microbiol. Ecol. 73, 190–196. 10.1111/j.1574-6941.2010.00880.x [DOI] [PubMed] [Google Scholar]
- Hommais F., Oger-Desfeux C., Van Gijsegem F., Castang S., Ligori S., Expert D., et al. (2008). PecS is a global regulator of the symptomatic phase in the phytopathogenic bacterium Erwinia chrysanthemi 3937. J. Bacteriol. 190, 7508–7522. 10.1128/JB.00553-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugouvieux-Cotte-Pattat N. (2004). The RhaS activator controls the Erwinia chrysanthemi 3937 genes rhiN, rhiT and rhiE involved in rhamnogalacturonan catabolism. Mol. Microbiol. 51, 1361–1374. 10.1046/j.1365-2958.2003.03908.x [DOI] [PubMed] [Google Scholar]
- Hugouvieux-Cotte-Pattat N. (2016). Metabolism and Virulence Strategies in Dickeya–Host Interactions. Host-Microbe Interact. 142, 93–129. 10.1016/bs.pmbts.2016.05.006 [DOI] [PubMed] [Google Scholar]
- Hugouvieux-Cotte-Pattat N., Condemine G., Nasser W., Reverchon S. (1996). Regulation of pectinolysis in Erwinia chrysanthemi. Annu. Rev. Microbiol. 50, 213–257. 10.1146/annurev.micro.50.1.213 [DOI] [PubMed] [Google Scholar]
- Hugouvieux-Cotte-Pattat N., Robert-Baudouy J. (1989). Isolation of Erwinia chrysanthemi mutants altered in pectinolytic enzyme production. Mol. Microbiol. 3, 1587–1597. 10.1111/j.1365-2958.1989.tb00144.x [DOI] [PubMed] [Google Scholar]
- Ionescu M., Zaini P. A., Baccari C., Tran S., da Silva A. M., Lindow S. E. (2014). Xylella fastidiosa outer membrane vesicles modulate plant colonization by blocking attachment to surfaces. Proc. Natl. Acad. Sci. U.S.A. 111, E3910–E3918. 10.1073/pnas.1414944111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji J., Hugouvieux-Cotte-Pattat N., Robert-Baudouy J. (1987). Use of Mu-lac insertions to study the secretion of pectate lyases by Erwinia chrysanthemi. Microbiology 133, 793–802. 10.1099/00221287-133-3-793 [DOI] [Google Scholar]
- Jiang X., Sobetzko P., Nasser W., Reverchon S., Muskhelishvili G. (2015). Chromosomal “stress-response” domains govern the spatiotemporal expression of the bacterial virulence program. MBio 6, e00353-15. 10.1128/mBio.00353-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khayi S., Blin P., Chong T. M., Chan K.-G., Faure D. (2016). Complete genome anatomy of the emerging potato pathogen Dickeya solani type strain IPO 2222T. Stand. Genomic Sci. 11:87. 10.1186/s40793-016-0208-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khayi S., Blin P., Pédron J., Chong T. M., Chan K. G., Moumni M., et al. (2015). Population genomics reveals additive and replacing horizontal gene transfers in the emerging pathogen Dickeya solani. BMC Genomics 16, 788–801. 10.1186/s12864-015-1997-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khayi S., Mondy S., Beury-Cirou A., Moumni M., Hélias V., Faure D. (2014). Genome sequence of the emerging plant pathogen Dickeya solani strain RNS 08.23.3.1A. Genome Announc. 2, e01270–e01213. 10.1128/genomeA.01270-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I., Kim Y. O., Park S. C., Chun J. (2016). OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 66, 1100–1103. 10.1099/ijsem.0.000760 [DOI] [PubMed] [Google Scholar]
- Lennox E. S. (1955). Transduction of linked genetic characters of the host by bacteriophage P1. Virology 1, 190–206. [DOI] [PubMed] [Google Scholar]
- Liao C.-T., Du S.-C., Lo H. H., Hsiao Y.-M. (2014). The galU gene of Xanthomonas campestris pv. campestris is involved in bacterial attachment, cell motility, polysaccharide synthesis, virulence, and tolerance to various stresses. Arch. Microbiol. 196, 729–738. 10.1007/s00203-014-1012-0 [DOI] [PubMed] [Google Scholar]
- Mandin P., Gottesman S. (2010). Integrating anaerobic/aerobic sensing and the general stress response through the ArcZ small RNA. EMBO J. 29, 3094–3107. 10.1038/emboj.2010.179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansfield J., Genin S., Magori S., Citovsky V., Sriariyanum M., Ronald P., et al. (2012). Top 10 plant pathogenic bacteria in molecular plant pathology. Mol. Plant Pathol. 13, 614–629. 10.1111/j.1364-3703.2012.00804.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J. H. (1992). A Short Course in Bacterial Genetics: A Laboratory Manual and Handbook for Escherichia Coli and Related Bacteria. New York, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Nair G. R., Liu Z., Binns A. N. (2003). Reexamining the role of the accessory plasmid pAtC58 in the virulence of Agrobacterium tumefaciens strain C58. Plant Physiol. 133, 989–999. 10.1104/pp.103.030262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasser W., Dorel C., Wawrzyniak J., Van Gijsegem F., Groleau M. C., Déziel E., et al. (2013). Vfm a new quorum sensing system controls the virulence of Dickeya dadantii. Environ. Microbiol. 15, 865–880. 10.1111/1462-2920.12049 [DOI] [PubMed] [Google Scholar]
- Parkinson N., Pritchard L., Bryant R., Toth I., Elphinstone J. (2014). Epidemiology of Dickeya dianthicola and Dickeya solani in ornamental hosts and potato studied using variable number tandem repeat analysis. Eur. J. Plant Pathol. 141, 63–70. 10.1007/s10658-014-0523-526811110 [DOI] [Google Scholar]
- Pédron J., Chapelle E., Alunni B., Van Gijsegem F. (2018). Transcriptome analysis of the Dickeya dadantii PecS regulon during the early stages of interaction with Arabidopsis thaliana. Mol. Plant Pathol. 19, 647–663. 10.1111/mpp.12549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pédron J., Mondy S., Gijsegem F., Van Faure, D. (2014). Genomic and metabolic comparison with Dickeya dadantii 3937 reveals the emerging Dickeya solani potato pathogen to display distinctive metabolic activities and T5SS/T6SS-related toxin repertoire. BMC Genomics 15, 283–296. 10.1186/1471-2164-15-283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perombelon M. C. M. (2002). Potato diseases caused by soft rot erwinias: an overview of pathogenesis. Plant Pathol. 51, 1–12. 10.1046/j.0032-0862.2001.Shorttitle.doc.x [DOI] [Google Scholar]
- Potrykus M., Golanowska M., Hugouvieux-Cotte-Pattat N., Lojkowska E. (2014). Regulators involved in Dickeya solani virulence, genetic conservation, and functional variability. Mol. Plant-Microbe Interact. 27, 700–711. 10.1094/MPMI-09-13-0270-R [DOI] [PubMed] [Google Scholar]
- Potrykus M., Golanowska M., Sledz W., Zoledowska S., Motyka A., Kolodziejska A., et al. (2016). Biodiversity of Dickeya spp. isolated from potato plants and water sources in temperate climate. Plant Dis. 100, 408–417. 10.1094/PDIS-04-15-0439-RE [DOI] [PubMed] [Google Scholar]
- Potrykus M., Hugouvieux-Cotte-Pattat N., Lojkowska E. (2018). Interplay of classic Exp and specific Vfm quorum sensing systems on the phenotypic features of Dickeya solani strains exhibiting different virulence levels. Mol. Plant Pathol. 19, 1238–1251. 10.1111/mpp.12614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard L., Humphris S., Baeyen S., Maes M., Van Vaerenbergh, J., Elphinstone J., et al. (2013). Draft genome sequences of four Dickeya dianthicola and four Dickeya solani strains. Genome Announc. 1, e00087–e00012. 10.1128/genomeA.00087-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Py B., Barras F., Harris S., Robson N., Salmond G. P. C. (1998). Extracellular enzymes and their role in Erwinia virulence. Methods Microbiol. 27, 157–168. 10.1016/S0580-9517(08)70279-4 [DOI] [Google Scholar]
- Reverchon S., Muskhelisvili G., Nasser W. (2016). Virulence program of a bacterial plant pathogen: the Dickeya model. Prog. Mol. Biol. Transl. Sci. 142, 51–92. 10.1016/bs.pmbts.2016.05.005 [DOI] [PubMed] [Google Scholar]
- Rodionov D. A., Gelfand M. S., Hugouvieux-Cotte-Pattat N. (2004). Comparative genomics of the KdgR regulon in Erwinia chrysanthemi 3937 and other gamma-proteobacteria. Microbiology 150, 3571–3590. 10.1099/mic.0.27041-0 [DOI] [PubMed] [Google Scholar]
- Rojas C. M., Ham J. H., Deng W. L., Doyle J. J., Collmer A. (2002). HecA, a member of a class of adhesins produced by diverse pathogenic bacteria, contributes to the attachment, aggregation, epidermal cell killing, and virulence phenotypes of Erwinia chrysanthemi EC16 on Nicotiana clevelandii seedlings. Proc. Natl. Acad. Sci. U.S.A. 99, 13142–13147. 10.1073/pnas.202358699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouanet C., Reverchon S., Rodionov D. A., Nasser W. (2004). Definition of a consensus DNA-binding site for PecS, a global regulator of virulence gene expression in Erwinia chrysanthemi and identification of new members of the PecS regulon. J. Biol. Chem. 279, 30158–30167. 10.1074/jbc.M403343200 [DOI] [PubMed] [Google Scholar]
- Sauvage C., Franza T., Expert D. (1996). Analysis of the Erwinia chrysanthemi ferrichrysobactin receptor gene: resemblance to the Escherichia coli fepA-fes bidirectional promoter region and homology with hydroxamate receptors. J. Bacteriol. 178, 1227–1231. 10.1128/jb.178.4.1227-1231.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. 10.1093/bioinformatics/btu153 [DOI] [PubMed] [Google Scholar]
- Slawiak M., van Beckhoven J. R. C. M., Speksnijder A. G. C. L., Czajkowski R., Grabe G., van der Wolf J. M. (2009). Biochemical and genetical analysis reveal a new clade of biovar 3 Dickeya spp. strains isolated from potato in Europe. Eur. J. Plant Pathol. 125, 245–261. 10.1007/s10658-009-9479-226811110 [DOI] [Google Scholar]
- Tardy F., Nasser W., Robert-Baudouy J., Hugouvieux-Cotte-Pattat N. (1997). Comparative analysis of the five major Erwinia chrysanthemi pectate lyases: enzyme characteristics and potential inhibitors. J. Bacteriol. 179, 2503–2511. 10.1128/jb.179.8.2503-2511.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson N. R., Thomas J. D., Salmond G. P. C. (1999). 12 virulence determinants in the bacterial phytopathogen Erwinia. Methods Microbiol. 29, 347–426. 10.1016/S0580-9517(08)70123-5 [DOI] [Google Scholar]
- Toth I. K., van der Wolf J. M., Saddler G., Lojkowska E., Helias V., Pirhonen M., et al. (2011). Dickeya species: an emerging problem for potato production in Europe. Plant Pathol. 60, 385–399. 10.1111/j.1365-3059.2011.02427.x24843434 [DOI] [Google Scholar]
- Tsror L., Ben-Daniel B., Chalupowicz L., van der Wolf J., Lebiush S., Erlich O., et al. (2013). Characterization of Dickeya strains isolated from potato grown under hot-climate conditions. Plant Pathol. 62, 1097–1105. 10.1111/ppa.1203024843434 [DOI] [Google Scholar]
- van der Wolf J. M., Nijhuis E. H., Kowalewska M. J., Saddler G. S., Parkinson N., Elphinstone J. G., et al. (2014). Dickeya solani sp. nov., a pectinolytic plant-pathogenic bacterium isolated from potato (Solanum tuberosum). Int. J. Syst. Evol. Microbiol. 64, 768–774. 10.1099/ijs.0.052944-0 [DOI] [PubMed] [Google Scholar]
- Varani A. M., Monteiro-Vitorello C. B., Nakaya H. I., Van Sluys M. A. (2013). The role of prophage in plant-pathogenic bacteria. Annu. Rev. Phytopathol. 51, 429–451. 10.1146/annurev-phyto-081211-173010 [DOI] [PubMed] [Google Scholar]
- Weller-Stuart T., Toth I., De Maayer P., Coutinho T. (2017). Swimming and twitching motility are essential for attachment and virulence of Pantoea ananatis in onion seedlings. Mol. Plant Pathol. 18, 734–745. 10.1111/mpp.12432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood P. J. (1980). Specificity in the interaction of direct dyes with polysaccharides. Carbohydr. Res. 85, 271–287. 10.1016/S0008-6215(00)84676-5 [DOI] [Google Scholar]
- Yang S., Peng Q., Zhang Q., Yi X., Choi C. J., Reedy R. M., et al. (2008). Dynamic regulation of gacA in type III secretion, pectinase gene expression, pellicle formation, and pathogenicity of Dickeya dadantii (Erwinia chrysanthemi 3937). Mol. Plant-Microbe Interact. 21, 133–142. 10.1094/MPMI-21-1-0133 [DOI] [PubMed] [Google Scholar]
- Yoon S. H., Ha S., Lim J., Kwon S., Chun J. (2017). A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 110, 1281–1286. 10.1007/s10482-017-0844-4 [DOI] [PubMed] [Google Scholar]
- Zeng Q., Sundin G. W. (2014). Genome-wide identification of Hfq-regulated small RNAs in the fire blight pathogen Erwinia amylovora discovered small RNAs with virulence regulatory function. BMC Genomics 15, 414–433. 10.1186/1471-2164-15-414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Liang Y., Lynch K. H., Dennis J. J., Wishart D. S. (2011). PHAST: a Fast Phage Search Tool. Nucleic Acids Res. 39, W347–W352. 10.1093/nar/gkr485 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.