

Abstract
Mutations of the DYSF gene leading to reduced dysferlin protein level causes limb girdle muscular dystrophy type 2B (LGMD2B). Dysferlin facilitates sarcolemmal membrane repair in healthy myofibers, thus its deficit compromises myofiber repair and leads to chronic muscle inflammation. An experimental therapeutic approach for LGMD2B is to protect damage or improve repair of myofiber sarcolemma. Here, we compared the effects of prednisolone and vamorolone (a dissociative steroid; VBP15) on dysferlin-deficient myofiber repair. Vamorolone, but not prednisolone, stabilized dysferlin-deficient muscle cell membrane and improved repair of dysferlin-deficient mouse (B6A/J) myofibers injured by focal sarcolemmal damage, eccentric contraction-induced injury or injury due to spontaneous in vivo activity. Vamorolone decreased sarcolemmal lipid mobility, increased muscle strength, and decreased late-stage myofiber loss due to adipogenic infiltration. In contrast, the conventional glucocorticoid prednisolone failed to stabilize dysferlin deficient muscle cell membrane or improve repair of dysferlinopathic patient myoblasts and mouse myofibers. Instead, prednisolone treatment increased muscle weakness and myofiber atrophy in B6A/J mice—findings that correlate with reports of prednisolone worsening symptoms of LGMD2B patients. Our findings showing improved cellular and pre-clinical efficacy of vamorolone compared to prednisolone and better safety profile of vamorolone indicates the suitability of vamorolone for clinical trials in LGMD2B.
Keywords: muscle injury, dysferlinopathy, steroid, VBP15, glucocorticoid, inflammation, LGMD2B, membrane repair, membrane lipids
Graphical Abstract
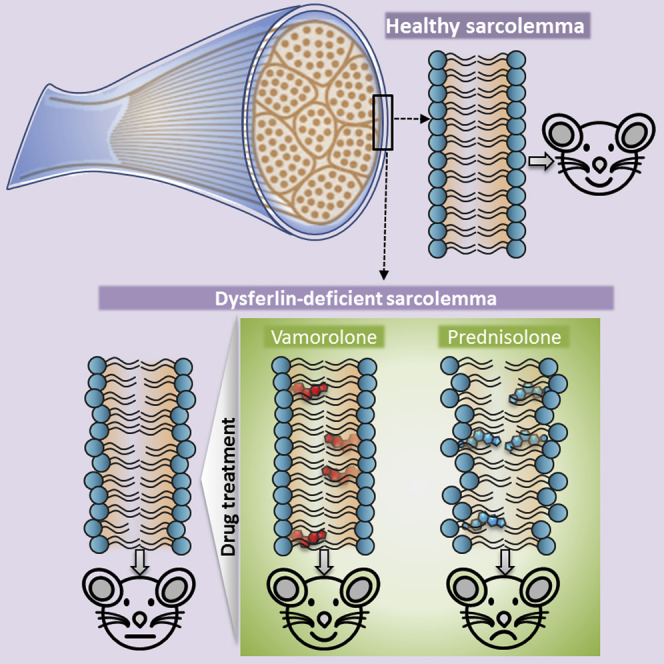
Glucocorticoids are ineffective at treating muscular dystrophy (LGMD2B) caused by mutations in dysferlin gene, which causes poor muscle cell membrane repair. Sreetama et al. show that glucocorticoids can destabilize the dysferlin-deficient muscle cell membrane and further compromise their repair. A novel dissociative steroid in clinical trial reverses this effect and demonstrates therapeutic benefits in pre-clinical studies.
Introduction
Lack of dysferlin protein causes muscular dystrophies known as dysferlinopathy, which includes limb girdle muscular dystrophy 2B (LGMD2B) and Miyoshi myopathy.1, 2 These diseases present sub-acutely during adolescence and lead to wasting and weakening of shoulder, pelvic girdle, and limb muscles. Mutations leading to the loss of dysferlin protein results in poor repair of muscle fibers, chronic muscle inflammation, and a progressive degeneration of muscle.3, 4
Currently, there is no therapy available for dysferlinopathy. Efforts to use gene therapy and drugs that target myofiber damage, calcium overload, muscle inflammation, and degeneration have resulted in mixed success thus far.5, 6, 7, 8, 9, 10 Inflammation—a hallmark of dysferlinopathy—has been targeted using glucocorticoids (GCs)—the mainstay for treating inflammatory muscle diseases. However, glucocorticoids are not effective in dysferlinopathic patients11 and also have significant safety concerns. Indeed, glucocorticoids have been reported to increase (not decrease) muscle weakness in the LGMD2B patients.10 Reducing corticosteroid drug dose and frequency in mouse models of dysferlinopathy reduces some side effects, but this approach has not yet been reported in human LGMD2B patients.12
Vamorolone (VBP15) is a dissociative steroid that aims to retain or improve the therapeutic benefit of traditional corticosteroids, while reducing side effects.13 Vamorolone has a delta 9-11 modification in the steroid backbone that retains transrepression activities of glucocorticoids (inhibition of innate immunity via activator protein 1 [AP1], and nuclear factor κB [NF-κB], while reducing or eliminating many side effects [transcriptional transactivation of genes due to binding the glucocorticoid response element [GRE]).13, 14 Vamorolone has shown efficacy similar to prednisolone, with improved safety profiles, in mouse models of Duchenne muscular dystrophy.15
Drugs with steroidal chemistries integrate into and change physicochemical properties of the cell membrane.16 Some delta 9-11 modified glucocorticoids (21-aminosteroids; lazaroids) have been shown to stabilize membranes and reduce membrane lipid peroxidation, leading to neuroprotection during traumatic brain injury.17, 18 Similarly, a recently developed delta 9-11-modified glucocorticoid, vamorolone (VBP15), has also been shown to facilitate repair of injured muscle cell membranes.15, 19
There are two drugs that are currently in clinical trials for DMD that have membrane stabilization as a mechanism of action—vamorolone (https://clinicaltrials.gov/; ClinicalTrials.gov: NCT02760264, NCT02760277, NCT03038399, and NCT03439670), and polaxamer (ClinicalTrials.gov: NCT03558958). In our study, we have focused on vamorolone due to the multiple mechanisms of action, over and above membrane stability, that may benefit LGMD2B patients, namely NF-κB inhibitory activity (anti-inflammatory), loss of many side effects seen with corticosteroid anti-inflammatories, and mineralocorticoid receptor antagonist activity15. As such, vamorolone has improved safety relative to corticosteroids and is in phase 2 clinical trials for Duchenne muscular dystrophy. We hypothesized that the potential of vamorolone to stabilize and improve repair of injured myofiber sarcolemma while inhibiting inflammation and avoiding the side effects of chronic glucocorticoid use might allow this drug to simultaneously address multiple muscle pathologies associated with dysferlin deficiency. Here, we have examined the potential of vamorolone by directly monitoring its role in stabilizing dysferlinopathic muscle cell membrane and evaluating the utility of this drug to treat LGMD2B by using a dysferlin-deficient mouse model.
Results
Vamorolone Improves Plasma Membrane Repair of LGMD2B Patient Myoblasts
Structurally, vamorolone is closely related to prednisolone—the major distinction being a delta 9-11 change leading to a loss of a hydroxyl group and double bond (Figure 1A). The efficiency of plasma membrane repair of healthy and LGMD2B patient muscle cells was tested using two complimentary injury approaches we previously described—glass-bead or laser injury.20 These assays allow monitoring the effectiveness and kinetics of plasma membrane repair. In glass-bead injury, the cells are injured in presence of fluorescein isothiocyanate (FITC) dextran (green), allowed to repair for 5 min, and then incubated with tetramethylrhodamine isothiocynate (TRITC) dextran to label unrepaired cells (red). We used this approach in conjunction with prednisolone and vamorolone, to assess the effect of acute treatment of patient cells with these drugs on their repair efficacy (Figures 1B and 1C). Injury and subsequent repair in calcium-containing media allowed over 86% of healthy human myoblasts to repair, but only 20% myoblasts managed to repair in media lacking calcium, demonstrating the known calcium dependence of this process.21 When LGMD2B patient myoblasts were allowed to repair in the presence of calcium, <60% of the cells repaired, demonstrating poor repair ability of these cells (Figure 1C). To test if vamorolone treatment can improve the ability of the LGMD2B patient cells, we treated these cells with three different doses of vamorolone (10, 50, and 100 μM) and monitored the effect of these drugs on the cell membrane repair ability. The different doses resulted in repair of 85.7% ± 2.1%, 91.2% ± 1.6%, and 92.3% ± 2.3% of cells, respectively, indicating that vamorolone improves dysferlinopathic patient myoblast repair ability in a dose-dependent manner. As 50 μM vamorolone improved patient myoblast repair similar to 100 μM vamorolone, for subsequent assays we used 50 μM vamorolone (Figures 1B and 1C). In contrast, prednisolone (50 μM) did not improve the repair of LGMD2B patient cells (60% for untreated versus 56% for prednisolone; Figures 1B and 1C). This glass-bead injury assay specifically monitors the repair ability of the injured cells (marked green), thus it reports on the plasma membrane reparative (facilitating repair) ability; it is not affected by any change in the susceptibility of the plasma membrane to injury caused by the drug treatment.
Figure 1.

Vamorolone Improves Repair of Injured LGMD2B Patient Cells
(A) Structure of the drugs used in this study; chemical differences between these drugs are marked in green. (B) Images showing injured myoblasts that repaired (green, FITC dextran) or failed to repair (red, TRITC dextran) following glass-bead injury. (C) Quantification of the number of healthy and dysferlin-deficient patient myoblasts that repaired successfully from glass-bead injury in the absence of calcium (−Ca2+) or the presence of calcium (+Ca2+). Additionally, myoblasts in media with calcium were treated for 20 min and then injured in the presence of 50 μM vamorolone (VAM), prednisolone (Pred), or the equivalent volume of the vehicle (DMSO). (D) Representative images of FM dye fluorescence prior to and 5 min following focal laser injury (site marked with arrow) of myoblasts that are treated as indicated. (E) Average intensity traces showing the kinetics of FM dye entry following laser injury of healthy or patient myoblasts treated as above with vehicle (DMSO), vamorolone, or prednisolone. (F) Plot showing the proportion of injured patient myoblasts treated with vehicle (DMSO) or with vamorolone that managed to repair following laser injury. Scale bars, 10 μm; p values are measured by unpaired Mann-Whitney t test and indicated by *p < 0.01, ***p < 0.001, ****p < 0.0001 (n > 50 uninjured cells and >30 injured cells each).
The kinetics of membrane repair were assessed in myoblasts as we have previously described, by monitoring the entry of the cell impermeant dye FM1-43 following focal laser injury.20 Compared to healthy myoblasts, dysferlin-deficient patient myoblasts showed greater dye entry that continued for longer following the injury (Figures 1D and 1E). Vamorolone and prednisolone both affected the kinetics of dye entry in patient myoblasts. In vamorolone-treated patient cells, dye entry following injury ceased significantly faster than the untreated patient cells and with the same kinetics as the healthy myoblasts, while prednisolone treatment did not improve patient cell repair kinetics (Figures 1D and 1E). Concomitant with the reduced dye entry in the injured patient cells, vamorolone increased the number of patient cells that repaired (Figure 1F).
Vamorolone Stabilizes Dysferlin-Deficient Cell Membrane by Reducing Lipid Mobility
Lipophilic molecules, such as steroidal drugs, can affect membrane rigidity as well as lateral diffusion of proteins and lipids in the plasma membranes. Lateral diffusion of membrane lipids can be directly measured by fluorescence recovery after photobleaching (FRAP).22 To monitor the plasma membrane lipid mobility, we incorporated fluorescent C12 sphingomyelin and imaged the fraction of this lipid that was freely mobile in plasma membranes by using total internal reflection microscopy (a method for high-resolution imaging of plasma membrane).23 Lipids in a small region of the plasma membrane of healthy human myoblasts were photobleached using laser irradiation (Figure 2A, white circle), and the extent to which the fluorescence in the bleached region is recovered was monitored to calculate the amount of mobile lipids (mobile fraction) in the plasma membrane (Figures 2A and 2B). Membrane injury resulted in a 9.1% increase in the mobile fraction of lipids, relative to uninjured cell membranes (Figure 2C). This injury-triggered increase in the mobile fraction of lipids in the healthy myoblasts was comparable to the 7.2% increase we observed in resting LGMD2B patient myoblasts (Figure 2C). Despite the greater mobile fraction of lipids in the plasma membrane of the patient myoblasts, injury increased this further (Figure 2C). Thus, compared to healthy myoblasts, LGMD2B patient myoblasts show increased plasma membrane lipid diffusion. Next, we tested the effect of vamorolone and prednisolone on lipid mobility—prednisolone increased the lipid mobile fraction, whereas vamorolone decreased it (Figures 2D and 2E). These changes suggest that the increased membrane lipid mobility is associated with slower and less effective repair of dysferlin-deficient muscle cell membrane, which is exacerbated by prednisolone treatment. In contrast, vamorolone treatment decreases membrane lipid mobility and improves repair of dysferlin-deficient muscle cell membrane. To establish if membrane lipid mobility is associated with membrane repair, we used a known membrane fluidizing agent methyl beta cyclodextrin (MβCD), which fluidizes membrane by extracting cholesterol-containing lipid microdomains.24 Using the same approach as above for FRAP analysis, we observed that while the vehicle (Hank’s buffered saline solution [HBSS])-treated C2C12 myoblasts show a 24.16% mobile fraction of lipids in their cell membrane, as expected this fraction increased to 36.67% in the cells pre-treated for 20 min with MβCD (Figure 2F). Concomitant with the increase in membrane lipid mobility, MβCD-treated cells were compromised in their ability to repair following laser injury—as compared to 83.82% of vehicle (HBSS)-treated cells, only 59.29% of the MβCD-treated cells repaired (Figure 2G). Together, these results show that increasing plasma membrane lipid mobility can compromise membrane repair.
Figure 2.

Vamorolone Stabilizes Injured and Dysferlin Deficient Plasma Membrane
Plasma membrane of healthy and patient myoblasts was labeled using BODIPY FL C12-sphingomyelin, and the lipid mobility was monitored by confocal FRAP microscopy. A region (white circle) was photobleached, and recovery of fluorescence in this region was monitored by real-time imaging. (A) Time-lapse images of an uninjured healthy myoblast and (B) corresponding plot showing raw fluorescence value (gray curve) and the corresponding curve fit for the fluorescence recovery (green) to determine the mobile fraction of lipids in the cell membrane. (C) Averaged plots for mobile fraction lipids in healthy and patient myoblasts prior to injury (uninj) or following injury (inj). (D) Time-lapse images of healthy LGMD2B patient myoblasts with vehicle (DMSO), VAM (50 μM), or Pred (50 μM) showing pre-bleach (−10 s), bleach (0 s), and during recovery (40 s) images. (E) Averaged plots for mobile fraction of lipids in myoblasts treated with vehicle, VAM, and Pred (treatments and drug concentration are the same as in Figure 1). (F) Averaged plots for mobile fraction of lipids in C2C12 cell membrane treated with vehicle (HBSS) or MβCD (20 mM). (G) Percentage of cells repaired when injured with glass beads in the presence of MβCD (20 mM). Scale bars, 10 μm. Data represent mean ± SEM (n > 16 cells each for C, >26 cells each for E, >20 cells each for F, and >170 cells for G), and p values are measured by unpaired Mann-Whitney t test are indicated by *p < 0.01, **p < 0.001, ***p < 0.0001, or ****p < 0.00001.
Acute Treatment with Vamorolone Rescues Dysferlin-Deficient Myofiber Repair Deficit
To assess if vamorolone can also improve repair of mature myofiber sarcolemma, we used muscles from WT (C57Bl6) and dysferlin-deficient (B6A/J) mice (Figure 3A). Bicep muscle from B6A/J mice was pre-treated ex vivo for 30 min with vamorolone (50 μM) and then focally injured by laser in the presence FM dye. Compared to vehicle (DMSO) treatment, vamorolone treatment significantly reduced the rate and the extent of FM dye entry in the injured muscle fibers (Figures 3B and 3C). Consequently, vamorolone treatment caused a two-fold increase in the number of repaired dysferlinopathic myofibers (Figure 3D; 93.8% versus 47.6%). To determine the potential benefit of vamorolone treatment of dysferlin-deficient mice in vivo, we treated the B6A/J mice with twice daily oral doses of 30 mg/kg vamorolone for 2 days, and then on the third day muscles were collected 2 hr after the last dose. Focal injury of sarcolemma of myofibers in the intact biceps muscle of vamorolone-treated mice resulted in reduced FM dye entry compared to the vehicle (cherry syrup)-treated mice (Figures 3E and 3F). This reduced dye entry in vamorolone-treated myofibers corroborated with a nearly two-fold increase in the number of myofibers that repaired from laser injury (Figure 3G).
Figure 3.

Ex Vivo and In Vivo Treatment with Vamorolone Improves Repair of Dysferlin-Deficient Myofibers
(A) Western blot analysis of dysferlin in WT (C57BL/6) and dysferlinopathic (B6A/J) mouse muscles. (B–D) Intact bicep muscles isolated from B6A/J mice were treated ex vivo with vehicle (DMSO) or 50 μM vamorolone and then focally injured by laser in presence of the drug or vehicle. (B) Representative images of muscle fibers (marked by dotted lines) before and 3 min after injury the site marked with the arrow. Plots showing (C) averaged kinetics of FM dye entry following injury and (D) proportion of myofibers that repaired from laser injury of muscles treated with DMSO (n = 15 fibers) or with vamorolone (n = 22 fibers). (E–G) Intact EDL muscle from WT and B6A/J mice treated orally for 3 days with vehicle (cherry syrup) or 30 mg/kg of vamorolone were isolated, and myofibers were focally injured by laser. (E) Images showing FM dye entry in myofibers focally injured at sites indicated by the arrow. Plot showing (F) averaged kinetics of FM dye entry and (G) proportion of myofibers that repaired following laser injury of myofibers from vehicle (n = 24 fibers) or vamorolone (n = 30 fibers) treated mice. (H) Plot showing EDL muscle force loss following lengthening contraction injury (n = 6 muscles). (I) Images of procion orange (PO)-labeled myofibers in an intact muscle section (boundary marked by white dotted line) following lengthening contraction injury. Scale bar, 10 μm for (B) and (E) and 200 μm for (I). p values measured by unpaired Mann-Whitney t test and indicated by *p < 0.01, ***p < 0.001, or ****p < 0.0001.
To test the effect of in vivo vamorolone treatment on repair of muscle injured due to eccentric contractions, we performed eccentric contraction-induced injury of extensor digitorum longus (EDL) muscles and monitored the resulting force loss. Muscle from vamorolone-treated mice trended toward 10% reduction in force drop, but this drop was not statistically significant (Figure 3H). Concomitantly, vamorolone-treated mouse EDL muscles did not show a change in the extent of their sarcolemmal integrity indicated by the entry of cell impermeant dye procion orange (PO) (Figure 3I).
Chronic Vamorolone Treatment of Mice Prevents Dysferlinopathic Muscle Pathology
We next analyzed the effects of 3 months of daily oral treatment of B6A/J mice with vamorolone on disease progression starting at 7 months of age. Up to eight mice per group were given daily oral doses of vamorolone (30 mg/kg), prednisolone (30 mg/kg), or equivalent volume of the vehicle (cherry syrup). At the end of the 3 months of treatment, mice were assessed for myofiber repair, limb grip strength, and muscle histopathology.
Myofibers in the bicep muscle of the vamorolone-treated mice showed reduced FM dye entry and formed significantly smaller repair “lip” at the site of injury (Figure 4A, arrows) compared to the vehicle or prednisolone-treated group. In vamorolone-treated mouse myofibers, FM dye entry stopped earlier than in the myofibers from vehicle-treated mice, but dye entry continued for significantly longer in myofibers from prednisolone-treated mice (Figure 4B). Consequently, compared to the vamorolone-treated muscles, fewer myofibers in the untreated and prednisolone-treated muscles showed repair following laser injury (Figure 4C). We next measured the effect of drug treatment on EDL muscle and observed that while the EDL muscles did not show a difference in their basal contractile ability (Figure 4D), following eccentric contraction-induced injury muscles from the vamorolone-treated mice showed slower force drop as compared to the vehicle or prednisolone treated mice (Figure 4E). Use of the membrane impermeable procion orange dye showed that slower force drop in vamorolone-treated muscles correlated with the reduced dye entry following eccentric injury of these muscles (Figures 4F and 4G). In vivo grip strength measurement showed that mice treated with vamorolone had greater hindlimb grip strength than the vehicle-treated mice, while prednisolone treatment reduced both the forelimb and hindlimb grip strength (Figures 4H and 4I). In view of the ability of vamorolone to inhibit NF-κB-dependent gene expression, we analyzed expression of two NF-κB-regulated genes CD209 (cluster of differentiation or dendritic cell-specific intercellular-adhesion molecule grabbing non-integrin)25 and IRF7 (interferon regulatory factor).26 Using qPCR analysis, we found that, as expected, the level of expression of both these genes in the gastrocnemius muscles of vamorolone-treated mice was lower than their expression in the vehicle-treated B6A/J mice (Figures 4J and 4K).
Figure 4.

Vamorolone, but Not Prednisolone, Improves Dysferlinopathic Mouse Muscle Function and Myofiber Repair
Dysferlinopathic mice (B6A/J) were treated for 3 months with daily dose of vehicle (cherry syrup) or 30 mg/kg of vamorolone or prednisolone. (A) Images of myofibers in intact bicep muscle before and after focal laser injury at the site marked by the arrow. (B) Plot showing averaged kinetics of FM dye entry following laser injury of myofibers and (C) quantification of myofibers that managed to repair following laser injury of muscles treated with vehicle (n = 30 fibers), VAM (n = 30 fibers), or Pred for (n = 30 fibers), and WT (n = 26 fibers). (D) Plot showing specific contractile force of EDL and (E) change in the contractile force (as a percentage of initial force) following sequential lengthening-contraction injury of EDL muscle (n = 18–23 muscles). (F) Representative images and (G) quantification of procion orange labeling of EDL after lengthening contraction injury (n = 6 muscles). Plots for average (H) hindlimb and (I) forelimb grip strength of the drug- or vehicle-treated animals (n = 8–10 animals). (J and K) Plots showing expression of inflammatory genes CD209 (J) and IRF7 (K) in the gastrocnemius muscle (n = 5 muscles). Scale bar, 10 μm for (A) and 200 μm for (F). p value measured by unpaired Mann-Whitney t test and *p < 0.01 or ****p < 0.0001.
Above results suggested that myofiber loss due to spontaneous in vivo injury may contribute to muscle force loss in B6A/J mice, which is reversed by vamorolone but not by prednisolone treatment. We thus scored spontaneous in vivo myofiber injury by monitoring the presence of serum immunoglobulin M (IgM) in the myofiber.27 We observed a 2-fold decrease in IgM-labeled myofibers in vamorolone-treated mice, while the prednisolone-treated mice showed two-fold increase in IgM-labeled myofibers (Figures 5A and 5B). This indicates that vamorolone treatment improves myofiber repair following spontaneous myofiber injury in vivo. While we find that vamorolone improves plasma membrane repair by stabilizing the membrane, intraperitoneal glucocorticoid injections have been suggested to regulate myofiber repair by regulating mRNA level of annexins A1 and A6.12 To examine if vamorolone may improve myofiber repair by regulating annexin level, we quantified the levels of Annexins A1 and A6. Additionally, with the known requirement of Annexin A2 to facilitate myofiber repair,28 we also examined the level of Annexin A2. Daily oral treatment for 3 months with vamorolone and prednisolone did not alter the level of Annexin A2 or A6 (Figures 5C–5E). Similarly, Annexin A1 protein level was unaffected by vamorolone treatment; however, it increased in prednisolone-treated mice (Figures 5F and 5G). Thus, opposing effects of vamorolone and prednisolone on sarcolemmal repair cannot be explained by their effect on the expression of annexin A1, A2, and A6 proteins. Instead, it correlates with the opposing effects of these drugs on plasma membrane stabilization.
Figure 5.

Vamorolone Reduces In Vivo Sarcolemmal Damage and Reduces Adipogenic Loss of Dysferlinopathic Muscle
(A) Gastrocnemius muscle cryosections from B6A/J animals treated for 3 months with vehicle (cherry syrup), or 30 mg/kg vamorolone or prednisolone were immunostained for immunoglobulin M (IgM; red) and wheat germ agglutinin (WGA; green) to identify myofibers with sarcolemmal injury (marked by IgM entry). (B) Quantification of IgM-labeled myofibers in gastrocnemius muscles from B6A/J mice treated as indicated (n = 6 muscles). (C–G) Western blot analysis for the expression of Annexin proteins in the gastrocnemius muscle of B6A/J mice treated as indicated. (C and F) Images and (D, E, and G) quantification of Annexin protein level normalized to the loading control (β-actin) (n = 3 muscles each). (H) Image of gastrocnemius muscle cross-section stained with H&E (I) quantification of the minimum Feret’s diameter in gastrocnemius myofiber (n > 200 myofibers from three muscles each). (J) Images of gastrocnemius muscle sections labeled for Perilipin-1 (red) and nuclei (DAPI, blue). (K) Quantification of perilipin-positive foci (n = 6 muscles each). All scale bars represent 50 μm, and all p values represent unpaired Mann-Whitney t test, *p < 0.01, **p < 0.005.
With the opposing effects of vamorolone and prednisolone on muscle grip strength, we examined the muscle histological changes due to these drug treatments. Prednisolone-treated mouse muscle showed increased myofiber loss and smaller fiber size (Figures 5H and 5I). Dysferlin deficit causes adipogenic replacement of myofibers,28, 29 and glucocorticoids enhance the adipogenic muscle replacement.12, 30 Thus, we examined if the increased myofiber loss due to prednisolone treatment was caused by greater adipogenic replacement of the myofibers. Using the lipid binding protein Perilipin-1 to quantify muscle adipogenic replacement,28 we found that compared to the vehicle-treated mice, prednisolone trended toward increased adipogenic replacement of myofibers, while vamorolone treatment decreased this (Figures 5J and 5K).
Discussion
Therapies targeting the basic cellular deficit, while avoiding the detrimental side effects, are required for treating LGMD2B patients, who presently lack any therapeutic avenue. As in other muscular dystrophies, LGMD2B patients have chronic muscle inflammation.31, 32 However, unlike other muscular dystrophies, glucocorticoid treatment results in little benefit and notable detriment to the LGMD2B patients.10 Using a conventional GC (prednisolone) we showed that their insertion into the patient muscle cell membrane destabilizes the membrane to an extent greater than the untreated patient cells (Figure 2E).This destabilization concomitantly worsens the repair ability of the dysferlin deficient myofibers (Figure 4B).
In healthy muscle cells, injury was associated with transient increases in lipid mobility (Figures 2C and 6). Comparison of dysferlin-deficient and healthy cells showed that dysferlin deficit increased lipid mobility even in uninjured cells, and prednisolone increased this mobility even further to levels greater than the injured healthy cells (Figures 2C–2E and 6). This increase in cell membrane lipid mobility by prednisolone likely contributes to further diminishing the already poor repair ability of the dysferlinopathic patient plasma membrane (Figure 1). Despite the clear importance of inflammation in the LGMD2B disease process and value of prednisolone as an anti-inflammatory drug, the above effect of prednisolone may contribute to the detrimental effect of glucocorticoids noted in case of the LGMD2B patients.10
Figure 6.

Mechanism for Improved Repair of LGMD2B Muscle Cell by Vamorolone
Healthy muscle cells show low lipid mobility at rest and injury causes the lipid mobility to increase. This increase is reversed by the endogenous cell membrane repair machinery, resulting in efficient repair of the injured healthy cells. Lack of dysferlin is associated with increased lipid mobility in the LGMD2B cell membrane, which is increased further by injury and prednisolone treatment, causing failure of these cells to undergo repair. In contrast, vamorolone treatment stabilizes the LGMD2B muscle cell membrane to near healthy cell level, enabling repair of the injured cells.
In contrast to prednisolone, we find that the delta 9-11 steroid vamorolone reversed the increased membrane lipid mobility in the resting dysferlinopathic cells (Figure 2). This finding is in agreement with the previous work with delta 9-11 modified (lazaroid) compounds that showed inhibition of the spread of membrane lipid peroxidation and are neuroprotective during injury.17, 18, 33 In muscle cells, membrane stabilization by lazaroids also inhibits calcium influx34 and stabilizes membrane proteins to attenuate cardiac ischemia-reperfusion injury.35 Our recently determined crystal structure of vamorolone demonstrates the organization of the lipophilic double bond of the delta 9-11 C-ring in vamorolone, which is in contrast to the hydrophilic hydroxyl or ketone group in the glucocorticoids.36 This seemingly minor structural difference appears to contribute to the differential physicochemical effect on the property of the cell membrane. Additional studies with reconstituted lipid bilayers should be undertaken to determine if vamorolone may also have additional cellular affects that can contribute to its ability to stabilize the dysferlinopathic membrane.
We and others have shown that dysferlin deficiency leads to activation of the adipogenic replacement of muscle.28, 29 But how vamorolone treatment reduces the adipogenic loss of muscle while prednisolone increases is not fully elucidated. One possibility is that the increased failure of dysferlinopathic myofibers to repair in prednisolone-treated mice causes increased myofiber degeneration and hence greater adipogenic replacement compared to vamorolone-treated mice, where better repair improves myofiber survival. Another possibility is that as glucocorticoids (e.g., dexamethasone) are known to facilitate adipogenic differentiation of pre-adipocytes37 and unlike conventional glucocorticoids, such as dexamethasone and prednisolone, vamorolone does not facilitate adipogenic conversion of the pre-adipocytes in vivo. Indeed, it has recently been shown that glucocorticoids work via GR to augment adipose tissue development in vivo.38 The differential GR binding and activation by vamorolone versus prednisolone could thus be responsible for the differential adipogenesis induced by these two drugs. If so, use of vamorolone may provide insights into specific gene regulatory pathways responsible for GR-mediated adipogenesis in vivo.
We have previously implicated plasma membrane lipid changes in membrane repair deficit observed in dysferlinopathic cells, such that lack of dysferlin reduces injury-triggered secretion of the lipid-modifying enzyme acid sphingomyelinase, and reversing this deficit improves membrane repair.39 We have also shown that dystrophin-deficient myofibers repair poorly. However, it is the mitochondrial dysfunction that causes poor repair of the dystrophin-deficient myofibers, while, in these myofibers, dysferlin and associated repair proteins are upregulated.40 It is thus plausible that the detrimental effect of prednisolone for dysferlinopathic muscle may be due to physiochemical instability of sarcolemma caused specifically by the lack of dysferlin. This possibility is supported by the protective effect of prednisolone on injury of healthy muscle.41 Low intraperitoneal glucocorticoid dosing appears to upregulate Annexin A6 and A1 transcript level and improve repair of isolated dysferlinopathic mouse myofibers.12 While we do not observe any effect of prednisolone treatment on Annexin A6 level, it does indeed increase Annexin A1 protein levels in the LGMD2B mice (Figure 5). However, increased Annexin A1 protein level in the prednisolone-treated B6A/J mice correlates with decreased myofiber membrane repair, suggesting Annexin A1 on its own is not sufficient to improve myofiber repair. This possibility is supported by our previous finding that knockout of Annexin A1 in mouse has no impact on the myofiber repair ability.42 In contrast, vamorolone-dependent improvement in myofiber repair occurs despite no change in the level of Annexin A1 or A6 protein levels, supporting a physicochemical mode of action of vamorolone for improving repair.
While lowering the dose and frequency of glucocorticoids is suggested to lower the side effects of glucocorticoids for LGMD2B,12 we find that vamorolone circumvents these detrimental effects even upon daily oral dosing at clinically relevant concentrations. As such, we have shown that vamorolone treatment in mdx mice treats muscular dystrophy without inhibiting growth or causing hormonal and immuno-suppressive side effects.15 While effective even at low doses, in human subjects, vamorolone is well tolerated even at high doses—20 mg/kg (mouse equivalent dose of 240 mg/kg).36 In our previous efforts to perform dose ranging study for the preclinical use of vamorolone in mice, we have shown that a daily dose of 30 mg/kg vamorolone (human equivalent dose of 2.4 mg/kg) is effective without GC-induced side effects.15 Thus, for our mouse study, we used vamorolone and prednisolone at this dose, which is also comparable to the 1-mg/kg dose of the glucocorticoid deflazacort tested in an independent human study analyzing the effect of glucocorticoids on LGMD2B patients.10 Further, pharmacokinetics (PK) and absorption, distribution, metabolism, and excretion (ADME) studies with vamorolone (VBP15) and its structurally related analogs in multiple pre-clinical animal models, including mice, has shown high (74.5%) bioavailability of vamorolone, a low-medium clearance 18.8 mL/min/kg, and terminal half-life of 0.35 hr.13
Beneficial effect of membrane stabilization for LGMD2B has been proposed through the use of agents such as recombinant Mitsugumin 53,43 and poloxamer;7 however, the nature of dysferlin-deficient myofiber cell membrane stabilization by these agents remains unexplored. With increasing recognition of the role of the cell membrane integrity and repair being recognized as critical for various tissues including muscle,44 heart,45 central nervous system,46, 47 and lungs,48 our findings of the mechanism and physicochemical ability of vamorolone as a membrane stabilizer identifies that chemical modification of glucocorticoids as a fruitful approach to develop glucocorticoid-based therapies to treat these diseases while minimizing glucocorticoid-use-associated side effects.
Materials and Methods
Cell Culture
The human cells used in this study were described in our previous study.39 These LGMD2B myoblasts were isolated from quadricep muscle of a dysferlinopathic 17-year-old male with a homozygous mutation in exon 44 that resulted in a c.4882G > A mutation, which led to loss of any detectable dysferlin protein by western blot analysis.39 Control myoblasts were isolated from the pectoralis major muscle of a 41-year-old male, and the cells were cultured in human myoblast culture media kit (Promocell) in standard humidified CO2 incubator at 37°C and 5% CO2. All experiments comparing the effect of prednisolone and vamorolone on patient myoblast plasma membrane stability and repair were done with the same dysferlinopathic patient cells and healthy (control) donor cells.
Cell Membrane Injury Assays
Laser Injury
Immortalized healthy and dysferlinopathic patient myoblasts cultured on coverslips were transferred to and incubated in cell imaging media (CIM)/PBS buffer with 1 mg/mL FM1-43 dye (Life Technologies) and placed in a Tokai Hit microscopy stage-top ZILCS incubator (Tokai Hit, Fujinomiya-shi, Japan) maintained at 37°C. For laser injury, a 1- to 5-μm2 area was irradiated for 10 ms with a pulsed laser (Ablate!, 3i Intelligent Imaging Innovations, Denver, CO, USA). Cells were imaged at 2-s intervals using IX81 Olympus microscope (Olympus America, Center Valley, PA, USA) as described.20 FM dye intensity (ΔF/F0, where F0 is the original value) was used to quantify the kinetics of cell membrane repair and represented with intervals of five frames. To quantify the dye intensity in the injured fiber, the area of dye entry was manually marked and the change in dye intensity (ΔF/F0) was quantified. In the laser injury assays, myoblasts or myofibers were categorized as having “repaired” if their final fluorescence intensity remained below the set intensity threshold—ΔF/F value < 1.2. Cells or myofibers that moved or hypercontracted during the course of imaging due their failure to repair could not be used for intensity measurement but were included in the quantification of percent repaired cells/fiber.
Glass-Bead Injury
Immortalized healthy and dysferlinopathic patient myoblasts cultured on coverslips were transferred to CIM or PBS (Sigma-Aldrich) containing 2 mg/mL of lysine-fixable FITC-dextran (Life Technologies). Cells were injured by rolling glass beads (Sigma-Aldrich) over the cells. They were allowed to heal at 37°C for 5 min and then incubated at 37°C for 5 min in CIM or PBS buffer containing 2 mg/mL of lysine-fixable TRITC dextran (Life Technologies). Cells were fixed in 4% paraformaldehyde (PFA) after thorough washing and nuclei were counterstained with Hoechst 33342; the cells were then mounted in fluorescence mounting medium (Dako) and imaged on IX81 Olympus microscope equipped with Lambda DG-4 (Sutter Instruments, Novato CA) widefield illumination system and Evolve 512 EMCCD (Photometrics, Tucson, AZ) camera using Slidebook 5.0 software (Intelligent Imaging Innovations, Denver, CO) in widefield mode using 40X/1.30NA oil objective (Olympus America, PA), as described in Defour et al.20 The number of FITC-positive cells (injured and repaired) and both FITC- and TRITC-positive cells (injured and not repaired) were counted. The number of injured cells that failed to be repaired was expressed as a percentage of the total injured cells.
Ex Vivo Cell Membrane Injury
Biceps were surgically isolated from euthanized wild-type (WT) (C57/Bl6) mice in Tyrode’s solution, and laser injury was carried out using the microscope and laser injury settings as described28 in the Tyrode’s buffer containing 1.33 mg/mL FM1-43 dye. The kinetics of repair was determined by measuring the cellular FM1-43 fluorescence. FM1-43-dye intensity (ΔF/F0, where F0 is the original value at time 0) was used to quantify the kinetics of cell membrane repair.
FRAP
Immortalized healthy and dysferlinopathy patient myoblasts were plated onto coverslips 24–48 hr prior to imaging at roughly 30,000–40,000 cells per coverslip, allowing for upward of 50% confluency by the time of imaging. For imaging, coverslips were placed in slide holders and washed in Dulbecco’s PBS (DPBS). Control cells were incubated for 30 min in DMEM with vehicle with 2 μM (final concentration) BODIPY-C12 sphingomyelin dye added, while in the drug-treated groups, vamorolone (VAM) (50 μM), prednisolone (50 μM), or MβCD (20 mM) were added in the incubation media 10 min after the dye incubation and continued for another 20 min. At the end of 30 min, the cells were rinsed and imaged in CIM. A 488-nm argon laser attached to Olympus confocal microscope was used for live cell imaging. Cells were photobleached, and fluorescence recovery was monitored. Results were analyzed using Slidebook 6.0. The data for each condition were recorded, and mobile fractions were further analyzed in excel.
Immunoblot and Immunostaining
Cells grown to 70%–80% confluence or cryopreserved muscle tissues lysed with radioimmunoprecipitation assay (RIPA) buffer (Sigma Aldrich, USA) containing protease inhibitor cocktail (Fisher Scientific, USA). Proteins resolved on a 4%–12% gradient gel (Life Technologies, USA) were transferred to nitrocellulose membrane and probed using the following antibodies: Anx1A (1:1000; Proteintech), Anx 2 (1:500; BD Biosciences), Anx6 (1:500; Abcam), Dys (1:500; NCL-Hamlet; Novacastra), and β-actin (1:2,000; Santa Cruz, USA) overnight at 4°C. This was followed by binding for an hour with horseradish peroxidase (HRP)-conjugated appropriate secondary antibody (Sigma Aldrich, MO, US).
Animal Care and Administration of Drugs
All experiments involving the use of mice were approved by the Institutional Animal Use and Care Committee (IAUCAC). B6A/J animals received VAM and prednisolone (Pred) suspended in cherry syrup (30 mg/kg) orally for short (acute; five feedings over 3 days) or long (chronic; once daily for 3 months) duration.
Grip-Strength Measurement
All experiments involving the use of mice were approved by the CRI animal care and use committee. Forelimb and hindlimb grip-strength measurement (GSM) were measured using a grip strength meter (Columbus Instruments, Columbus, OH, USA) as previously described.49 The animals were acclimatized for 3 days before actual data collection. The forelimb and hindlimb grip-strength data were then collected over 5 consecutive days. Data were represented as averaged grip strength/kg body weight over 5 days.
Contraction-Induced Myofiber and Sarcolemmal Injury
After anesthetizing the treated B6A/J mice (with different treatments) with a mixture of ketamine and xylazine, EDL muscles from the right hindlimb of the mice were carefully exposed and 6-0 silk sutures and firmly attached to proximal and distal tendons. The EDL was dissected and placed in a bath containing buffered mammalian Ringer’s solution (137 mM NaCl, 24 mM NaHCO3, 11 mM glucose, 5 mM KCl, 2 mM CaCl2, 1 mM MgSO4, 1 mM NaH2PO4, and 0.025 mM tubocurarine chloride) at 25°C and bubbled with 95% O2–5% CO2 to stabilize the pH at 7.4. In the bath, the distal tendon was securely connected to a fixed bottom plate, and the proximal tendon was attached to the arm of a servomotor (800A in vitro muscle apparatus, Aurora Scientific). The vertically aligned EDL muscle was flanked by two stainless steel plate electrodes. Using single 0.2-mm square simulation pulses, the muscle was adjusted to the optimal muscle length for force generation. At optimal length, with isometric tetanic contractions 300 ms in duration at frequencies up to 250 Hz separated by 2 min of rest intervals, the maximal force was determined. Specific force, defined as maximal force normalized for the muscle cross sectional area, was calculated as the maximal force/(muscle mass * (density of muscle tissue * fiber length)−1). The fiber length was based on the fiber to muscle length ratio of 0.45. The muscle tissue density is 1.056 kg/L. The data were represented as mean ± SD. To study contraction-induced sarcolemmal injury, EDL muscles were subjected to nine lengthening contractions with 10% strain at a velocity of two fiber lengths per second. Each contraction was separated by a 1-min rest interval. Lengthening contraction (LC)-induced force deficits were expressed as percentage of first contraction. The values were represented as mean ± SD.
Procion Orange and Muscle Fiber Membrane Damage
Plasma membrane (PM) injury was assessed using microscopic analysis of muscle cross sections that reveal the presence of procion orange dye, known to be excluded from cells with intact membranes, within the sarcoplasm of muscle fibers. Immediately after the LC assay, muscles were trimmed of tendons, blotted, weighed, and then incubated while held at Lo in a 0.2% procion orange solution at room temperature for 30 min, washed in Ringer solution, and quickly frozen in isopentane. Frozen cross-sections of 10 μm thickness were cut, mounted with fluorescent mounting medium (Dako), and imaged by wide-field fluorescence microscope for procion orange. Fields, containing the majority of the muscle cross-sections, were photographed under identical conditions of brightness and contrast. Unlabeled fibers were masked by thresholding the image using an unstained muscle section. From each muscle cross-section, the area that contained procion orange-labeled fibers ranging from minimal to maximal dye uptake was measured and expressed as procion orange intensity/unit area percentage of the complete muscle-section area. Procion orange-labeled fibers at the edge of the sections were considered as artifact and so were excluded from analysis.
Histology and Immunohistochemistry
Skeletal muscles were dissected out and frozen in isopentane cooled in liquid nitrogen. Transverse cryosections (7-μm thickness) were prepared from frozen muscles and were processed for H&E. Digital images were captured with a VS120 virtual slide microscope, and images were processed and quantified using CellSens software. The antibodies used for immuno-staining were Perilipin (1:500; Sigma) or IgM (1:100; Invitrogen). After thawing, sections were rehydrated in PBS at room temperature, blocked in Tween 20 (0.05%), normal goat serum (NGS) (10%), and BSA (2%) in PBS for a half-hour, and incubated in primary antibody for overnight at 4°C. Primary antibody incubation was followed by labeling with Alexa 488 or 594 secondary antibodies (Invitrogen, 1/100). Sections were mounted in Prolong diamond (Invitrogen, USA) and examined through VS120 virtual slide scanner (Olympus), and the images were processed by CellSens (Olympus) software. For quantification of in vivo injured myofibers, the total number of wheat germ agglutinin (WGA)-labeled fibers from randomly chosen areas of entire gastrocnemius cross-sections was scored for fibers that were positive for IgM. These were then presented as the number if IgM-positive fibers across 1 mm2 cross-sectional area of the muscle. For the quantification of adipogenic deposits, perilipin-positive structures in the gastrocnemius muscle cross-section were counted and presented as the number of structures across a 1 mm2 cross-sectional area of the muscle.
Statistical Analysis
The statistical analysis was carried out using the GraphPad Prism Software, where the data were tested by nonparametric Mann-Whitney test. For membrane repair analysis, the data are presented as averaged values for all of the cells used for that analysis, and individual time points were compared across all cells in a given treatment to determine significance by unpaired t test.
Author Contributions
The study was conceptualized and designed by J.K.J. and K.N. with inputs from E.P.H. S.C.S. performed the study with help from G.C. for in vivo studies, from J.H.V.M. for ex vivo muscle physiology, with P.S. for MβCD treatment studies and M.M.A. and S.B. for the acquisition and analysis of histology data. J.K.J. obtained funding, supervised the study, and helped with data acquisition and analysis. S.C.S., J.K.J., and E.P.H. wrote the manuscript with inputs from all the authors.
Conflicts of Interest
K.N. and E.P.H. are co-founders, shareholders, and part-time employees of ReveraGen BioPharma, owner of the intellectual property and sponsor for vamorolone (VBP15).
Acknowledgments
This work is supported by a research grant award by the Clark Charitable Foundation, MDA (MDA277389) and NIAMS (R01AR055686) to J.K.J. K.N. acknowledges financial support by the NIH (K26OD011171 and R24HD050846). Microscopy was assisted by the CRI Cellular Imaging Core, which is supported by the CRI and District of Columbia Intellectual and Developmental Disabilities Research Center Award (1U54HD090257) from the NIH.
References
- 1.Bashir R., Britton S., Strachan T., Keers S., Vafiadaki E., Lako M., Richard I., Marchand S., Bourg N., Argov Z. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat. Genet. 1998;20:37–42. doi: 10.1038/1689. [DOI] [PubMed] [Google Scholar]
- 2.Liu J., Aoki M., Illa I., Wu C., Fardeau M., Angelini C., Serrano C., Urtizberea J.A., Hentati F., Hamida M.B. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat. Genet. 1998;20:31–36. doi: 10.1038/1682. [DOI] [PubMed] [Google Scholar]
- 3.Fanin M., Angelini C. Muscle pathology in dysferlin deficiency. Neuropathol. Appl. Neurobiol. 2002;28:461–470. doi: 10.1046/j.1365-2990.2002.00417.x. [DOI] [PubMed] [Google Scholar]
- 4.Cenacchi G., Fanin M., De Giorgi L.B., Angelini C. Ultrastructural changes in dysferlinopathy support defective membrane repair mechanism. J. Clin. Pathol. 2005;58:190–195. doi: 10.1136/jcp.2004.018978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krahn M., Wein N., Bartoli M., Lostal W., Courrier S., Bourg-Alibert N., Nguyen K., Vial C., Streichenberger N., Labelle V. A naturally occurring human minidysferlin protein repairs sarcolemmal lesions in a mouse model of dysferlinopathy. Sci. Transl. Med. 2010;2:50ra69. doi: 10.1126/scitranslmed.3000951. [DOI] [PubMed] [Google Scholar]
- 6.Kerr J.P., Ziman A.P., Mueller A.L., Muriel J.M., Kleinhans-Welte E., Gumerson J.D., Vogel S.S., Ward C.W., Roche J.A., Bloch R.J. Dysferlin stabilizes stress-induced Ca2+ signaling in the transverse tubule membrane. Proc. Natl. Acad. Sci. USA. 2013;110:20831–20836. doi: 10.1073/pnas.1307960110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suzuki N., Akiyama T., Takahashi T., Komuro H., Warita H., Tateyama M., Itoyama Y., Aoki M. Continuous administration of poloxamer 188 reduces overload-induced muscular atrophy in dysferlin-deficient SJL mice. Neurosci. Res. 2012;72:181–186. doi: 10.1016/j.neures.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 8.Weisleder N., Takizawa N., Lin P., Wang X., Cao C., Zhang Y., Tan T., Ferrante C., Zhu H., Chen P.J. Recombinant MG53 protein modulates therapeutic cell membrane repair in treatment of muscular dystrophy. Sci. Transl. Med. 2012;4:139ra85. doi: 10.1126/scitranslmed.3003921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lerario A., Cogiamanian F., Marchesi C., Belicchi M., Bresolin N., Porretti L., Torrente Y. Effects of rituximab in two patients with dysferlin-deficient muscular dystrophy. BMC Musculoskelet. Disord. 2010;11:157. doi: 10.1186/1471-2474-11-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walter M.C., Reilich P., Thiele S., Schessl J., Schreiber H., Reiners K., Kress W., Müller-Reible C., Vorgerd M., Urban P. Treatment of dysferlinopathy with deflazacort: a double-blind, placebo-controlled clinical trial. Orphanet J. Rare Dis. 2013;8:26. doi: 10.1186/1750-1172-8-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoffman E.P., Rao D., Pachman L.M. Clarifying the boundaries between the inflammatory and dystrophic myopathies: insights from molecular diagnostics and microarrays. Rheum. Dis. Clin. North Am. 2002;28:743–757. doi: 10.1016/s0889-857x(02)00031-5. [DOI] [PubMed] [Google Scholar]
- 12.Quattrocelli M., Salamone I.M., Page P.G., Warner J.L., Demonbreun A.R., McNally E.M. Intermittent Glucocorticoid Dosing Improves Muscle Repair and Function in Mice with Limb-Girdle Muscular Dystrophy. Am. J. Pathol. 2017;187:2520–2535. doi: 10.1016/j.ajpath.2017.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reeves E.K.M., Hoffman E.P., Nagaraju K., Damsker J.M., McCall J.M. VBP15: preclinical characterization of a novel anti-inflammatory delta 9,11 steroid. Bioorg. Med. Chem. 2013;21:2241–2249. doi: 10.1016/j.bmc.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Newton R., Holden N.S. Separating transrepression and transactivation: a distressing divorce for the glucocorticoid receptor? Mol. Pharmacol. 2007;72:799–809. doi: 10.1124/mol.107.038794. [DOI] [PubMed] [Google Scholar]
- 15.Heier C.R., Damsker J.M., Yu Q., Dillingham B.C., Huynh T., Van der Meulen J.H., Sali A., Miller B.K., Phadke A., Scheffer L. VBP15, a novel anti-inflammatory and membrane-stabilizer, improves muscular dystrophy without side effects. EMBO Mol. Med. 2013;5:1569–1585. doi: 10.1002/emmm.201302621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arriaga L.R., Rodríguez-García R., Moleiro L.H., Prévost S., López-Montero I., Hellweg T., Monroy F. Dissipative dynamics of fluid lipid membranes enriched in cholesterol. Adv. Colloid Interface Sci. 2017;247:514–520. doi: 10.1016/j.cis.2017.07.007. [DOI] [PubMed] [Google Scholar]
- 17.Bracken M.B., Shepard M.J., Holford T.R., Leo-Summers L., Aldrich E.F., Fazl M., Fehlings M., Herr D.L., Hitchon P.W., Marshall L.F. Administration of methylprednisolone for 24 or 48 hours or tirilazad mesylate for 48 hours in the treatment of acute spinal cord injury. Results of the Third National Acute Spinal Cord Injury Randomized Controlled Trial. National Acute Spinal Cord Injury Study. JAMA. 1997;277:1597–1604. [PubMed] [Google Scholar]
- 18.Cahill L., Hall E.D. Is it time to resurrect “lazaroids”? J. Neurosci. Res. 2017;95:17–20. doi: 10.1002/jnr.23842. [DOI] [PubMed] [Google Scholar]
- 19.Baudy A.R., Reeves E.K., Damsker J.M., Heier C., Garvin L.M., Dillingham B.C., McCall J., Rayavarapu S., Wang Z., Vandermeulen J.H. Δ-9,11 modification of glucocorticoids dissociates nuclear factor-κB inhibitory efficacy from glucocorticoid response element-associated side effects. J. Pharmacol. Exp. Ther. 2012;343:225–232. doi: 10.1124/jpet.112.194340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Defour A., Sreetama S.C., Jaiswal J.K. Imaging cell membrane injury and subcellular processes involved in repair. J. Vis. Exp. 2014;85:e51106. doi: 10.3791/51106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McNeil P.L., Steinhardt R.A. Plasma membrane disruption: repair, prevention, adaptation. Annu. Rev. Cell Dev. Biol. 2003;19:697–731. doi: 10.1146/annurev.cellbio.19.111301.140101. [DOI] [PubMed] [Google Scholar]
- 22.Chen Y., Lagerholm B.C., Yang B., Jacobson K. Methods to measure the lateral diffusion of membrane lipids and proteins. Methods. 2006;39:147–153. doi: 10.1016/j.ymeth.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 23.Jaiswal J.K., Simon S.M. Total internal reflection fluorescence microscopy for high-resolution imaging of cell-surface events. Curr. Protoc. Cell Biol. 2003;4 doi: 10.1002/0471143030.cb0412s20. 10.1002/0471143030.cb0412s20. [DOI] [PubMed] [Google Scholar]
- 24.Larbi A., Douziech N., Khalil A., Dupuis G., Gheraïri S., Guérard K.P., Fülöp T., Jr. Effects of methyl-beta-cyclodextrin on T lymphocytes lipid rafts with aging. Exp. Gerontol. 2004;39:551–558. doi: 10.1016/j.exger.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 25.Liu H., Yu W., Liou L.Y., Rice A.P. Isolation and characterization of the human DC-SIGN and DC-SIGNR promoters. Gene. 2003;313:149–159. doi: 10.1016/s0378-1119(03)00674-7. [DOI] [PubMed] [Google Scholar]
- 26.Lu R., Moore P.A., Pitha P.M. Stimulation of IRF-7 gene expression by tumor necrosis factor alpha: requirement for NFkappa B transcription factor and gene accessibility. J. Biol. Chem. 2002;277:16592–16598. doi: 10.1074/jbc.M111440200. [DOI] [PubMed] [Google Scholar]
- 27.McNeil P.L., Khakee R. Disruptions of muscle fiber plasma membranes. Role in exercise-induced damage. Am. J. Pathol. 1992;140:1097–1109. [PMC free article] [PubMed] [Google Scholar]
- 28.Defour A., Medikayala S., Van der Meulen J.H., Hogarth M.W., Holdreith N., Malatras A., Duddy W., Boehler J., Nagaraju K., Jaiswal J.K. Annexin A2 links poor myofiber repair with inflammation and adipogenic replacement of the injured muscle. Hum. Mol. Genet. 2017;26:1979–1991. doi: 10.1093/hmg/ddx065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grounds M.D., Terrill J.R., Radley-Crabb H.G., Robertson T., Papadimitriou J., Spuler S., Shavlakadze T. Lipid accumulation in dysferlin-deficient muscles. Am. J. Pathol. 2014;184:1668–1676. doi: 10.1016/j.ajpath.2014.02.005. [DOI] [PubMed] [Google Scholar]
- 30.Dong Y., Silva K.A., Dong Y., Zhang L. Glucocorticoids increase adipocytes in muscle by affecting IL-4 regulated FAP activity. FASEB J. 2014;28:4123–4132. doi: 10.1096/fj.14-254011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fanin M., Nascimbeni A.C., Angelini C. Muscle protein analysis in the detection of heterozygotes for recessive limb girdle muscular dystrophy type 2B and 2E. Neuromuscul. Disord. 2006;16:792–799. doi: 10.1016/j.nmd.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 32.Gallardo E., Rojas-García R., de Luna N., Pou A., Brown R.H., Jr., Illa I. Inflammation in dysferlin myopathy: immunohistochemical characterization of 13 patients. Neurology. 2001;57:2136–2138. doi: 10.1212/wnl.57.11.2136. [DOI] [PubMed] [Google Scholar]
- 33.Taylor B.M., Fleming W.E., Benjamin C.W., Wu Y., Mathews W.R., Sun F.F. The mechanism of cytoprotective action of lazaroids I: Inhibition of reactive oxygen species formation and lethal cell injury during periods of energy depletion. J. Pharmacol. Exp. Ther. 1996;276:1224–1231. [PubMed] [Google Scholar]
- 34.Passaquin A.C., Lhote P., Rüegg U.T. Calcium influx inhibition by steroids and analogs in C2C12 skeletal muscle cells. Br. J. Pharmacol. 1998;124:1751–1759. doi: 10.1038/sj.bjp.0702036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Durmaz R., Ertilav K., Akyüz F., Kanbak G., Bildirici K., Tel E. Lazaroid U-74389G attenuates edema in rat brain subjected to post-ischemic reperfusion injury. J. Neurol. Sci. 2003;215:87–93. doi: 10.1016/s0022-510x(03)00207-7. [DOI] [PubMed] [Google Scholar]
- 36.Hoffman E.P., Riddle V., Siegler M.A., Dickerson D., Backonja M., Kramer W.G., Nagaraju K., Gordish-Dressman H., Damsker J.M., McCall J.M. Phase 1 trial of vamorolone, a first-in-class steroid, shows improvements in side effects via biomarkers bridged to clinical outcomes. Steroids. 2018;134:43–52. doi: 10.1016/j.steroids.2018.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gregoire F.M., Smas C.M., Sul H.S. Understanding adipocyte differentiation. Physiol. Rev. 1998;78:783–809. doi: 10.1152/physrev.1998.78.3.783. [DOI] [PubMed] [Google Scholar]
- 38.Bauerle K.T., Hutson I., Scheller E.L., Harris C.A. Glucocorticoid Receptor Signaling Is Not Required for In Vivo Adipogenesis. Endocrinology. 2018;159:2050–2061. doi: 10.1210/en.2018-00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Defour A., Van der Meulen J.H., Bhat R., Bigot A., Bashir R., Nagaraju K., Jaiswal J.K. Dysferlin regulates cell membrane repair by facilitating injury-triggered acid sphingomyelinase secretion. Cell Death Dis. 2014;5:e1306. doi: 10.1038/cddis.2014.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vila M.C., Rayavarapu S., Hogarth M.W., Van der Meulen J.H., Horn A., Defour A., Takeda S., Brown K.J., Hathout Y., Nagaraju K., Jaiswal J.K. Mitochondria mediate cell membrane repair and contribute to Duchenne muscular dystrophy. Cell Death Differ. 2017;24:330–342. doi: 10.1038/cdd.2016.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jacobs S.C., Bootsma A.L., Willems P.W., Bär P.R., Wokke J.H. Prednisone can protect against exercise-induced muscle damage. J. Neurol. 1996;243:410–416. doi: 10.1007/BF00869001. [DOI] [PubMed] [Google Scholar]
- 42.Leikina E., Defour A., Melikov K., Van der Meulen J.H., Nagaraju K., Bhuvanendran S., Gebert C., Pfeifer K., Chernomordik L.V., Jaiswal J.K. Annexin A1 Deficiency does not Affect Myofiber Repair but Delays Regeneration of Injured Muscles. Sci. Rep. 2015;5:18246. doi: 10.1038/srep18246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gushchina L.V., Bhattacharya S., McElhanon K.E., Choi J.H., Manring H., Beck E.X., Alloush J., Weisleder N. Treatment with Recombinant Human MG53 Protein Increases Membrane Integrity in a Mouse Model of Limb Girdle Muscular Dystrophy 2B. Mol. Ther. 2017;25:2360–2371. doi: 10.1016/j.ymthe.2017.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cooper S.T., McNeil P.L. Membrane Repair: Mechanisms and Pathophysiology. Physiol. Rev. 2015;95:1205–1240. doi: 10.1152/physrev.00037.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chase T.H., Cox G.A., Burzenski L., Foreman O., Shultz L.D. Dysferlin deficiency and the development of cardiomyopathy in a mouse model of limb-girdle muscular dystrophy 2B. Am. J. Pathol. 2009;175:2299–2308. doi: 10.2353/ajpath.2009.080930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hendricks B.K., Shi R. Mechanisms of neuronal membrane sealing following mechanical trauma. Neurosci. Bull. 2014;30:627–644. doi: 10.1007/s12264-013-1446-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sreetama S.C., Takano T., Nedergaard M., Simon S.M., Jaiswal J.K. Injured astrocytes are repaired by Synaptotagmin XI-regulated lysosome exocytosis. Cell Death Differ. 2016;23:596–607. doi: 10.1038/cdd.2015.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cong X., Hubmayr R.D., Li C., Zhao X. Plasma membrane wounding and repair in pulmonary diseases. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017;312:L371–L391. doi: 10.1152/ajplung.00486.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spurney C.F., Gordish-Dressman H., Guerron A.D., Sali A., Pandey G.S., Rawat R., Van Der Meulen J.H., Cha H.J., Pistilli E.E., Partridge T.A. Preclinical drug trials in the mdx mouse: assessment of reliable and sensitive outcome measures. Muscle Nerve. 2009;39:591–602. doi: 10.1002/mus.21211. [DOI] [PMC free article] [PubMed] [Google Scholar]