

Abstract
Pancreatic cancer is one of the most aggressive and lethal malignancies worldwide, with a very poor prognosis and a five-year survival rate less than 8%. This dismal outcome is largely due to delayed diagnosis, early distant dissemination and resistance to conventional chemo-therapies. Kras mutation is a well-defined hallmark of pancreatic cancer, with over 95% of cases harbouring Kras mutations that give rise to constitutively active forms of Kras. As important down-stream effectors of Kras, p21-activated kinases (PAKs) are involved in regulating cell proliferation, apoptosis, invasion/migration and chemo-resistance. Immunotherapy is now emerging as a promising treatment modality in the era of personalized anti-cancer therapeutics. In this review, basic knowledge of PAK structure and regulation is briefly summarised and the pivotal role of PAKs in Kras-driven pancreatic cancer is highlighted in terms of tumour biology and chemo-resistance. Finally, the involvement of PAKs in immune modulation in the tumour microenvironment is discussed and the potential advantages of targeting PAKs are explored.
Keywords: Pancreatic cancer, Kras, p21-activated kinases, Cell signalling, Chemo-resistance, Immune response, Tumour microenvironment
Core tip: Pancreatic cancer is still one of the most lethal malignancies, with a five-year survival of less than 8%. The dismal prognosis is largely the result of reprogramming of the tumour microenvironment, which leads to chemo-resistance and high aggressiveness. So far, combination chemotherapies can only marginally improve patients’ survival, but with high toxicity. Therefore, alternative treatment targeting protein kinase signalling has been proposed. As downstream effectors of Kras signalling, p21-activated kinases (PAKs) are positioned at the nexus of multiple oncogenic signalling pathways. Here, the importance of PAKs as therapeutic targets in Kras signalling is discussed, and their essential role in tumour biology and immune modulation within the tumour microenvironment is highlighted.
INTRODUCTION
Pancreatic cancer is a highly aggressive and lethal malignancy with a dismal prognosis. In contrast to the improvements in therapies and the consequent increasing long-term survival rate for most other cancers, few advances have been achieved in pancreatic cancer, for which the overall five-year survival rate is still less than 8%[1]. The death rate from pancreatic cancer continues to increase by 0.3% per annum, and it is estimated that this malignancy will become the second most common cause of cancer-related death in the United States by 2030[2].
Although surgery remains the only curative treatment, chemotherapy is still an important and indispensable treatment in maximizing the life span for both resectable and unresectable patients. Currently gemcitabine-based combination therapies and FOLFIRINOX (irinotecan, oxaliplatin, fluorouracil, and leucovorin) are the mainstream approaches for patients with local advanced and metastatic pancreatic cancer, with an increased survival compared to gemcitabine alone[3-6]. However, the modest improvement in survival, the highly toxic side effects and chemo-resistance have become major challenges in the clinical setting. Therefore, there is an urgent need to develop more effective and less toxic therapeutic strategies to treat this malignancy.
Progression of pancreatic cancer is marked by an accumulation of multiple genetic mutations, of which mutation in the Kras oncogene is the most frequent, with over 95% of pancreatic cancers harbouring a Kras mutation[7]. The presence of missense mutations at codons 12, 13 or 61 within the Kras gene disrupts the physiological inactivation cycle of the Kras protein, resulting in a constitutively activated state even in the presence of GTPase activating protein.
The Kras protein is notable for the absence of a well-defined drug-binding domain on its surface[8]. So far, despite over thirty years of intensive biomedical research, no drug directly targeting the Kras protein has proved to be an effective cancer treatment in the clinic[7,9]. While some exciting and promising results have appeared for treatments that targeted important downstream effectors of Kras such as PI3K, AKT and MEK, resistance developed rapidly in almost all cases, making these molecular targets less effective[10]. In order to overcome this challenge, approaches targeting novel downstream effectors of the Kras protein are urgently needed. Recently, the National Cancer Institute in the United States has proposed a new project to fight against Ras-driven cancers, with the stated aim that new therapeutic strategies interfering with Ras-dependent signalling pathways should be given priority in cancer research[11]. One such family of novel effectors is the p21-activated kinases (PAKs), which are activated by Kras and by other small GTPases like Cdc42 and Rac by both direct and indirect mechanisms. PAKs are positioned at the nexus of multiple oncogenic signalling pathways that mediate a variety of hallmark processes in pancreatic cancer.
Pancreatic cancer has its own unique immune response during tumour development. The Kras oncogene can mediate the inflammatory process and establish within the tumour microenvironment an immune-privileged condition, which is responsible for the suppression of effector cells and the stimulation of immunosuppressive cells[12]. Additionally, the extensive desmoplastic reaction in pancreatic cancer also functions as a physiological barrier against immune surveillance, leading to evasion of the anti-tumoural immune response and tumour progression[13].
In this review, basic knowledge of PAK structure and regulation is briefly summarised, and the importance of PAKs as a therapeutic target in Kras signalling is highlighted. The essential role of PAKs in regulating tumour biology and stromal re-programming, especially of the immune response within the tumour microenvironment, is also discussed.
STRUCTURE AND ACTIVATION OF PAKS
PAKs are a family of serine/threonine kinases that are the downstream effector proteins of Ras, and of other small GTPases such as Cdc42 and Rac. The six known members of the PAK family can be categorized by similarities in their sequence and structure into two groups: group I (PAK1-3) and group II (PAK4-6)[14]. All PAKs are characterized by an N-terminal regulatory domain and a conserved C-terminal serine/threonine kinase domain with a single phosphorylation site (Figure 1), but the activation of group I and group II PAKs is regulated through completely different mechanisms[15,16].
Figure 1.
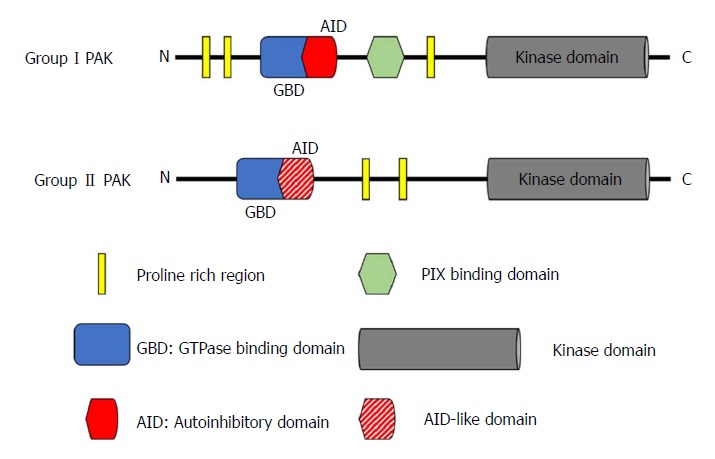
Structure of p21-activated kinases. The six members of the PAK family can be divided by sequence and structural differences into two groups: Group I (PAK1-3) and group II (PAK4-6). All PAKs have an N-terminal regulatory domain and a conserved C-terminal serine/threonine kinase domain. In group I PAKs, the regulatory domain contains an AID, whereas group II PAKs (with the possible exception of PAK5) do not have a well-defined AID, but instead an AID-like domain. PIX: PAK-interactive exchange factor; PAK: p21-activated kinases; AID: Autoinhibitory domain; GBD: GTPase-binding domain.
The group I PAKs share a high level of structural homology with over 88% identity in the GTPase-binding domain (GBD) that is responsible for binding Cdc42 or Rac, and more than 93% identity in the kinase domain[14]. However, their tissue specific distribution is quite different from each other. PAK1 can be found in various organs including brain, mammary gland, muscle, and spleen; PAK2 is ubiquitously expressed; whereas PAK3 is only expressed in the nervous system[17]. The N-terminal regulatory domain of group I PAKs contains an autoinhibitory domain (AID) that overlaps with the GBD. In the inactivated state, group I PAKs form homodimers, with the AID domain of one PAK molecule binding to the kinase domain of its companion. When Cdc42/Rac binds to the GBD, binding of the AID to its partner PAK is disrupted. This critical process generates two PAK monomers, and allows the subsequent autophosphorylation at the Thr423 site, which is important for maintaining PAK1 activation[18-21].
Group II PAKs have a quite different structure from group I PAKs, but the three members are still similar to each other. They share at least 60% identity in the N-terminal GBD and over 75% identity in the kinase domain. However, the identities between Group I and Group II PAKs are less than 40% in the GBD and only about 54% in the kinase domain[14]. PAK4 is highly expressed throughout embryonic development, and ubiquitously expressed in all adult tissue at a low level[22,23]. PAK5 is specifically expressed in the brain[24]. PAK6 is not only found in the adult nervous system, but also in the male reproductive system (e.g., testes and prostate). This distribution correlates with its important role in the androgen receptor signalling pathway[25,26]. Although group II PAKs (with the possible exception of PAK5) have no well-defined AID in the N-terminal domain, later studies have reported AID-like domains[27,28]. Unlike group I PAKs, group II PAKs are monomers and are constitutively phosphorylated, even in their inactivated state[15]. Although there is still much debate on the exact activation mechanism of group II PAKs, two different activation models have been proposed over the last decade. In the first model, the AID-like domain binds to the kinase domain of the same molecule, which results in an inactive confirmation regardless of the constitutive autophosphorylation. When Cdc42 binds to the GBD, binding of the AID-like domain to the kinase domain is disrupted, leading to an active conformation[15]. In the second model, an autoinhibitory pseudo-substrate domain, next to the GBD but distinct from the AID, interacts with the kinase domain, reducing activity. The binding of Cdc42 to GBD translocates the group II PAK to a subcellular region where a Src homology 3 domain-containing protein binds to the autoinhibitory pseudo-substrate domain, preventing its interaction with the kinase domain and hence increasing activity[28].
ROLE OF PAKS IN KRAS-DRIVEN ONCOGENIC PATHWAYS
Kras is the most frequently mutated isoform observed in all types of human cancer compared to NRas and Hras. Kras mutation is a key oncogenic driver in the development of pancreatic, colorectal and lung cancer[29]. It acts as a regulatory switch in diverse sub-cellular signal transduction networks, which are responsible for stem-cell like features, cell survival, proliferation, invasion and migration[30].
A study of a genetically engineered mouse model for pancreatic cancer, the KPC (LSL-KrasG12D; LSL-Trp53R172H; Pdx1-Cre) model, has revealed that expression of the KrasG12D mutation is sufficient to induce pancreatic intraepithelial neoplasia (PanIN), followed by advanced carcinoma[31]. Similarly, Ying and colleagues demonstrated that the KrasG12D mutation was necessary for the maintenance of pancreatic cancer as Kras depletion resulted in rapid tumoural regression and stromal degeneration in an oncogenic Kras-induced tumour model[32]. Mutated Kras can cause phosphorylation and activation of other p21 proteins such as Rac1 and Cdc42, through both canonical and alternative pathways[33]. Then the interaction between Rac1/Cdc42 and PAKs can increase PAK activity, leading to persistent activation of downstream signalling pathways such as the RAF/MEK/ERK and PI3K/PDK1/AKT pathways[34-37] (Figure 2).
Figure 2.
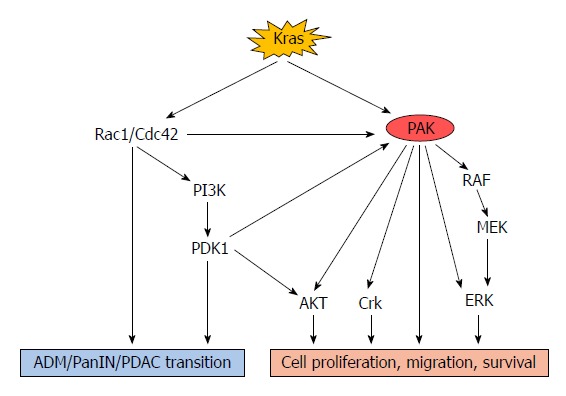
Role of p21-activated kinases in Kras-driven oncogenic signalling pathways. Rac1 is the 4th best validated effector in Kras signalling and is a well-defined upstream protein of PAKs. Rac1 plays an important role in the ADM/PanIN/PDAC transition. In addition, Rac1/Cdc42 mediates this pathological process via the PI3K-PDK1 signalling pathway. PDK1 can also interact with PAK1, leading to its phosphorylation. The Kras oncogene activates PAKs through direct and indirect pathways. Activated PAKs can increase cancer cell proliferation, migration and survival through activation of AKT, Crk and RAF-MEK-ERK pathways. PAK: p21-activated kinases; ADM: Acinar-ductal metaplasia; PDAC: Pancreatic ductal adenocarcinoma; PDK1: Phosphoinositide-dependent kinase-1.
A recent study of non-small cell lung cancer reported that Kras-mutated tumours expressed more Thr423-phosphorylated PAK1 than Kras-wild type tumours, and that the Kras/PAK1/Crk axis played an essential role in the oncogenesis of Kras-mutated lung cancer[38]. Activation of PAK1 could also be mediated by multiple Kras-dependent pathways via different cell surface receptors. Dominant-negative Ras, Rac, and Cdc42 suppressed PAK1 activation whereas activated Rac1 and Cdc42 were able to stimulate PAK1 even in the absence of any agonists[39]. As a potent activator of PAK1, Rac1 is the 4th best validated effector in the Kras-driven cell signalling cascade[40]. In an early study, Rac1 was found to be associated with pancreatic acinar plasticity and Rac1 inhibition reduced acinar cell damage induced by pathological inflammation[41]. The important role of Rac1 in early metaplasia and neoplasia-related actin rearrangements has been revealed in a Kras-driven mouse model of pancreatic cancer[42]. Rac1 ablation in this model reduced the incidence of acinar-ductal metaplasia (ADM), PanIN and tumour formation and significantly improved animal survival. Interestingly, this study also found that Rac1 was not indispensable in pancreas development[42]. Similarly, Zheng et al[43] reported that Rac1 and Cdc42 could mediate the activation of PI3K by interacting with its 85-kDa regulatory domain. Another study documented that PI3K together with PDK1 acted as critical downstream effectors of oncogenic Kras signalling in mediating ADM and formation of pancreatic cancer[44]. PDK1 was also reported to interact with PAK1 both in vitro and in vivo, leading to increased phosphorylation at the Thr423 site and hence activation of PAK1[45].
There is increasing evidence for a key role of PAK1 in regulating Kras-dependent signalling pathways. PAK1 can phosphorylate c-RAF at Ser338 in NIH3T3 cells (murine fibroblast cell line), and inhibition of group I PAK kinase activity significantly reduced the phosphorylation of MEK1 at Ser298 and the activation of ERK in response to different growth factors (e.g., platelet-derived growth factor or epidermal growth factor) in NIH3T3 and HeLa cells (human cervical cancer cell line)[46]. Huynh et al[47] demonstrated that PAK1 stimulates colon cancer cell proliferation, migration/invasion, and survival via ERK- and AKT-dependent pathways. Inhibition of PAK1 effectively inhibits both ERK and AKT, to an extent which cannot be achieved by inhibition of either alone. Another study also showed that genetic deletion of PAK1, followed by decreased ERK and AKT activity, suppressed tumourigenesis and progression in a Kras-mediated skin cancer model[48]. In contrast, Tabusa and colleagues found that knockdown of PAK1 or PAK4 inhibited the proliferation of Kras-mutated colorectal cancer cells via non-canonical pathways independent of RAF/MEK/ERK and PI3K/AKT signalling[49].
A relationship between PAK4 and Kras has been identified through genetic analysis of human pancreatic cancer cell lines and patients’ samples[50]. By sequencing the Kras gene in PAK4-amplified tumour samples, mutations in codon 12 were observed in 4 out of the 5 samples. Furthermore, genomic amplification and overexpression of Kras occurred in 3 samples. Interestingly, no mutations were detected in Kras or PAK4 in the fifth sample, but the observation of increased PAK4 expression suggests that PAK4 could be up-regulated and activated through some Kras-independent pathways. Taken together, the above evidence suggests that PAKs play an important role in interacting with and transmitting Kras-driven oncogenic signals in different kinds of human cancer.
PAK SIGNALLING IN PANCREATIC CANCER
Amplification of the PAK1 gene within chromosomal region 11q13 was reported to be linked to both tumourigenesis and poor prognosis of different human cancers[51,52]. Amplification of the PAK4 gene within chromosomal region 19q13.2 was also identified in a variety of human malignancies, especially pancreatic, breast, and ovarian cancer[50,53,54]. By using fluorescent in situ hybridization on tumour microarrays, Kimmelman et al[55] found PAK4 amplification occurred in 14 of 63 (22%) pancreatic cancer samples. In addition, RT-qPCR and Western blots showed increased PAK4 expression in multiple pancreatic cancer cell lines regardless of gene amplification, implying different underlying mechanisms mediating PAK4 expression. Interestingly, the observation that the CCND1 (Cyclin D1) and CCNE1 (Cyclin E1) genes were co-amplified within the same chromosomal region as PAK1 and PAK4, respectively[16,56], suggests that co-amplification of PAK1 with CCND1 and PAK4 with CCNE1 may have synergistic effects on the initiation and progression of pancreatic cancer.
Role of PAK1 in tumour proliferation, migration and tumour-stroma interaction
A study of 304 human primary pancreatic cancer samples found that 262 (86%) cases were positive for cytoplasmic PAK1 staining, and approximately one-third of all samples showed moderate (2+) to strong (3+) intensity in the malignant cells and nuclear localization of PAK1[57]. Two more recent studies have also shown increased PAK1 expression in resected human pancreatic cancer tissues and cell lines, when compared to the adjacent normal pancreas and an immortalized normal pancreatic ductal epithelial cell line, respectively[58,59]. A PAK1 knock-down pancreatic cancer cell line failed to develop tumours in nude mice[58] and showed markedly reduced proliferation[59]. Furthermore, Jagadeeshan and colleagues demonstrated that fibronectin was a transcriptional target of PAK1 signalling via the NF-κB-p65-fibronectin axis, which modulates cell transformation and the invasive EMT phenotype of pancreatic cancer cells (Figure 3). Early studies also showed the localization of activated PAK1 to the cell nucleus[60] and its involvement in the activation of NF-κB, by demonstrating that active Ras or Rac1 stimulated NF-κB in a PAK1-dependent manner and that active PAK1 itself could stimulate NF-κB as well[61]. An important transmembrane mucin (MUC13) was also reported to be involved in PAK1 signalling in the development of pancreatic cancer[62]. Overexpression of MUC13 promoted cancer cell proliferation, invasion/migration and anchorage-dependent or -independent colony formation in vitro and led to increased xenograft tumour growth and decreased animal survival in vivo. These tumourigenic properties were closely associated with the up-regulated expression and phosphorylation of PAK1, ERK and AKT, and suppression of p53. Wei et al[57] screened a panel of pancreatic cancer cell lines and characterized PAK1 as a key downstream effector of cell motility triggered by multiple growth factors. In their study, PAK1 inhibition not only restored sensitivity to a hepatocyte growth factor/Met antagonist (onartuzumab) in the presence of exogenous growth factors or PAK1-amplification in vitro, but also suppressed tumour growth and metastasis in vivo.
Figure 3.
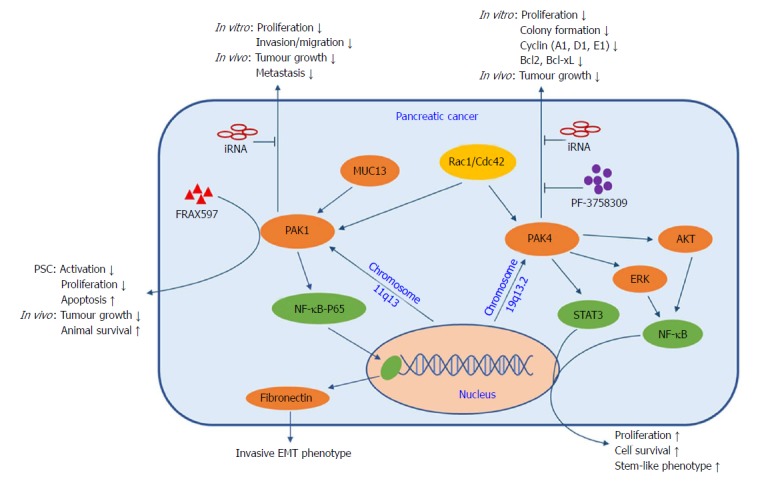
p21-activated kinases signalling in the development of pancreatic cancer. PAK signalling is involved in several pathobiological processes in pancreatic cancer, including proliferation, migration/invasion, apoptosis and maintenance of stem cell-like properties. Amplification of the PAK1 and PAK4 genes, present within the chromosomal regions 11q13 and 19q13.2, respectively, has been observed. Activated PAK1 regulates cell transformation and the invasive EMT phenotype of pancreatic cancer cells via the NF-κB-p65-fibronectin axis. Additionally, MUC13 promotes cancer cell growth and invasion/migration, and reduces animal survival, by up-regulating expression and phosphorylation of PAK1. Furthermore, PAK4 modulates proliferation and survival by mediating the activity of NF-κB via AKT- and ERK-dependent pathways, and cancer stem cell-like properties via STAT3 signalling. Pharmacological or genetic inhibition of PAK1 or PAK4 leads to decreased cancer cell proliferation, invasion/migration and PSC activation in vitro, and reduced tumour growth and metastasis, and increased animal survival in vivo. PAK: p21-activated kinases; PSC: Pancreatic stellate cells.
In recent years, the interaction between pancreatic cancer cells and pancreatic stellate cells (PSCs) has become the focus of pancreatic cancer research. Activation of PSCs by cancer cells is predominately responsible for fibrosis and stromal remodelling. The role of PAK1 in modulating EMT markers (e.g., fibronectin, E-cadherin, and vimentin) has been established in early studies[63]. A recent study revealed the role of PAK1 in PSC modulation for the first time by showing that inhibition of PAK1 by FRAX597 (a potent group I PAK inhibitor) reduced the activation and proliferation of PSC and increased apoptosis in vitro. In an orthotopic pancreatic cancer mouse model, survival in the PAK1 knockout group was significantly increased compared to the PAK1 wildtype group, and depletion of PAK1 in the pancreatic stroma also reduced PAK1 expression and activity in the tumour[64]. Similarly, survival was prolonged in the group treated with FRAX597 plus gemcitabine in the same orthotopic pancreatic cancer model[59]. These results pave the way to a detailed investigation of the role of PAK1 in tumour-stroma interactions in order to improve therapeutic response by using targeted inhibitors.
Role of PAK4 in tumour proliferation, migration, survival and stemness maintenance
PAK4 expression is reported to be correlated with pancreatic cancer pathology. Tyagi et al[65] found 54 out of 56 tumour samples from patients with pancreatic cancer had positive PAK4 staining, with no PAK4 positive staining in normal pancreatic tissue. Furthermore, PAK4 promoted cancer cell proliferation and survival by stimulating the nuclear accumulation and transcriptional activity of NF-κB via AKT- and ERK-dependent pathways (Figure 3). PAK4 knockdown in pancreatic cancer cells caused suppression of growth and colony formation associated with reduced expression of cell-cycle (cyclin A1, D1, E1) and anti-apoptosis (Bcl2, Bcl-xL) proteins. Similarly, inhibition of PAK4 by PF-3758309 (a potent pan-PAK inhibitor) suppressed cancer cell proliferation and migration both in vitro and in vivo[66]. Kimmelman et al[55] identified Rio Kinase 3 and PAK4 as amplified genes in highly recurrent and focal amplifications in pancreatic cancer. Rio Kinase 3 can activate the small GTPase protein Rac, which can subsequently promote cell motility and invasion via PAK4-mediated signalling. In addition, overexpression of activated PAK4 resulted in increased invasion/migration in a gain-of-function experiment, while PAK4 knockdown by shRNA significantly reduced anchorage independent growth in a loss-of-function experiment. Recently, PAK4 has been shown to modulate STAT3 signalling in the maintenance of pancreatic cancer stem cells, which are considered to be responsible for high aggressiveness and chemo-resistance. Pancreatic cancer stem-like cells (CD24+/CD44+/EpCAM+) showed higher PAK4 expression as compared to triple negative cells (CD24-/CD44-/EpCAM-). PAK4 expression enhanced the expression of stem cell-associated transcription factors (Oct4/Nanog/Sox2 and KLF4), whereas PAK4 silencing caused reduced nuclear accumulation and transcriptional activity of STAT3 and loss of stem cell phenotypes[67,68]. The accumulated evidence, which suggests that PAKs are positioned at the convergence point of numerous oncogenic pathways, highlights their potential as promising therapeutic targets in the treatment of pancreatic cancer.
PAKS AND CHEMO-RESISTANCE IN PANCREATIC CANCER
Currently, surgical resection is the only curative treatment for pancreatic cancer. However, due to the lack of biomarkers for early diagnosis, low surgical resectability (only 15%-20% of patients are considered to be eligible candidates)[69], and high recurrence rate (up to 60%), the overall median survival is still less than 20 mo in patients undergoing resection of pancreatic cancer with curative intent[70]. Therefore, chemotherapy remains a crucial alternative or adjuvant treatment for patients with resectable or unresectable tumours[71].
Two decades ago, gemcitabine emerged as the standard of care for pancreatic cancer patients[72]. So far, gemcitabine + nab-paclitaxel and FOLFIRINOX have been approved by the United States Food and Drug Administration as first-line therapies, especially for patients with locally advanced and metastatic pancreatic cancer[3,6]. Although previous clinical studies reported several advantages of gemcitabine over 5-FU, including prolonged median survival, improved tumour-related symptoms and lower systemic toxicity[73-75], the results were still unsatisfactory, with effective responses in less than 10% of patients. Therefore, various modifications of gemcitabine treatment have been designed to overcome resistance and increase drug delivery into the tumour. These modifications include CO-101[76] (a lipid-conjugated gemcitabine, which can be transported into tumour cells independently of the human equilibrative nucleoside transporter, and Acelarin[77] (an aryloxy phosphoramidate derivative of gemcitabine with greater lipophilicity, which accumulates in cancer cells by passive diffusion independently of the nucleoside transporter). However, intrinsic and acquired gemcitabine resistance occurs in most patients, and its underlying molecular mechanism is still not fully understood.
Mechanisms involved in chemo-resistance of cancer cells
In a prospective randomized clinical study, the human equilibrative nucleoside transporter 1(hENT1), which is the principal cellular transporter of gemcitabine, was found to be a valuable predictive marker of gemcitabine sensitivity in patients with resected pancreatic cancer[78]. In addition, a comparative study also indicated that decreased hENT1 expression was associated with gemcitabine resistance and poorer overall survival in patients with pancreatic cancer[79]. However, an early study reported that up-regulated hENT1 expression was also observed in some gemcitabine-resistant pancreatic cancer cell lines[80]. This evidence implicated hENT1 as the predominant, but not the only, metabolic protein mediating resistance. To some extent, hENT2 and the human concentrative nucleoside transporters hCNT1 and hCNT3 may also contribute to the development of acquired and intrinsic gemcitabine resistance[81,82]. Moon and colleagues have shown that gemcitabine-resistant cell lines express more PAK4 and less hENT1. PAK4 knockdown in gemcitabine-resistant cell lines induces the up-regulation of hENT1 and restoration of sensitivity to gemcitabine[83]. In contrast, one recent retrospective clinical study, which analyzed 160 resected human pancreatic cancer samples by immunohistochemistry, reported that higher expression of PAK4 was correlated with higher expression of hENT1[84]. Therefore, the controversial role of PAK4 in regulating hENT1 should be further explored.
Furthermore, Jagadeeshan et al[85] revealed that PAK1 plays a pivotal role in mediating gemcitabine resistance by altering apoptosis and survival signals, and suppressing DNA damage via the NF-κB pathway. Phosphorylation of PAK1 and ribonucleotide reductase M1 was elevated in patient samples when compared with normal tissue. Combination treatment with a PAK1 inhibitor synergistically improved gemcitabine efficacy and led to tumour regression in animal models. In agreement with this finding, inhibition of PAK1 or/and PAK4 by shRNA knockdown or PAK inhibitors enhanced gemcitabine sensitivity both in vitro and in vivo[59,66].
Other potential PAK-associated signalling pathways also contribute to chemo-resistance. Higher expression of HIF-1α was observed in gemcitabine-resistant pancreatic cancer cell lines[86] and inhibition of HIF-1α can sensitize cancer cells to gemcitabine treatment[87]. The increased activity of HIF-1α was associated with inhibition of the transcription of hENT1 and hENT2, leading to reduced expression of transporter proteins followed by decreased gemcitabine uptake[88,89]. The critical role of PAKs in regulating HIF-1α has been demonstrated in our previous studies, which showed that PAK1 could enhance cancer cell survival by up-regulation of HIF-1α and that inhibition of PAK1 caused decreased expression of HIF-1α and tumour growth[59,90]. Another recent study also found that PAK4 inhibition reduced expression of HIF-1α via the AKT-mTOR-4E-BP1 axis[91].
Additionally, the important transcription factor NF-κB is also a critical regulator closely associated with gemcitabine resistance in pancreatic cancer. As discussed above, localization of active PAK1 to the nucleus is involved in activation of NF-κB[60,61], and both PAK1 and PAK4 contribute to cell transformation, proliferation and survival via NF-κB-dependent signalling pathways in pancreatic cancer[58,65]. Over a decade ago, Arlt and colleagues revealed that resistant cell lines (BxPC-3, Capan-1 and PancTu-1) showed higher expression of NF-κB, comparing to sensitive cell lines (PT45-P1 and T3M4). Treatment of these five pancreatic cancer cell lines with gemcitabine induced NF-κB activity in a dose-dependent manner[92]. Inhibition of the p65 subunit of NF-κB by siRNA can improve gemcitabine sensitivity to suppress proliferation and induce apoptosis both in vitro and in vivo[93]. In agreement with this conclusion, Skrypek et al[94] also showed that decreased activation of NF-κB pathway was correlated with an alteration of hCNT1 expression and increased gemcitabine sensitivity in MUC4-knockdown pancreatic cancer cell lines.
Furthermore, both expression and activity of PAK4 have also been reported to be up-regulated in cisplatin-resistant cancer cells as compared with parental cells. Inhibition of PAK4 diminished cisplatin resistance via PI3K/AKT and MEK/ERK-dependent signalling pathways[95].
Stromal remodelling
The extensive desmoplastic reaction, which is a hallmark of pancreatic cancer, is reported to result in a dense stroma, deficient vascularization and inefficient drug delivery, eventually leading to chemo-resistance[96,97]. As mentioned above, PAK1 is responsible for PSC activation, leading to stromal fibrosis[64] in pancreatic cancer similar to its pivotal role in liver fibrogenic pathways[98].
The importance of Hedgehog (Hh) signalling in tumourigenesis and desmoplasia has been well established. Hh can modify the extracellular matrix component via regulation of the differentiation and motility of PSCs and fibroblasts[99,100]. The observation, in an early global genomic analysis of 24 human pancreatic cancers, that Hh signalling was one of the core set of 12 most commonly altered cellular signalling pathways and was present in 100% of cases suggests a significant contribution of Hh signalling to the development of pancreatic cancer[101]. A number of previous studies also demonstrated that depletion of the Hh signalling pathway could partly diminish desmoplasia-associated resistance and synergistically enhance gemcitabine efficacy both in vitro and in vivo[102-104]. Hiroshi et al[105] found that activation of NF-κB resulted in the aberrant activation of Hh signalling via up-regulation of sonic Hh (a ligand of Hh signalling) in pancreatic cancer.
Interaction of the C-X-C motif chemokine 12 (CXCL12), which is also known as stromal cell-derived factor 1, with its receptor, the C-X-C motif chemokine receptor 4 (CXCR4), can induce activation of downstream signalling pathways related to tumour progression and metastasis[106]. Singh et al[107] have identified the essential role of the CXCL12/CXCR4 axis in stimulation of Hh signalling in a dose- and time-dependent manner. CXCL12-induced Hh up-regulation is due to the increased nuclear accumulation and activation of NF-κB mediated by AKT and ERK signalling pathways. The involvement of PAK1 and PAK4 in NF-κB signalling in pancreatic cancer has been clearly identified[58,61,65]. Although the interaction between PAKs and Hh-mediated chemo-resistance is still not clear, the above findings indicate that PAKs might regulate Hh signalling in a NF-κB-dependent manner and further investigation is needed.
EMERGING ROLE OF PAKS IN IMMUNE MODULATION IN THE TUMOUR MICROENVIRONMENT
As an important component of the stromal microenvironment, infiltrating immune cells (IICs) have been characterized as valuable markers in predicting prognosis. Generally, IICs exhibit both pro-tumour and anti-tumour effects. The former class of IICs include regulatory T cells, myeloid-derived suppressor cells (MDSC), and tumour-associated macrophages (TAM) which suppress anti-tumour immunity and promote tumour growth, whereas the latter class include CD8+ T cells, Th1-type CD4+ T cells, and natural killer cells[108,109]. Immunosuppressive cells can ward off the host immune defence, prevent tumour cells from being recognized and further lead to immune evasion, even in pre-cancerous lesions such as PanINs and intraductal papillary mucinous neoplasm (IPMN)[110]. Therefore, a great deal of attention has been paid to targeting the aberrant immune regulation of the tumour microenvironment, with the intention of reversing the suppression of active anti-tumour immunity. A good example is the conversion of pancreatic cancer from “a non-immunogenic malignancy” into “an immunogenic malignancy” by treatment with a novel immunomodulatory vaccine, which blockaded the immune checkpoint (PD-1/PD-L1, CTLA-4) and made therapy more effective in vaccine-treated patients than in untreated patients[111].
Myeloid-derived suppressor cells
MDSCs, which include both granulocytic and monocytic subsets, are a heterogeneous mixture of activated immature myeloid cells, which can stimulate angiogenesis, promote tumour invasion and migration, and suppress T-cell activation[112]. Both circulating and tumour-infiltrating MDSCs, of the granulocytic subset (Lin-HLA-DR-CD33+CD11b+CD15+), but not the monocytic subset (Lin-HLA-DR-CD14+), are markedly elevated in patients with pancreatic cancer compared to the healthy population. Moreover, MDSCs can also serve as an independent prognostic factor for patients’ survival as one unit increase in MDSC percentage leads to a 22% greater risk of mortality[113,114]. The report by Thomas et al[115] of a non-canonical role of the mammalian target of rapamycin (mTOR) protein in recruiting tumourigenic MDSC suggests that cancer cells can stimulate intra-tumoural MDSC accumulation by promoting granulocyte colony-stimulating factor (G-CSF) production via an mTOR-dependent pathway. He and colleagues demonstrated the critical role of PAK4 in regulating mTOR signalling through the PI3K/AKT axis in breast cancer[116]. In addition, PAK1 can be activated by the mTOR/p70 S6 kinase pathway, and treatment with rapamycin, a mTOR inhibitor, leads to reduced PAK1 expression[117]. Interestingly, an early study on vascular smooth muscle cells indicated that G-CSF was involved in activation of the GTPase Rac1, a potent upstream activator of PAK1, and inhibition of Rac1 suppressed G-CSF-driven migration of vascular smooth muscle cells[118]. Previous studies also found that pancreatic cancer cells or stellate cells can attract and transform peripheral blood monocytes into MDSCs via STAT3 activation, which in turn will increase the stem-cell properties and mesenchymal features of tumour cells[119,120]. The role of PAK1 and PAK4 in regulating STAT3 signalling in pancreatic cancer cells has been clearly identified[67,121]. Although there is as yet little direct evidence linking PAK to MDSC modulation, the above findings indicate that PAK might orchestrate multiple signalling pathways to mediate MDSC recruitment and activation.
Tumour-associated macrophages
TAMs can be divided into two subtypes: M1 (pro-inflammatory macrophages) and M2 (anti-inflammatory macrophages). Like MDSCs, the majority of TAMs are derived from circulating monocytes[122]. M1 TAMs can suppress tumour development by stimulating a T-cell-mediated anti-tumour response, whereas the crosstalk of M2 TAMs with tumour and stellate cells can stimulate secretion of various anti-inflammatory cytokines, and reprogram immune surveillance within the tumour microenvironment to facilitate tumour progression[123].
Stephen et al[124] have identified a role of PAK1 in regulating macrophage spreading and lamellipodial dynamics through the activation of ERK1/2. However, they also found that PAK1 knockout had no impact on migration or chemotaxis of macrophages, whereas another study reported that absence of Rac1 or Rac2 could promote macrophage migration[125]. These observations suggest either that PAK2 might compensate for the lack of PAK1, or that other Rac down-stream effectors are involved in regulating cell migration[126]. In addition, Gringel et al[127] found that PAK4 functioned as a physiological regulator of podosomes, which are involved in the migration of human macrophages. Up-regulated expression and activity of PAK4 and its regulator α-PIX (PAK-interacting exchange factor) enhanced the number and size of macrophage podosomes.
There are some additional potential mechanisms linking PAKs to macrophage migration and chemotaxis. The interaction between PAK and HIF-1α has been well established from previous studies[59,90,91]. Recently, HIF-1α was reported to be involved in the recruitment of TAMs in pancreatic cancer through promoting C-C motif chemokine ligand 2 (CCL2) secretion, which stimulated monocyte infiltration into the tumour microenvironment by binding to its receptor CCR2[128]. In agreement with this report, Sanford et al[129] revealed an important role of CCL2/CCR2 in TAM recruitment by showing that a CCR2 antagonist (PF-04136309) was able to block the migration of circulating CCR2+ monocytes toward the tumour with a consequent depletion of TAMs in a mouse model of pancreatic cancer. Their clinical data also indicated that pancreatic cancer patients with a higher level of CCL2 expression and greater infiltration of immunosuppressive CCR2+ TAMs were significantly more likely to have decreased survival. Additionally, Allen and colleagues revealed the importance of Rho family proteins in regulating actin organization and cell adhesion in macrophages[130]. Using a colony-stimulating factor-1 -dependent murine macrophage cell line (Bac1.2F5), they demonstrated that constitutively activated Rac1 or Cdc42, which are both well-defined up-stream activators of PAKs, could stimulate formation of lamellipodia or filopodia, whereas dominant negative Rac1 or Cdc42 inhibited colony-stimulating factor-1-induced formation of lamellipodia or filopodia.
Macrophage polarization is induced by different stimuli via interferon-regulatory factor/signal transducer and activator of transcription (IRF/STAT) signalling pathways, NF-κB pathways and HIF stabilization. IRF/STAT factors (IRF3, IRF5, STAT1 and STAT5), HIF-1 and the active NF-κB heterodimer (p50-p65) contribute to M1 polarization, while IRF/STAT factors (IRF4, STAT3 and STAT6), HIF-2 and the inhibitory NF-κB heterodimer (p50-p50) trigger an M2 response[131]. The involvement of PAK1 in macrophage polarization has recently been characterized by Zhang and colleagues, who reported that pharmacological or genetic inhibition of PAK1 diminished M1 macrophage polarization. This observation suggested that the up-regulation of PAK1 induced by inflammatory stimuli may contribute to M1 polarization via NF-κB-mediated transcriptional activation. PAK1 was also observed to play a key role in suppressing M2 macrophage polarization[132]. Blockade of the M2 response is an important approach to treatment involving TAM reprogramming. As mentioned above, STAT3 and STAT6 have been reported to be important regulators of M2 polarization. Pharmacological inhibition of STAT3 and STAT6 with specific inhibitors resulted in suppression of M2 polarization, increased production of pro-inflammatory cytokines and stimulated a T cell response[133-135]. Since PAK1 and PAK4 are closely associated with the STAT3 and NF-κB signalling pathway[67,121], it is likely that PAK may interact with STAT3 or NF-κB signalling pathways to block M2 polarization.
Tumour-infiltrating lymphocytes
Tumour-infiltrating lymphocytes (TILs), including CD4+ T cells, CD8+ T cells, regulatory T cells, B cells and natural killer cells, are another class of immune cells, which are critical in modulating the tumour microenvironment in pancreatic cancer[136]. Although CD8+ T cells are also referred to as cytotoxic T cells, with the capability of recognizing and killing tumour cells, infiltration of CD8+ T cells into the tumour microenvironment is very rare. In contrast, a large number of CD4+ T cells, which can promote the development of PanINs via inhibition of the anti-tumour response, are observed in the stromal compartment[108,137]. Indeed, an increasing number of studies have revealed the predictive value of stromal TILs in patients with resectable pancreatic cancer. Two of the latest studies have demonstrated that negative stromal TIL patients had larger tumour at a more advanced stage and showed worse overall survival and liver metastasis[138], and that an increased number of tumour-infiltrated CD8+ lymphocytes were significantly and independently related to improved disease-free survival and overall survival[139]. Recently, it was reported that pharmacological or genetic depletion of PAK1 up-regulated the immune response to tumours in a colorectal mouse model (APC14/+ mouse)[140].Similarly, the role of PAK in regulating the tumour-associated immune response was investigated in an murine orthotopic pancreatic cancer model[66]. In agreement with the colorectal model, removal of PAK1 by knock-out or inhibition of PAK1 by PF-3758309 not only suppressed tumour growth in vivo, but also stimulated the immune response by increasing the numbers of splenic CD3+ and CD8+ T lymphocytes as well as by promoting tumour-infiltrating CD3+ T lymphocytes. In contrast, gemcitabine did not significantly change the tumour-associated immune response. Furthermore, it has been reported that granulocyte-macrophage colony-stimulating factor (GM-CSF) secreted by tumour cells can recruit and stimulate the development of stromal myeloid cells (Gr-1+ CD11b+ cells), which can suppress the anti-tumour effect of CD8+ T cells[141]. So far, the mechanism by which PAK regulates the production of GM-CSF has not been fully elucidated. However, Kras activation is found to be positively associated with GM-CSF expression in cancer patients when compared to normal controls[142], and this observation is consistent with an early study indicating that oncogenic Kras-dependent secretion of GM-CSF can promote the development of pancreatic neoplasia via immunosuppression mediated by Gr-1+ CD11b+ myeloid cells[143]. As Kras is the most important oncogenic mutation in pancreatic cancer and a potent up-stream regulator of PAK, these studies provided possible evidence implicating the involvement of PAK in a pathway linking aberrantly activated Kras to GM-CSF-induced immuno-evasion. The mechanisms underlying the connection between PAKs and GM-CSF should be investigated further.
CONCLUSION
More than a decade ago, an expert consensus proposed precise targeting of protein kinase signalling pathways as a potential weapon against cancers[144,145]. As an important down-stream effector of Kras, PAK is overexpressed and hyperactivated in different types of cancer, especially pancreas, colorectal and lung cancer. This review highlights the key role of PAK in Kras signalling pathways in pancreatic cancer, and summarizes that PAK mediates the biological behaviour of pancreatic cancer cells by orchestrating multiple oncogenic pathways, such as NF-κB, STAT3, RAF/MEK/ERK, PI3K/PDK1/AKT etc., PAK inhibition (FRAX597, PF-3758309 etc.), not only suppresses tumour growth and synergistically improves chemotherapeutic efficacy, but also plays a critical role in mediating tumour-stroma crosstalk. More importantly, immunotherapy is now emerging as a promising approach for cancer treatment and immune modulation within the tumour microenvironment has become a hot spot in pancreatic cancer research. The potential role of PAK in the anti-tumour immune response has been unveiled by showing that pharmacological and genetic depletion of PAK leads to an increased number of tumour infiltrated T cells in pancreatic and colorectal cancer. In this regard, PAK may become a novel target for reprogramming the tumour microenvironment.
Pancreatic cancer is still one of most lethal malignancies and, in contrast to other types of cancers (e.g., melanoma, breast cancer, prostate cancer etc.), the poor survival of pancreatic cancer patients has been only marginally improved over past decades. Therapeutic breakthroughs in pancreatic cancer still require a more comprehensive understanding of its biology and of the intrinsic mechanisms involved in tumour progression. Further study of PAKs holds the promise of developing more effective and less toxic treatments for this devastating malignancy.
ACKNOWLEDGMENTS
The authors would like to acknowledge Pancare Foundation (https://www.pancare.org.au) for supporting the pancreatic cancer research program in the Department of Surgery, University of Melbourne. Kai Wang was supported by Melbourne International Fee Remission Scholarship (MIFRS), Melbourne International Research Scholarship (MIRS) and the Moshe Sambor Scholarship (Pancare Foundation).
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Australia
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B, B, B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Conflict-of-interest statement: No potential conflicts of interest.
Peer-review started: May 23, 2018
First decision: June 11, 2018
Article in press: June 27, 2018
P- Reviewer: Chandra D, Fusai G, Guo JC S- Editor: Wang XJ L- Editor: A E- Editor: Huang Y
Contributor Information
Kai Wang, Department of Surgery, University of Melbourne, Melbourne 3084, Australia.
Graham S Baldwin, Department of Surgery, University of Melbourne, Melbourne 3084, Australia.
Mehrdad Nikfarjam, Department of Surgery, University of Melbourne, Melbourne 3084, Australia.
Hong He, Department of Surgery, University of Melbourne, Melbourne 3084, Australia. hong.he@unimelb.edu.au.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–2921. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 3.Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardière C, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 4.Cunningham D, Chau I, Stocken DD, Valle JW, Smith D, Steward W, Harper PG, Dunn J, Tudur-Smith C, West J, et al. Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. J Clin Oncol. 2009;27:5513–5518. doi: 10.1200/JCO.2009.24.2446. [DOI] [PubMed] [Google Scholar]
- 5.Colucci G, Giuliani F, Gebbia V, Biglietto M, Rabitti P, Uomo G, Cigolari S, Testa A, Maiello E, Lopez M. Gemcitabine alone or with cisplatin for the treatment of patients with locally advanced and/or metastatic pancreatic carcinoma: a prospective, randomized phase III study of the Gruppo Oncologia dell’Italia Meridionale. Cancer. 2002;94:902–910. [PubMed] [Google Scholar]
- 6.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov. 2014;13:828–851. doi: 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lazo JS, Sharlow ER. Drugging Undruggable Molecular Cancer Targets. Annu Rev Pharmacol Toxicol. 2016;56:23–40. doi: 10.1146/annurev-pharmtox-010715-103440. [DOI] [PubMed] [Google Scholar]
- 9.Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging ras back in the ring. Cancer Cell. 2014;25:272–281. doi: 10.1016/j.ccr.2014.02.017. [DOI] [PubMed] [Google Scholar]
- 10.Baines AT, Xu D, Der CJ. Inhibition of Ras for cancer treatment: the search continues. Future Med Chem. 2011;3:1787–1808. doi: 10.4155/fmc.11.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thompson H. US National Cancer Institute’s new Ras project targets an old foe. Nat Med. 2013;19:949–950. doi: 10.1038/nm0813-949. [DOI] [PubMed] [Google Scholar]
- 12.Collins MA, Bednar F, Zhang Y, Brisset JC, Galbán S, Galbán CJ, Rakshit S, Flannagan KS, Adsay NV, Pasca di Magliano M. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest. 2012;122:639–653. doi: 10.1172/JCI59227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18:4266–4276. doi: 10.1158/1078-0432.CCR-11-3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jaffer ZM, Chernoff J. p21-activated kinases: three more join the Pak. Int J Biochem Cell Biol. 2002;34:713–717. doi: 10.1016/s1357-2725(01)00158-3. [DOI] [PubMed] [Google Scholar]
- 15.Baskaran Y, Ng YW, Selamat W, Ling FT, Manser E. Group I and II mammalian PAKs have different modes of activation by Cdc42. EMBO Rep. 2012;13:653–659. doi: 10.1038/embor.2012.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Radu M, Semenova G, Kosoff R, Chernoff J. PAK signalling during the development and progression of cancer. Nat Rev Cancer. 2014;14:13–25. doi: 10.1038/nrc3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manser E, Leung T, Salihuddin H, Zhao ZS, Lim L. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature. 1994;367:40–46. doi: 10.1038/367040a0. [DOI] [PubMed] [Google Scholar]
- 18.Pirruccello M, Sondermann H, Pelton JG, Pellicena P, Hoelz A, Chernoff J, Wemmer DE, Kuriyan J. A dimeric kinase assembly underlying autophosphorylation in the p21 activated kinases. J Mol Biol. 2006;361:312–326. doi: 10.1016/j.jmb.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 19.Lei M, Lu W, Meng W, Parrini MC, Eck MJ, Mayer BJ, Harrison SC. Structure of PAK1 in an autoinhibited conformation reveals a multistage activation switch. Cell. 2000;102:387–397. doi: 10.1016/s0092-8674(00)00043-x. [DOI] [PubMed] [Google Scholar]
- 20.Buchwald G, Hostinova E, Rudolph MG, Kraemer A, Sickmann A, Meyer HE, Scheffzek K, Wittinghofer A. Conformational switch and role of phosphorylation in PAK activation. Mol Cell Biol. 2001;21:5179–5189. doi: 10.1128/MCB.21.15.5179-5189.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parrini MC, Lei M, Harrison SC, Mayer BJ. Pak1 kinase homodimers are autoinhibited in trans and dissociated upon activation by Cdc42 and Rac1. Mol Cell. 2002;9:73–83. doi: 10.1016/s1097-2765(01)00428-2. [DOI] [PubMed] [Google Scholar]
- 22.Qu J, Li X, Novitch BG, Zheng Y, Kohn M, Xie JM, Kozinn S, Bronson R, Beg AA, Minden A. PAK4 kinase is essential for embryonic viability and for proper neuronal development. Mol Cell Biol. 2003;23:7122–7133. doi: 10.1128/MCB.23.20.7122-7133.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Callow MG, Clairvoyant F, Zhu S, Schryver B, Whyte DB, Bischoff JR, Jallal B, Smeal T. Requirement for PAK4 in the anchorage-independent growth of human cancer cell lines. J Biol Chem. 2002;277:550–558. doi: 10.1074/jbc.M105732200. [DOI] [PubMed] [Google Scholar]
- 24.Pandey A, Dan I, Kristiansen TZ, Watanabe NM, Voldby J, Kajikawa E, Khosravi-Far R, Blagoev B, Mann M. Cloning and characterization of PAK5, a novel member of mammalian p21-activated kinase-II subfamily that is predominantly expressed in brain. Oncogene. 2002;21:3939–3948. doi: 10.1038/sj.onc.1205478. [DOI] [PubMed] [Google Scholar]
- 25.Yang F, Li X, Sharma M, Zarnegar M, Lim B, Sun Z. Androgen receptor specifically interacts with a novel p21-activated kinase, PAK6. J Biol Chem. 2001;276:15345–15353. doi: 10.1074/jbc.M010311200. [DOI] [PubMed] [Google Scholar]
- 26.Schrantz N, da Silva Correia J, Fowler B, Ge Q, Sun Z, Bokoch GM. Mechanism of p21-activated kinase 6-mediated inhibition of androgen receptor signaling. J Biol Chem. 2004;279:1922–1931. doi: 10.1074/jbc.M311145200. [DOI] [PubMed] [Google Scholar]
- 27.Ching YP, Leong VY, Wong CM, Kung HF. Identification of an autoinhibitory domain of p21-activated protein kinase 5. J Biol Chem. 2003;278:33621–33624. doi: 10.1074/jbc.C300234200. [DOI] [PubMed] [Google Scholar]
- 28.Ha BH, Davis MJ, Chen C, Lou HJ, Gao J, Zhang R, Krauthammer M, Halaban R, Schlessinger J, Turk BE, et al. Type II p21-activated kinases (PAKs) are regulated by an autoinhibitory pseudosubstrate. Proc Natl Acad Sci USA. 2012;109:16107–16112. doi: 10.1073/pnas.1214447109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72:2457–2467. doi: 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morris JP 4th, Wang SC, Hebrok M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat Rev Cancer. 2010;10:683–695. doi: 10.1038/nrc2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 32.Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–670. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yeo D, He H, Baldwin GS, Nikfarjam M. The role of p21-activated kinases in pancreatic cancer. Pancreas. 2015;44:363–369. doi: 10.1097/MPA.0000000000000276. [DOI] [PubMed] [Google Scholar]
- 34.Eser S, Schnieke A, Schneider G, Saur D. Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer. 2014;111:817–822. doi: 10.1038/bjc.2014.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci. 2014;39:91–100. doi: 10.1016/j.tibs.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Réjiba S, Wack S, Aprahamian M, Hajri A. K-ras oncogene silencing strategy reduces tumor growth and enhances gemcitabine chemotherapy efficacy for pancreatic cancer treatment. Cancer Sci. 2007;98:1128–1136. doi: 10.1111/j.1349-7006.2007.00506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11:761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mortazavi F, Lu J, Phan R, Lewis M, Trinidad K, Aljilani A, Pezeshkpour G, Tamanoi F. Significance of KRAS/PAK1/Crk pathway in non-small cell lung cancer oncogenesis. BMC Cancer. 2015;15:381. doi: 10.1186/s12885-015-1360-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Menard RE, Mattingly RR. Cell surface receptors activate p21-activated kinase 1 via multiple Ras and PI3-kinase-dependent pathways. Cell Signal. 2003;15:1099–1109. doi: 10.1016/s0898-6568(03)00087-1. [DOI] [PubMed] [Google Scholar]
- 40.Malliri A, van der Kammen RA, Clark K, van der Valk M, Michiels F, Collard JG. Mice deficient in the Rac activator Tiam1 are resistant to Ras-induced skin tumours. Nature. 2002;417:867–871. doi: 10.1038/nature00848. [DOI] [PubMed] [Google Scholar]
- 41.Binker MG, Binker-Cosen AA, Gaisano HY, Cosen-Binker LI. Inhibition of Rac1 decreases the severity of pancreatitis and pancreatitis-associated lung injury in mice. Exp Physiol. 2008;93:1091–1103. doi: 10.1113/expphysiol.2008.043141. [DOI] [PubMed] [Google Scholar]
- 42.Heid I, Lubeseder-Martellato C, Sipos B, Mazur PK, Lesina M, Schmid RM, Siveke JT. Early requirement of Rac1 in a mouse model of pancreatic cancer. Gastroenterology. 2011;141:719–730, 730.e1-730.e7. doi: 10.1053/j.gastro.2011.04.043. [DOI] [PubMed] [Google Scholar]
- 43.Zheng Y, Bagrodia S, Cerione RA. Activation of phosphoinositide 3-kinase activity by Cdc42Hs binding to p85. J Biol Chem. 1994;269:18727–18730. [PubMed] [Google Scholar]
- 44.Eser S, Reiff N, Messer M, Seidler B, Gottschalk K, Dobler M, Hieber M, Arbeiter A, Klein S, Kong B, et al. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell. 2013;23:406–420. doi: 10.1016/j.ccr.2013.01.023. [DOI] [PubMed] [Google Scholar]
- 45.King CC, Gardiner EM, Zenke FT, Bohl BP, Newton AC, Hemmings BA, Bokoch GM. p21-activated kinase (PAK1) is phosphorylated and activated by 3-phosphoinositide-dependent kinase-1 (PDK1) J Biol Chem. 2000;275:41201–41209. doi: 10.1074/jbc.M006553200. [DOI] [PubMed] [Google Scholar]
- 46.Beeser A, Jaffer ZM, Hofmann C, Chernoff J. Role of group A p21-activated kinases in activation of extracellular-regulated kinase by growth factors. J Biol Chem. 2005;280:36609–36615. doi: 10.1074/jbc.M502306200. [DOI] [PubMed] [Google Scholar]
- 47.Huynh N, Liu KH, Baldwin GS, He H. P21-activated kinase 1 stimulates colon cancer cell growth and migration/invasion via ERK- and AKT-dependent pathways. Biochim Biophys Acta. 2010;1803:1106–1113. doi: 10.1016/j.bbamcr.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 48.Chow HY, Jubb AM, Koch JN, Jaffer ZM, Stepanova D, Campbell DA, Duron SG, O’Farrell M, Cai KQ, Klein-Szanto AJ, et al. p21-Activated kinase 1 is required for efficient tumor formation and progression in a Ras-mediated skin cancer model. Cancer Res. 2012;72:5966–5975. doi: 10.1158/0008-5472.CAN-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tabusa H, Brooks T, Massey AJ. Knockdown of PAK4 or PAK1 inhibits the proliferation of mutant KRAS colon cancer cells independently of RAF/MEK/ERK and PI3K/AKT signaling. Mol Cancer Res. 2013;11:109–121. doi: 10.1158/1541-7786.MCR-12-0466. [DOI] [PubMed] [Google Scholar]
- 50.Chen S, Auletta T, Dovirak O, Hutter C, Kuntz K, El-ftesi S, Kendall J, Han H, Von Hoff DD, Ashfaq R, et al. Copy number alterations in pancreatic cancer identify recurrent PAK4 amplification. Cancer Biol Ther. 2008;7:1793–1802. doi: 10.4161/cbt.7.11.6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brown LA, Kalloger SE, Miller MA, Shih IeM, McKinney SE, Santos JL, Swenerton K, Spellman PT, Gray J, Gilks CB, Huntsman DG. Amplification of 11q13 in ovarian carcinoma. Genes Chromosomes Cancer. 2008;47:481–489. doi: 10.1002/gcc.20549. [DOI] [PubMed] [Google Scholar]
- 52.Rane CK, Minden A. P21 activated kinases: structure, regulation, and functions. Small GTPases. 2014:5. doi: 10.4161/sgtp.28003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shrestha Y, Schafer EJ, Boehm JS, Thomas SR, He F, Du J, Wang S, Barretina J, Weir BA, Zhao JJ, et al. PAK1 is a breast cancer oncogene that coordinately activates MAPK and MET signaling. Oncogene. 2012;31:3397–3408. doi: 10.1038/onc.2011.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mahlamäki EH, Kauraniemi P, Monni O, Wolf M, Hautaniemi S, Kallioniemi A. High-resolution genomic and expression profiling reveals 105 putative amplification target genes in pancreatic cancer. Neoplasia. 2004;6:432–439. doi: 10.1593/neo.04130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kimmelman AC, Hezel AF, Aguirre AJ, Zheng H, Paik JH, Ying H, Chu GC, Zhang JX, Sahin E, Yeo G, et al. Genomic alterations link Rho family of GTPases to the highly invasive phenotype of pancreas cancer. Proc Natl Acad Sci USA. 2008;105:19372–19377. doi: 10.1073/pnas.0809966105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lundgren K, Holm K, Nordenskjöld B, Borg A, Landberg G. Gene products of chromosome 11q and their association with CCND1 gene amplification and tamoxifen resistance in premenopausal breast cancer. Breast Cancer Res. 2008;10:R81. doi: 10.1186/bcr2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou W, Jubb AM, Lyle K, Xiao Q, Ong CC, Desai R, Fu L, Gnad F, Song Q, Haverty PM, et al. PAK1 mediates pancreatic cancer cell migration and resistance to MET inhibition. J Pathol. 2014;234:502–513. doi: 10.1002/path.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jagadeeshan S, Krishnamoorthy YR, Singhal M, Subramanian A, Mavuluri J, Lakshmi A, Roshini A, Baskar G, Ravi M, Joseph LD, et al. Transcriptional regulation of fibronectin by p21-activated kinase-1 modulates pancreatic tumorigenesis. Oncogene. 2015;34:455–464. doi: 10.1038/onc.2013.576. [DOI] [PubMed] [Google Scholar]
- 59.Yeo D, He H, Patel O, Lowy AM, Baldwin GS, Nikfarjam M. FRAX597, a PAK1 inhibitor, synergistically reduces pancreatic cancer growth when combined with gemcitabine. BMC Cancer. 2016;16:24. doi: 10.1186/s12885-016-2057-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Singh RR, Song C, Yang Z, Kumar R. Nuclear localization and chromatin targets of p21-activated kinase 1. J Biol Chem. 2005;280:18130–18137. doi: 10.1074/jbc.M412607200. [DOI] [PubMed] [Google Scholar]
- 61.Frost JA, Swantek JL, Stippec S, Yin MJ, Gaynor R, Cobb MH. Stimulation of NFkappa B activity by multiple signaling pathways requires PAK1. J Biol Chem. 2000;275:19693–19699. doi: 10.1074/jbc.M909860199. [DOI] [PubMed] [Google Scholar]
- 62.Chauhan SC, Ebeling MC, Maher DM, Koch MD, Watanabe A, Aburatani H, Lio Y, Jaggi M. MUC13 mucin augments pancreatic tumorigenesis. Mol Cancer Ther. 2012;11:24–33. doi: 10.1158/1535-7163.MCT-11-0598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jagadeeshan S, Venkatraman G, Rayala SK. Targeting p21 activated kinase 1 (Pak1) to PAKup Pancreatic Cancer. Expert Opin Ther Targets. 2016;20:1283–1285. doi: 10.1080/14728222.2016.1239719. [DOI] [PubMed] [Google Scholar]
- 64.Yeo D, Phillips P, Baldwin GS, He H, Nikfarjam M. Inhibition of group 1 p21-activated kinases suppresses pancreatic stellate cell activation and increases survival of mice with pancreatic cancer. Int J Cancer. 2017;140:2101–2111. doi: 10.1002/ijc.30615. [DOI] [PubMed] [Google Scholar]
- 65.Tyagi N, Bhardwaj A, Singh AP, McClellan S, Carter JE, Singh S. p-21 activated kinase 4 promotes proliferation and survival of pancreatic cancer cells through AKT- and ERK-dependent activation of NF-κB pathway. Oncotarget. 2014;5:8778–8789. doi: 10.18632/oncotarget.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang K, Huynh N, Wang X, Baldwin G, Nikfarjam M, He H. Inhibition of p21 activated kinase enhances tumour immune response and sensitizes pancreatic cancer to gemcitabine. Int J Oncol. 2018;52:261–269. doi: 10.3892/ijo.2017.4193. [DOI] [PubMed] [Google Scholar]
- 67.Tyagi N, Marimuthu S, Bhardwaj A, Deshmukh SK, Srivastava SK, Singh AP, McClellan S, Carter JE, Singh S. p-21 activated kinase 4 (PAK4) maintains stem cell-like phenotypes in pancreatic cancer cells through activation of STAT3 signaling. Cancer Lett. 2016;370:260–267. doi: 10.1016/j.canlet.2015.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nuche-Berenguer B, Ramos-Álvarez I, Jensen RT. The p21-activated kinase, PAK2, is important in the activation of numerous pancreatic acinar cell signaling cascades and in the onset of early pancreatitis events. Biochim Biophys Acta. 2016;1862:1122–1136. doi: 10.1016/j.bbadis.2016.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Konstantinidis IT, Warshaw AL, Allen JN, Blaszkowsky LS, Castillo CF, Deshpande V, Hong TS, Kwak EL, Lauwers GY, Ryan DP, et al. Pancreatic ductal adenocarcinoma: is there a survival difference for R1 resections versus locally advanced unresectable tumors? What is a “true” R0 resection? Ann Surg. 2013;257:731–736. doi: 10.1097/SLA.0b013e318263da2f. [DOI] [PubMed] [Google Scholar]
- 70.Oettle H, Post S, Neuhaus P, Gellert K, Langrehr J, Ridwelski K, Schramm H, Fahlke J, Zuelke C, Burkart C, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA. 2007;297:267–277. doi: 10.1001/jama.297.3.267. [DOI] [PubMed] [Google Scholar]
- 71.Hammel P, Huguet F, van Laethem JL, Goldstein D, Glimelius B, Artru P, Borbath I, Bouché O, Shannon J, André T, et al. Effect of Chemoradiotherapy vs Chemotherapy on Survival in Patients With Locally Advanced Pancreatic Cancer Controlled After 4 Months of Gemcitabine With or Without Erlotinib: The LAP07 Randomized Clinical Trial. JAMA. 2016;315:1844–1853. doi: 10.1001/jama.2016.4324. [DOI] [PubMed] [Google Scholar]
- 72.Burris HA 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, Nelson R, Dorr FA, Stephens CD, Von Hoff DD. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 73.Rothenberg ML, Moore MJ, Cripps MC, Andersen JS, Portenoy RK, Burris HA 3rd, Green MR, Tarassoff PG, Brown TD, Casper ES, Storniolo AM, Von Hoff DD. A phase II trial of gemcitabine in patients with 5-FU-refractory pancreas cancer. Ann Oncol. 1996;7:347–353. doi: 10.1093/oxfordjournals.annonc.a010600. [DOI] [PubMed] [Google Scholar]
- 74.Di Costanzo F, Carlini P, Doni L, Massidda B, Mattioli R, Iop A, Barletta E, Moscetti L, Recchia F, Tralongo P, et al. Gemcitabine with or without continuous infusion 5-FU in advanced pancreatic cancer: a randomised phase II trial of the Italian oncology group for clinical research (GOIRC) Br J Cancer. 2005;93:185–189. doi: 10.1038/sj.bjc.6602640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Min YJ, Joo KR, Park NH, Yun TK, Nah YW, Nam CW, Park JH. Gemcitabine therapy in patients with advanced pancreatic cancer. Korean J Intern Med. 2002;17:259–262. doi: 10.3904/kjim.2002.17.4.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Poplin E, Wasan H, Rolfe L, Raponi M, Ikdahl T, Bondarenko I, Davidenko I, Bondar V, Garin A, Boeck S, et al. Randomized, multicenter, phase II study of CO-101 versus gemcitabine in patients with metastatic pancreatic ductal adenocarcinoma: including a prospective evaluation of the role of hENT1 in gemcitabine or CO-101 sensitivity. J Clin Oncol. 2013;31:4453–4461. doi: 10.1200/JCO.2013.51.0826. [DOI] [PubMed] [Google Scholar]
- 77.Adamska A, Domenichini A, Falasca M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int J Mol Sci. 2017:18. doi: 10.3390/ijms18071338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Farrell JJ, Elsaleh H, Garcia M, Lai R, Ammar A, Regine WF, Abrams R, Benson AB, Macdonald J, Cass CE, et al. Human equilibrative nucleoside transporter 1 levels predict response to gemcitabine in patients with pancreatic cancer. Gastroenterology. 2009;136:187–195. doi: 10.1053/j.gastro.2008.09.067. [DOI] [PubMed] [Google Scholar]
- 79.Giovannetti E, Del Tacca M, Mey V, Funel N, Nannizzi S, Ricci S, Orlandini C, Boggi U, Campani D, Del Chiaro M, et al. Transcription analysis of human equilibrative nucleoside transporter-1 predicts survival in pancreas cancer patients treated with gemcitabine. Cancer Res. 2006;66:3928–3935. doi: 10.1158/0008-5472.CAN-05-4203. [DOI] [PubMed] [Google Scholar]
- 80.Nakano Y, Tanno S, Koizumi K, Nishikawa T, Nakamura K, Minoguchi M, Izawa T, Mizukami Y, Okumura T, Kohgo Y. Gemcitabine chemoresistance and molecular markers associated with gemcitabine transport and metabolism in human pancreatic cancer cells. Br J Cancer. 2007;96:457–463. doi: 10.1038/sj.bjc.6603559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ritzel MW, Ng AM, Yao SY, Graham K, Loewen SK, Smith KM, Hyde RJ, Karpinski E, Cass CE, Baldwin SA, et al. Recent molecular advances in studies of the concentrative Na+-dependent nucleoside transporter (CNT) family: identification and characterization of novel human and mouse proteins (hCNT3 and mCNT3) broadly selective for purine and pyrimidine nucleosides (system cib) Mol Membr Biol. 2001;18:65–72. doi: 10.1080/09687680010026313. [DOI] [PubMed] [Google Scholar]
- 82.Mackey JR, Mani RS, Selner M, Mowles D, Young JD, Belt JA, Crawford CR, Cass CE. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res. 1998;58:4349–4357. [PubMed] [Google Scholar]
- 83.Moon SU, Kim JW, Sung JH, Kang MH, Kim SH, Chang H, Lee JO, Kim YJ, Lee KW, Kim JH, et al. p21-Activated Kinase 4 (PAK4) as a Predictive Marker of Gemcitabine Sensitivity in Pancreatic Cancer Cell Lines. Cancer Res Treat. 2015;47:501–508. doi: 10.4143/crt.2014.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Park S, Kim JW, Kim H, Kim JW, Kim YJ, Lee KW, Kim JH, Kim JH, Hwang JH, Choi YR, et al. Prognostic value of p21-activated kinase 4 in resected pancreatic cancer. APMIS. 2017;125:699–707. doi: 10.1111/apm.12705. [DOI] [PubMed] [Google Scholar]
- 85.Jagadeeshan S, Subramanian A, Tentu S, Beesetti S, Singhal M, Raghavan S, Surabhi RP, Mavuluri J, Bhoopalan H, Biswal J, et al. P21-activated kinase 1 (Pak1) signaling influences therapeutic outcome in pancreatic cancer. Ann Oncol. 2016;27:1546–1556. doi: 10.1093/annonc/mdw184. [DOI] [PubMed] [Google Scholar]
- 86.Wang R, Cheng L, Xia J, Wang Z, Wu Q, Wang Z. Gemcitabine resistance is associated with epithelial-mesenchymal transition and induction of HIF-1α in pancreatic cancer cells. Curr Cancer Drug Targets. 2014;14:407–417. doi: 10.2174/1568009614666140226114015. [DOI] [PubMed] [Google Scholar]
- 87.Zhao T, Ren H, Jia L, Chen J, Xin W, Yan F, Li J, Wang X, Gao S, Qian D, et al. Inhibition of HIF-1α by PX-478 enhances the anti-tumor effect of gemcitabine by inducing immunogenic cell death in pancreatic ductal adenocarcinoma. Oncotarget. 2015;6:2250–2262. doi: 10.18632/oncotarget.2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Eltzschig HK, Abdulla P, Hoffman E, Hamilton KE, Daniels D, Schönfeld C, Löffler M, Reyes G, Duszenko M, Karhausen J, et al. HIF-1-dependent repression of equilibrative nucleoside transporter (ENT) in hypoxia. J Exp Med. 2005;202:1493–1505. doi: 10.1084/jem.20050177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Morote-Garcia JC, Rosenberger P, Nivillac NM, Coe IR, Eltzschig HK. Hypoxia-inducible factor-dependent repression of equilibrative nucleoside transporter 2 attenuates mucosal inflammation during intestinal hypoxia. Gastroenterology. 2009;136:607–618. doi: 10.1053/j.gastro.2008.10.037. [DOI] [PubMed] [Google Scholar]
- 90.Liu KH, Huynh N, Patel O, Shulkes A, Baldwin G, He H. P21-activated kinase 1 promotes colorectal cancer survival by up-regulation of hypoxia-inducible factor-1α. Cancer Lett. 2013;340:22–29. doi: 10.1016/j.canlet.2013.06.024. [DOI] [PubMed] [Google Scholar]
- 91.Kim H, Woo DJ, Kim SY, Yang EG. p21-activated kinase 4 regulates HIF-1α translation in cancer cells. Biochem Biophys Res Commun. 2017;486:270–276. doi: 10.1016/j.bbrc.2017.03.024. [DOI] [PubMed] [Google Scholar]
- 92.Arlt A, Gehrz A, Müerköster S, Vorndamm J, Kruse ML, Fölsch UR, Schäfer H. Role of NF-kappaB and Akt/PI3K in the resistance of pancreatic carcinoma cell lines against gemcitabine-induced cell death. Oncogene. 2003;22:3243–3251. doi: 10.1038/sj.onc.1206390. [DOI] [PubMed] [Google Scholar]
- 93.Kong R, Sun B, Jiang H, Pan S, Chen H, Wang S, Krissansen GW, Sun X. Downregulation of nuclear factor-kappaB p65 subunit by small interfering RNA synergizes with gemcitabine to inhibit the growth of pancreatic cancer. Cancer Lett. 2010;291:90–98. doi: 10.1016/j.canlet.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 94.Skrypek N, Duchêne B, Hebbar M, Leteurtre E, van Seuningen I, Jonckheere N. The MUC4 mucin mediates gemcitabine resistance of human pancreatic cancer cells via the Concentrative Nucleoside Transporter family. Oncogene. 2013;32:1714–1723. doi: 10.1038/onc.2012.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fu X, Feng J, Zeng D, Ding Y, Yu C, Yang B. PAK4 confers cisplatin resistance in gastric cancer cells via PI3K/Akt- and MEK/ERK-dependent pathways. Biosci Rep. 2014:34. doi: 10.1042/BSR20130102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Neesse A, Michl P, Frese KK, Feig C, Cook N, Jacobetz MA, Lolkema MP, Buchholz M, Olive KP, Gress TM, et al. Stromal biology and therapy in pancreatic cancer. Gut. 2011;60:861–868. doi: 10.1136/gut.2010.226092. [DOI] [PubMed] [Google Scholar]
- 97.Vonlaufen A, Joshi S, Qu C, Phillips PA, Xu Z, Parker NR, Toi CS, Pirola RC, Wilson JS, Goldstein D, et al. Pancreatic stellate cells: partners in crime with pancreatic cancer cells. Cancer Res. 2008;68:2085–2093. doi: 10.1158/0008-5472.CAN-07-2477. [DOI] [PubMed] [Google Scholar]
- 98.Martin K, Pritchett J, Llewellyn J, Mullan AF, Athwal VS, Dobie R, Harvey E, Zeef L, Farrow S, Streuli C, et al. PAK proteins and YAP-1 signalling downstream of integrin beta-1 in myofibroblasts promote liver fibrosis. Nat Commun. 2016;7:12502. doi: 10.1038/ncomms12502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, Qi YP, Gysin S, Fernández-del Castillo C, Yajnik V, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425:851–856. doi: 10.1038/nature02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bailey JM, Swanson BJ, Hamada T, Eggers JP, Singh PK, Caffery T, Ouellette MM, Hollingsworth MA. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin Cancer Res. 2008;14:5995–6004. doi: 10.1158/1078-0432.CCR-08-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Huang FT, Zhuan-Sun YX, Zhuang YY, Wei SL, Tang J, Chen WB, Zhang SN. Inhibition of hedgehog signaling depresses self-renewal of pancreatic cancer stem cells and reverses chemoresistance. Int J Oncol. 2012;41:1707–1714. doi: 10.3892/ijo.2012.1597. [DOI] [PubMed] [Google Scholar]
- 104.Xu M, Li L, Liu Z, Jiao Z, Xu P, Kong X, Huang H, Zhang Y. ABCB2 (TAP1) as the downstream target of SHH signaling enhances pancreatic ductal adenocarcinoma drug resistance. Cancer Lett. 2013;333:152–158. doi: 10.1016/j.canlet.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 105.Nakashima H, Nakamura M, Yamaguchi H, Yamanaka N, Akiyoshi T, Koga K, Yamaguchi K, Tsuneyoshi M, Tanaka M, Katano M. Nuclear factor-kappaB contributes to hedgehog signaling pathway activation through sonic hedgehog induction in pancreatic cancer. Cancer Res. 2006;66:7041–7049. doi: 10.1158/0008-5472.CAN-05-4588. [DOI] [PubMed] [Google Scholar]
- 106.Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. 2010;16:2927–2931. doi: 10.1158/1078-0432.CCR-09-2329. [DOI] [PubMed] [Google Scholar]
- 107.Singh AP, Arora S, Bhardwaj A, Srivastava SK, Kadakia MP, Wang B, Grizzle WE, Owen LB, Singh S. CXCL12/CXCR4 protein signaling axis induces sonic hedgehog expression in pancreatic cancer cells via extracellular regulated kinase- and Akt kinase-mediated activation of nuclear factor κB: implications for bidirectional tumor-stromal interactions. J Biol Chem. 2012;287:39115–39124. doi: 10.1074/jbc.M112.409581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67:9518–9527. doi: 10.1158/0008-5472.CAN-07-0175. [DOI] [PubMed] [Google Scholar]
- 109.Roghanian A, Fraser C, Kleyman M, Chen J. B Cells Promote Pancreatic Tumorigenesis. Cancer Discov. 2016;6:230–232. doi: 10.1158/2159-8290.CD-16-0100. [DOI] [PubMed] [Google Scholar]
- 110.Inman KS, Francis AA, Murray NR. Complex role for the immune system in initiation and progression of pancreatic cancer. World J Gastroenterol. 2014;20:11160–11181. doi: 10.3748/wjg.v20.i32.11160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lutz ER, Wu AA, Bigelow E, Sharma R, Mo G, Soares K, Solt S, Dorman A, Wamwea A, Yager A, et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol Res. 2014;2:616–631. doi: 10.1158/2326-6066.CIR-14-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhan HX, Zhou B, Cheng YG, Xu JW, Wang L, Zhang GY, Hu SY. Crosstalk between stromal cells and cancer cells in pancreatic cancer: New insights into stromal biology. Cancer Lett. 2017;392:83–93. doi: 10.1016/j.canlet.2017.01.041. [DOI] [PubMed] [Google Scholar]
- 113.Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother. 2011;60:1419–1430. doi: 10.1007/s00262-011-1028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Khaled YS, Ammori BJ, Elkord E. Increased levels of granulocytic myeloid-derived suppressor cells in peripheral blood and tumour tissue of pancreatic cancer patients. J Immunol Res. 2014;2014:879897. doi: 10.1155/2014/879897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Welte T, Kim IS, Tian L, Gao X, Wang H, Li J, Holdman XB, Herschkowitz JI, Pond A, Xie G, et al. Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumour initiation. Nat Cell Biol. 2016;18:632–644. doi: 10.1038/ncb3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.He LF, Xu HW, Chen M, Xian ZR, Wen XF, Chen MN, Du CW, Huang WH, Wu JD, Zhang GJ. Activated-PAK4 predicts worse prognosis in breast cancer and promotes tumorigenesis through activation of PI3K/AKT signaling. Oncotarget. 2017;8:17573–17585. doi: 10.18632/oncotarget.7466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Khare V, Dammann K, Asboth M, Krnjic A, Jambrich M, Gasche C. Overexpression of PAK1 promotes cell survival in inflammatory bowel diseases and colitis-associated cancer. Inflamm Bowel Dis. 2015;21:287–296. doi: 10.1097/MIB.0000000000000281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chen X, Kelemen SE, Autieri MV. Expression of granulocyte colony-stimulating factor is induced in injured rat carotid arteries and mediates vascular smooth muscle cell migration. Am J Physiol Cell Physiol. 2005;288:C81–C88. doi: 10.1152/ajpcell.00322.2004. [DOI] [PubMed] [Google Scholar]
- 119.Panni RZ, Sanford DE, Belt BA, Mitchem JB, Worley LA, Goetz BD, Mukherjee P, Wang-Gillam A, Link DC, Denardo DG, et al. Tumor-induced STAT3 activation in monocytic myeloid-derived suppressor cells enhances stemness and mesenchymal properties in human pancreatic cancer. Cancer Immunol Immunother. 2014;63:513–528. doi: 10.1007/s00262-014-1527-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, Young GS, Fuchs JR, Eubank TD, Frankel WL, Bekaii-Saab T, et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res. 2013;73:3007–3018. doi: 10.1158/0008-5472.CAN-12-4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gan J, Ke X, Jiang J, Dong H, Yao Z, Lin Y, Lin W, Wu X, Yan S, Zhuang Y, et al. Growth hormone-releasing hormone receptor antagonists inhibit human gastric cancer through downregulation of PAK1-STAT3/NF-κB signaling. Proc Natl Acad Sci USA. 2016;113:14745–14750. doi: 10.1073/pnas.1618582114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Engblom C, Pfirschke C, Pittet MJ. The role of myeloid cells in cancer therapies. Nat Rev Cancer. 2016;16:447–462. doi: 10.1038/nrc.2016.54. [DOI] [PubMed] [Google Scholar]
- 123.Hu H, Jiao F, Han T, Wang LW. Functional significance of macrophages in pancreatic cancer biology. Tumour Biol. 2015;36:9119–9126. doi: 10.1007/s13277-015-4127-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Smith SD, Jaffer ZM, Chernoff J, Ridley AJ. PAK1-mediated activation of ERK1/2 regulates lamellipodial dynamics. J Cell Sci. 2008;121:3729–3736. doi: 10.1242/jcs.027680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wheeler AP, Wells CM, Smith SD, Vega FM, Henderson RB, Tybulewicz VL, Ridley AJ. Rac1 and Rac2 regulate macrophage morphology but are not essential for migration. J Cell Sci. 2006;119:2749–2757. doi: 10.1242/jcs.03024. [DOI] [PubMed] [Google Scholar]
- 126.Weiss-Haljiti C, Pasquali C, Ji H, Gillieron C, Chabert C, Curchod ML, Hirsch E, Ridley AJ, Hooft van Huijsduijnen R, Camps M, et al. Involvement of phosphoinositide 3-kinase gamma, Rac, and PAK signaling in chemokine-induced macrophage migration. J Biol Chem. 2004;279:43273–43284. doi: 10.1074/jbc.M402924200. [DOI] [PubMed] [Google Scholar]
- 127.Gringel A, Walz D, Rosenberger G, Minden A, Kutsche K, Kopp P, Linder S. PAK4 and alphaPIX determine podosome size and number in macrophages through localized actin regulation. J Cell Physiol. 2006;209:568–579. doi: 10.1002/jcp.20777. [DOI] [PubMed] [Google Scholar]
- 128.Li N, Li Y, Li Z, Huang C, Yang Y, Lang M, Cao J, Jiang W, Xu Y, Dong J, et al. Hypoxia Inducible Factor 1 (HIF-1) Recruits Macrophage to Activate Pancreatic Stellate Cells in Pancreatic Ductal Adenocarcinoma. Int J Mol Sci. 2016:17. doi: 10.3390/ijms17060799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sanford DE, Belt BA, Panni RZ, Mayer A, Deshpande AD, Carpenter D, Mitchem JB, Plambeck-Suess SM, Worley LA, Goetz BD, et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin Cancer Res. 2013;19:3404–3415. doi: 10.1158/1078-0432.CCR-13-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Allen WE, Jones GE, Pollard JW, Ridley AJ. Rho, Rac and Cdc42 regulate actin organization and cell adhesion in macrophages. J Cell Sci. 1997;110:707–720. doi: 10.1242/jcs.110.6.707. [DOI] [PubMed] [Google Scholar]
- 131.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889–896. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 132.Zhang W, Liu H, Liu W, Liu Y, Xu J. Polycomb-mediated loss of microRNA let-7c determines inflammatory macrophage polarization via PAK1-dependent NF-κB pathway. Cell Death Differ. 2015;22:287–297. doi: 10.1038/cdd.2014.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hussain SF, Kong LY, Jordan J, Conrad C, Madden T, Fokt I, Priebe W, Heimberger AB. A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res. 2007;67:9630–9636. doi: 10.1158/0008-5472.CAN-07-1243. [DOI] [PubMed] [Google Scholar]
- 134.Dong R, Gong Y, Meng W, Yuan M, Zhu H, Ying M, He Q, Cao J, Yang B. The involvement of M2 macrophage polarization inhibition in fenretinide-mediated chemopreventive effects on colon cancer. Cancer Lett. 2017;388:43–53. doi: 10.1016/j.canlet.2016.11.029. [DOI] [PubMed] [Google Scholar]
- 135.Sun L, Chen B, Jiang R, Li J, Wang B. Resveratrol inhibits lung cancer growth by suppressing M2-like polarization of tumor associated macrophages. Cell Immunol. 2017;311:86–93. doi: 10.1016/j.cellimm.2016.11.002. [DOI] [PubMed] [Google Scholar]
- 136.Nielsen MF, Mortensen MB, Detlefsen S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J Gastroenterol. 2016;22:2678–2700. doi: 10.3748/wjg.v22.i9.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Neesse A, Algül H, Tuveson DA, Gress TM. Stromal biology and therapy in pancreatic cancer: a changing paradigm. Gut. 2015;64:1476–1484. doi: 10.1136/gutjnl-2015-309304. [DOI] [PubMed] [Google Scholar]
- 138.Lianyuan T, Dianrong X, Chunhui Y, Zhaolai M, Bin J. The predictive value and role of stromal tumor-infiltrating lymphocytes in pancreatic ductal adenocarcinoma (PDAC) Cancer Biol Ther. 2018;19:296–305. doi: 10.1080/15384047.2017.1416932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lohneis P, Sinn M, Bischoff S, Jühling A, Pelzer U, Wislocka L, Bahra M, Sinn BV, Denkert C, Oettle H, et al. Cytotoxic tumour-infiltrating T lymphocytes influence outcome in resected pancreatic ductal adenocarcinoma. Eur J Cancer. 2017;83:290–301. doi: 10.1016/j.ejca.2017.06.016. [DOI] [PubMed] [Google Scholar]
- 140.Huynh N, Wang K, Yim M, Dumesny CJ, Sandrin MS, Baldwin GS, Nikfarjam M, He H. Depletion of p21-activated kinase 1 up-regulates the immune system of APC ∆14/+ mice and inhibits intestinal tumorigenesis. BMC Cancer. 2017;17:431. doi: 10.1186/s12885-017-3432-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, Vonderheide RH. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21:822–835. doi: 10.1016/j.ccr.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Petanidis S, Anestakis D, Argyraki M, Hadzopoulou-Cladaras M, Salifoglou A. Differential expression of IL-17, 22 and 23 in the progression of colorectal cancer in patients with K-ras mutation: Ras signal inhibition and crosstalk with GM-CSF and IFN-γ. PLoS One. 2013;8:e73616. doi: 10.1371/journal.pone.0073616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21:836–847. doi: 10.1016/j.ccr.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Keen N, Taylor S. Aurora-kinase inhibitors as anticancer agents. Nat Rev Cancer. 2004;4:927–936. doi: 10.1038/nrc1502. [DOI] [PubMed] [Google Scholar]
- 145.Melnikova I, Golden J. Targeting protein kinases. Nat Rev Drug Discov. 2004;3:993–994. doi: 10.1038/nrd1600. [DOI] [PubMed] [Google Scholar]