

Abstract
AIM
To explore the correlation of metabolomics profiles of gastric cancer (GC) with its chromosomal instability (CIN) status.
METHODS
Nineteen GC patients were classified as CIN and non-CIN type by The Cancer Genome Atlas Research Group system, based on 409 oncogenes and tumor suppressor genes sequenced. The aqueous metabolites of the GC tumor and its surrounding adjacent healthy tissues were identified through liquid chromatography-mass spectrometry. Groups were compared by defining variable importance in projection score of > 1.2, a fold change value or its reciprocal of > 1.2, and a P value of < 0.05 as a significant difference.
RESULTS
In total, twelve men and seven women were enrolled, with a median age of 66 years (range, 47-87 years). The numbers of gene alterations in the CIN GC group were significantly higher than those in the non-CIN GC (32-218 vs 2-17; P < 0.0005). Compared with the adjacent healthy tissues, GC tumors demonstrated significantly higher aspartic acid, citicoline, glutamic acid, oxidized glutathione, succinyladenosine, and uridine diphosphate-N-acetylglucosamine levels, but significantly lower butyrylcarnitine, glutathione hydroxyhexanoycarnitine, inosinic acid, isovalerylcarnitine, and threonine levels (all P < 0.05). CIN tumors contained significantly higher phosphocholine and uridine 5’-monophosphate levels but significantly lower beta-citryl-L-glutamic acid levels than did non-CIN tumors (all P < 0.05). CIN GC tumors demonstrated additional altered pathways involving alanine, aspartate, and glutamate metabolism, glyoxylate and dicarboxylate metabolism, histidine metabolism, and phenylalanine, tyrosine, and tryptophan biosynthesis.
CONCLUSION
Metabolomic profiles of GC tumors and the adjacent healthy tissue are distinct, and the CIN status is associated with downstream metabolic alterations in GC.
Keywords: Gastric cancer, Metabolomics, Oncogene, Copy-number, Chromosomal instability, Liquid chromatography-mass spectrometry
Core tip: We studied the correlation of the comprehensive metabolomic profiles of gastric cancer with its chromosomal instability (CIN) status. In this disease landscape study with no pre-specified hypothesis, we combined a gene molecule classification method with a metabolomics method to discover metabolic information for accurate tumor classification. CIN status-based metabolomic profiling has demonstrated translational potential in biomarker discovery and novel therapeutics development.
INTRODUCTION
Gastric cancer (GC) is one of the most common malignancies worldwide[1,2], with the highest incidence rates in Asia[3]. Most patients with GC are diagnosed in the advanced stage and thus have poor prognosis and limited treatment options[4]. GC can be histologically classified using the Lauren classification system, developed in 1965, which divides GC into diffuse and intestinal subtypes[5]. Although this classification system can suggest surgical choices, it cannot provide precise information of treatments suitable for individual patients[6]. In addition to histological subtypes, the clinicopathological characteristics of GC vary from case to case, making it difficult to identify detailed subtypes and to choose a subtype-optimized therapeutic approach[7].
The Cancer Genome Atlas (TCGA) Research Group has developed molecular classification systems based on gene expression profiling[8]. The TCGA network has provided sequencing- and array-based approaches to investigate exome sequences, copy-number alterations, gene expression, DNA methylation, and protein activities in GC[7,9] and classified GC into four subtypes: Epstein Barr Virus positive, microsatellite unstable, chromosomal instability (CIN), and genomically stable (GS)[8]. The GS and CIN subtypes are the most common and are distinguished by low-vs-high somatic copy-number variation. In general, GS tumors are of the Lauren diffuse subtype; they are typically diagnosed at a young age, and thus have insufficient time to accumulate mutations[10]. By definition, CIN tumors have a high degree of somatic copy-number variation, and they account for nearly half of all GCs, making them the predominant cancer subtype in the gastroesophageal junction or cardia[11]. CIN GCs are the most common Lauren intestinal subtype; however, molecularly and histologically, CIN GCs comprise a highly heterogeneous group of tumors. In addition, an easy-to-use biomarker for CIN is still not available[12].
Metabolomics may offer practical solutions to the traditional methods for GC detection and treatment[13]. Metabolites are not merely the end product of gene expression; they are the result of the interaction of the system’s genome with its environment. They are an integral part of any cellular regulatory system[9]. Liquid chromatography-mass spectrometry (LC-MS) is the most common method of analysis. Several biomarkers have been proposed for GC diagnosis, prognosis, and surveillance[14-16]. Systematic reviews have demonstrated variation in the relative abundance of the metabolites of glycolysis, lactic acid fermentation, de novo lipid, and amino acid synthesis in biological samples of patients with GC compared with controls[13,17,18]. Glutamine is the most consistent biomarker, showing upregulation in the serum, urine, and tumor tissues of patients with GC[18]. Thus far, numerous biomarkers discovered from metabolomic studies may play a noteworthy role in GC with regard to early-stage detection, diagnosis, prognosis, drug development, and chemosensitivity predictions[13], but the evidence of metabolomics’ association with CIN status remains lacking.
We hypothesize that metabolic alternations reflect the CIN or GS status of GC and aim to study the comprehensive metabolomic profiles of GC and correlate them to CIN status.
MATERIALS AND METHODS
Patients and histopathology
This was a disease landscape study with no pre-specified hypothesis. The institutional review board approved the protocol of this prospective study (IRB103-7448B), and informed consents were obtained in a tertiary referral center with a dedicated GC interdisciplinary team to screen patient enrollment. From May 2015 to April 2017, we screened a consecutive cohort of patients with GC. The inclusion criteria were (1) histologically confirmed adenocarcinoma of the stomach, (2) surgical resection of primary GC, (3) age of 20-80 years, and complete pathological, surgical, treatment, and follow-up data. Exclusion criteria were (1) patients received neoadjuvant chemotherapy or chemoradiation therapy, (2) tumor size < 1 cm on computed tomography (CT), (3) prior gastric surgery, (4) anti-Helicobacter pylori eradication therapy, and (5) taking nonsteroidal anti-inflammatory drugs within 1 wk prior to surgery. A gastrointestinal pathologist (RCW) reviewed hematoxylin and eosin stain slides to select cases with estimated carcinoma. We used primary GC tissues for the genomic analysis and reevaluated the pathological diagnosis, histological Lauren subtype, invasion depth, and lymphovascular invasion in all tumors.
Genomics
Patient samples were classified as CIN or non-CIN type by the TCGA system. Genomic DNA was extracted from formalin-fixed paraffin-embedded (FFPE) tumor samples using the QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany), and quantified using the Quant-iT dsDNA HS Assay (Invitrogen, Waltham, MA, United States). Genomic DNA (80 ng) was amplified using four pools of 15992 primer pairs (Ion AmpliSeq Comprehensive Cancer Panel, Life Technologies, Carlsbad, CA, United States) to target the coding exon regions of 409 cancer-related genes, which covered TP53/cell cycle, JAK/STAT, Ras/PI3K, Wnt, receptor tyrosine kinase, chromatin remodeling, DNA repair, TGF, and cadherin signaling. We classified patients with GC by tumor using the high and low proportion of the altered genes. The 409 oncogene and tumor suppressor genes in the GC tumor tissue were sequenced (Supplementary Table 1).
Hydrophilic metabolite extraction
Here, a modified Folch’s method was employed[19]. In brief, 50 mg of homogenized tissue was transferred to a glass tube, to which 6 mL of chloroform/methanol (2:1, v/v) solution and 1.5 mL of water were added. The sample was vortexed four times for 30 s each and subsequently centrifuged at 700 × g for 30 min at 4 °C. The upper phase (the hydrophilic phase and water soluble phase) and lower phase (the hydrophobic phase and lipid layer) were transferred to new glass tubes and then dried using nitrogen gas. The dried samples were stored at -80 °C. Prior to analysis, the sample was dissolved in 200 μL of 40% methanol.
Global analysis of hydrophilic metabolites by LC-TOF-MS
Liquid chromatographic separation was achieved on a 100 × 2.1 mm2 Acquity 1.7 μm C8 column (Waters, Milford, MA, United States) using an ACQUITY Ultra Performance Liquid Chromatography system (Waters, Milford, MA, United States). The column was maintained at 45 °C and at a flow rate of 0.5 mL/min. Samples were eluted from the liquid chromatography column using a linear gradient of: 0-2 min: 1%-80% B; 2-6.5 min: 80%-99% B; 6.5-8.0 min: 99% B; 8.1-10 min: 1% B for re-equilibration. Solvent A was water and solvent B was acetonitrile, and both contained 0.1% formic acid. The lyophilized sample was diluted with 200 μL of water/acetonitrile (95:5, v/v). Each sample was analyzed six times. MS was performed on Waters Q TOF-MS (SYNAPT HDMS; Waters MS Technologies, United Kingdom) operated in electrospray ionization (ESI)-positive ion mode. The desolvation gas was set to 700 L/h at 300 °C, the cone gas at 25 L/h, and the source temperature at 80 °C. The capillary and cone voltages were set to 3000 and 35 V, respectively. The micro-channel plate detector voltage was set to 1700 V. All analyses were acquired using a lock spray to ensure accuracy and reproducibility; sulfadimethoxine was used as the lock mass.
Metabolite identification
We analyzed the aqueous metabolites of the GC tumor and its surrounding adjacent healthy tissues through ESI+/− LC-MS by applying an untargeted metabolic approach to screen all potential metabolomic biomarkers[20]. Identified metabolites demonstrating notable differences between the control and test groups were searched against the METLIN database and the Human Metabolome Database (HMDB) by the m/z of their features. Potential metabolites were confirmed by local database according to the m/z and retention time under identical chromatographic conditions. MS and MS/MS analyses were performed under identical conditions. MS/MS spectra were collected at 10 spectra/s, with a medium isolation window of approximately 4 m/z. The collision energy was set from 5 to 35 V. Several metabolites for the MS/MS analysis were confirmed through chemical standards under chromatographic conditions identical to those of the profiling experiment.
Data processing and statistical analysis
All MS data, namely retention times, m/z, and ion intensities, were extracted using MarkerLynx XS software (Waters, Milford, MA, United States) and inserted into a matrix. Metabolites were searched against the HMDB (http://www.hmdb.ca) or confirmed by in-house data (standards based on both retention times and MS spectra). The data were then analyzed through principal component analysis and partial least squares discriminate analysis (PLS-DA) using Markerlyn XS (Waters, Milford, MA, United States) and Metaboanalyst 3 (http://www.metaboanalyst.ca).
The variable importance in the projection (VIP) value of each variable in the model was calculated to indicate its contribution to the classification. A higher VIP value represented a stronger contribution to discrimination among the groups. VIP values > 1.2 were considered significant. The results are expressed as the mean ± standard deviation for continuous variables and as the number (percent) for categorical variables. Data were compared by 2-sample or paired Student’s t test, analysis of variance, or chi-square test, when appropriate. A P value of < 0.05 was considered significant (Supplementary Tables 2 and 3).
RESULTS
Patient demographics
In total, 19 patients with GC enrolled in this study (median age, 66 years; range, 47-87 years) were divided into CIN and non-CIN types by using a 5% frequency of genetic variation as a demarcation point. We prospectively enrolled these 19 patients in a continuous cohort. No noteworthy difference in demographics was observed between the two groups (Table 1).
Table 1.
Clinical characteristics of the study
| Term |
TCGA system |
P value |
|
| CIN | non-CIN | (CIN vs non-CIN) | |
| Number | 9 | 10 | |
| Age (median yr, range) | 68.1 (56-79) | 64.7 (47-87) | 0.485 |
| Sex (male/female) | 7/2 | 5/5 | 0.233 |
| Size (cm) | 4.0 (1.8-6.9) | 5.4 (2.3-11.6) | 0.297 |
| Stage | |||
| I | 1 | 0 | |
| II | 2 | 1 | |
| III | 5 | 8 | |
| IV | 1 | 1 | |
TCGA: The Cancer Genome Atlas; CIN: Chromosomal instability.
Metabolic alterations of GC tumors vs adjacent healthy tissues
The PLS-DA results of metabolite concentration distribution in the GC tumors and their surrounding healthy tissues are illustrated in Figure 1A and B. Compared with adjacent healthy tissue, GC tumors demonstrated significantly higher levels of butyrylcarnitine, hydroxyhexanoycarnitine, inosinic acid, isovalerylcarnitine, and threonine, but significantly lower levels of aspartic acid, citicoline, glutamic acid, glutamine, isoleucine, oxidized glutathione, proline, succinyladenosine, and xanthine (all P < 0.05; Table 2).
Figure 1.
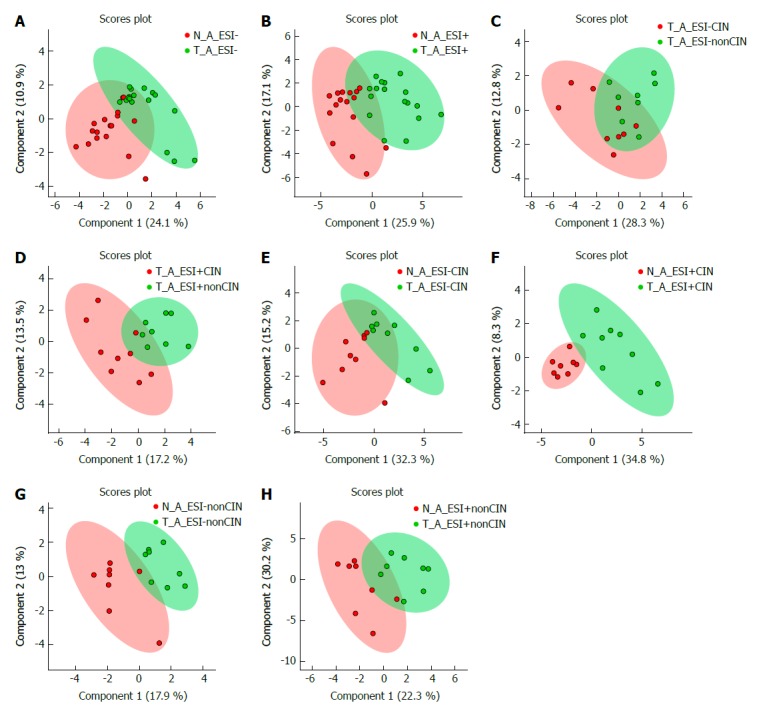
Metabolite-concentration distribution of gastric cancer tumor and its surrounding healthy tissue using the partial least squares discriminate analysis statistical method. A and B: Tumor (green, n = 19) vs healthy tissue (red, n = 19); C: and D: Tumor between chromosomal instability (CIN) type (red, n = 9) and non-CIN type (green, n = 10); E and F: Tumor (green, n = 9) and healthy tissue (red, n = 9) in the CIN type; G and H: Tumor (green, n = 10) and healthy tissue (red, n = 10) in the non-CIN type. A/C/E/G and B/D/F/H defined metabolites from ESI- and ESI+ LC-MS. Arrows indicate the variation in the two groups of metabolite distribution. The straight line indicates the difference of metabolite distribution between two patient types. Within the ellipse was the 95% confidence region. CIN: Chromosomal instability; ESI: Electrospray ionization; LC-MS: Liquid chromatography-mass spectrometry.
Table 2.
Altered metabolites with statistical significance between gastric cancer tumor tissue and its adjacent healthy tissue
| Metabolites | VIP score (VIP > 1.2) | Fold change (tumor/healthy > 1.2 or < 0.8) | P value (P < 0.05) | |
| Increased | Butyrylcarnitine | 1.204 | 1.498 | 0.002 |
| Hydroxyhexanoycarnitine | 2.021 | 2.342 | 0.002 | |
| Inosinic acid | 1.218 | 1.720 | 0.042 | |
| Isovalerylcarnitine | 1.424 | 2.265 | 0.006 | |
| Threonine | 1.316 | 1.911 | 0.019 | |
| Decreased | Aspartic acid | 1.534 | 0.544 | 0.004 |
| Citicoline | 1.973 | 0.371 | 0.005 | |
| Glutamic acid | 1.356 | 0.536 | 0.001 | |
| Glutamine | 1.221 | 0.518 | 0.005 | |
| Isoleucine | 1.323 | 0.593 | 0.015 | |
| Oxidized glutathione | 1.591 | 0.54 | 0.008 | |
| Proline | 1.231 | 0.574 | 0.001 | |
| Succinyladenosine | 1.855 | 0.293 | 0.001 | |
| Xanthine | 1.966 | 0.443 | 0.002 | |
VIP: Variable importance in the projection.
Metabolomic profiling of CIN vs non-CIN GC tumors
Among the 409 genes, GC patients exhibited had the highest proportion (53%) of mutations in TP53, followed by JAK2, PSIP1, and PTPRO (47%; Table 3). In the TCGA classification, the proportion of TP53 mutations in the CIN-type population was 1.6-fold higher than that in the non-CIN type population. In the CIN GC group, all patients had alterations in JAK2, PSIP1, and PTPRD (Table 3).
Table 3.
Altered genes in patients with gastric cancer
| Patients (%) | Alteration gene | |
| All | 53 | TP53 |
| 47 | JAK2, PSIP1, PTPRO | |
| 37 | ARID1A, CDH20, CDKN2A, DCC,ITGB2, MALT1, SYNE1, KMT2C | |
| 32 | AFF1, CDH1, CDH11, CDH2, CDH5, CDKN2B, LRP1B, MBD1, MMP2, SMAD2, SMAD4, ZNF521 | |
| 26 | AKAP9, APC, ATM, BCL2, CYLD, EP400, ERG, FANCA, FN1, IL2, LTF, MAF, MTOR, PBRM1, RUNX1, SAMD9, SMARCA4, TAF1L, TCF12, TLR4, TNK2 | |
| < 21 | 364 genes | |
| CIN tumor | 100 | JAK2, PSIP1, PTPRD |
| 78 | CDH20, CDKN2A, DCC, MALT1 | |
| 67 | TP53, AFF1, CDH1, CDH11, CDH2, CDH5, CDKN2B, MBD1, MMP2, SMAD2 | |
| 56 | ITGB2, ZNF521, ATM, BCL2, CYLD, ERG, FANCA, IL2, MAF, MTOR, PBRM1, RUNX1, TCF12, TLR4 | |
| 44 | 43 genes | |
| 33 | 70 genes | |
| < 22 | 265 genes | |
| Non-CIN tumor | > 50 | None |
| 40 | TP53, SYNE1 | |
| 30 | ARID1A | |
| 20 | ITGB2, KMT2C, LRP1B, AKAP9, SAMD9, SMARCA4, TNK2, RNF213, EPHB6, ERBB3 | |
| 10 | 50 genes | |
| 0 | 346 genes |
GC: Gastric cancer; CIN: Chromosomal instability.
The numbers of gene alterations in the CIN GC group were significantly higher than those in the non-CIN GC (32-218 vs 2-17; P < 0.0005). CIN GC tumors contained significantly higher levels of phosphocholine and uridine 5′-monophosphate (P < 0.05) and a significantly lower level of beta-citryl-L-glutamic acid (P < 0.05) than non-CIN GC tumors (Table 4). Compared with adjacent healthy tissues, 12 and six metabolites in CIN and non-CIN GC were significantly different, respectively (Table 5). CIN and non-CIN tumors demonstrated changes in glutamic acid, oxidized glutathione, and succinyladenosine levels, indicating alterations in aminoacyl-tRNA biosynthesis, arginine and proline metabolism, glutamine and glutamate metabolism, and glutathione metabolism. The metabolite distributions in GC tumors between CIN and non-CIN GC could be clearly distinguished (Figure 1C and D). CIN GC tumors demonstrated additional altered pathways involving alanine, aspartate, and glutamate metabolism, glyoxylate and dicarboxylate metabolism, histidine metabolism, and phenylalanine, tyrosine, and tryptophan biosynthesis.
Table 4.
Altered metabolites with statistical significance between tumor and non-cancerous tissue
| Metabolites | VIP score (VIP > 1.2) | Fold change (CIN/non-CIN > 1.2 or < 0.8) | P value (P < 0.05) | |
| Tumor tissue | Beta-citryl-L-glutamic acid | 1.859 | 0.567 | 0.040 |
| Phosphocholine | 1.998 | 2.093 | 0.017 | |
| Uridine 5'-monophosphate | 1.677 | 1.632 | 0.032 | |
| Non-cancerous tissue | NAD | 1.649 | 0.607 | 0.027 |
VIP: Variable importance in the projection; CIN: Chromosomal instability; NAD: Nicotinamide adenine dinucleotide.
Table 5.
Altered metabolites with statistical significance between chromosomal instability and non-chromosomal instability types
| Metabolites | VIP score (VIP > 1.2) | Fold change (tumor/healthy > 1.2 or < 0.8) | P value (P < 0.05) | ||
| CIN tumor | Increased | Aspartic acid | 1.309 | 2.029 | 0.043 |
| Citicoline | 1.751 | 2.886 | 0.033 | ||
| Glutamic acid | 1.219 | 2.101 | 0.011 | ||
| Oxidized glutathione | 1.642 | 2.249 | 0.018 | ||
| Succinyladenosine | 1.671 | 3.434 | 0.011 | ||
| Uridine diphosphate-N-acetylglucosamine | 1.264 | 2.006 | 0.036 | ||
| Decreased | Butyrylcarnitine | 1.218 | 0.590 | 0.014 | |
| Glutathione | 1.214 | 0.582 | 0.042 | ||
| Hydroxyhexanoycarnitine | 1.883 | 0.397 | 0.013 | ||
| Inosinic acid | 1.572 | 0.506 | 0.008 | ||
| Isovalerylcarnitine | 1.657 | 0.346 | 0.004 | ||
| Threonine | 1.714 | 0.394 | 0.006 | ||
| non-CIN tumor | Increased | Glutamic acid | 1.424 | 1.688 | 0.031 |
| Oxidized glutathione | 1.678 | 1.676 | 0.019 | ||
| Proline | 1.542 | 1.569 | 0.003 | ||
| Succinyladenosine | 1.929 | 3.207 | 0.031 | ||
| Xanthine | 2.360 | 2.085 | 0.007 | ||
| Decreased | NAD | 1.584 | 0.599 | 0.047 | |
VIP: Variable importance in the projection; CIN: Chromosomal instability; NAD: Nicotinamide adenine dinucleotide.
Metabolic transformation of CIN GC tumors vs adjacent healthy tissues
The CIN tumors had 31 genetic alterations (alteration frequency > 56%). JAK2, PSIP1, and PTPRD changed the gene copy-number in all patients with CIN GC. CDH20, CDKN2A, DCC, and MALT1 demonstrated gene mutations and were changed in copy-number in 78% of patients. TP53, AFF1, CDH1, CDH11, CDH2, CDH5, CDKN2B, MBD1, MMP2, and SMAD2 were altered in 67% of the patients. ITGB2, ZNF521, ATM, BCL2, CYLD, ERG, FANCA, IL2, MAF, MTOR, PBRM1, RUNX1, TCF12, and TLR4 were additionally altered in 56% of the patients (Table 3).
The metabolite distributions in tumor and healthy tissue could be clearly distinguished in CIN GC (Figure 1E and F). In the GC tumors, the levels of aspartic acid, citicoline, glutamic acid, oxidized glutathione, succinyladenosine, and uridine diphosphate-N-acetylglucosamine significantly increased, whereas those of butyrylcarnitine, glutathione hydroxyhexanoycarnitine, inosinic acid, isovalerylcarnitine, and threonine significantly decreased (P < 0.05) compared with the adjacent healthy tissues (Table 5).
These metabolic alterations indicated alterations in eight pathways (aminoacyl-tRNA biosynthesis, glutamine and glutamate metabolism, alanine, aspartate, and glutamate metabolism, glutathione metabolism, arginine and proline metabolism, histidine metabolism, glyoxylate and dicarboxylate metabolism, and phenylalanine, tyrosine, and tryptophan biosynthesis; P < 0.05).
Metabolic transformation of non-CIN GC tumors vs adjacent healthy tissues
The non-CIN tumors demonstrated most alterations in TP53 and SYNE1 (40%), followed by ARID1A (30%). ITGB2, KMT2C, LRP1B, AKAP9, SAMD9, SMARCA4, TNK2, RNF213, EPHB6, and ERBB3 were altered in 20% of the non-CIN tumors. The other 346 genes demonstrated no alterations (Table 3). The metabolite distributions in tumor and healthy tissue in the non-CIN GC could be clearly distinguished (Figure 1G and H). In the non-CIN tumors, the level of glutamic acid, oxidized glutathione, proline, succinyladenosine, and xanthine significantly increased (P < 0.05), whereas those of nicotinamide adenine dinucleotide (NAD) significantly decreased (P = 0.047), compared with the adjacent healthy tissues. The six metabolites and 13 genes were associated with four pathways in the non-CIN GC tumors: glutathione metabolism, aminoacyl-tRNA biosynthesis, arginine and proline metabolism, and glutamine and glutamate metabolism.
Metabolomic profiling of healthy tissues: CIN vs non-CIN type
The metabolite distributions in healthy tissue between the CIN and non-CIN type tumors could be clearly distinguished, and the decreased NAD level was significantly different (P = 0.027). The concentration of NAD in the healthy tissue was 0.607 times that in the non-CIN type.
Nonspecific metabolic patterns based on the Lauren classifications
In the study, 87.5% of the Lauren intestinal-type tumors belonged to the CIN GC group, whereas all of the Lauren diffuse-type tumors belonged to the non-CIN GC group. The Lauren mixed-type tumors belonged to both CIN (50%) and non-CIN type (50%) (Supplementary Table 4). The intestinal-type tumors demonstrated a high alteration rate of 92.2% (377 genes), particularly those with copy-number changes, whereas the diffuse-type tumors indicated a low alteration rate of 8.56% (35 genes). Therefore, the metabolite distribution of tumors could not be distinguished based on the Lauren classification system.
DISCUSSION
In the present study, we used the TCGA molecular classification method rather than using the traditional Lauren histological classification. In line with our study, CIN tumors were 80% intestinal type, 12% diffuse type, and 7% mixed type in a TCGA report[21]. The CIN phenotype can be induced by dysfunctions of different cellular processes, which can be categorized into (1) inaccurate chromosome segregation during mitosis, (2) cell-cycle checkpoint defects, (3) oncogene-induced mitotic stress, and (4) replication stress[12]. Accelerated loss of heterozygosity in tumor suppressor genes or accelerated gain of oncogene copies due to chromosomal duplication results from CIN, which leads to cancer[22]. To our knowledge, the present study is the first report combining the CIN status of gastric cancer with the metabolomics data. According to our data, we revealed the metabolomic profiling of GC directly correlated with the genetic CIN status, but not with the traditional Lauren’s classification. We demonstrated that the CIN was associated with downstream biochemical alterations.
Genetic alterations continue to accumulate over time, resulting in serious disruptive changes in cell biochemical metabolism and finally leading to GC tumor formation. Through mRNA expression analysis, Carvalho et al[14] discovered that upregulated gene sets associated with CIN were also associated with GC compared with benign adenomas. Disruption of specific biological processes, such as CIN maintenance and altered metabolism, is the key factor in the progression from adenoma to carcinoma. In the present study, CIN GC tumors contained significantly higher levels of phosphocholine and uridine 5′-monophosphate and significantly lower levels of beta-citryl-L-glutamic acid compared with the non-CIN GC tumors. Eight metabolic pathways were involved in GC tumor formation. Pathways involving glutathione metabolism, aminoacyl-tRNA biosynthesis, arginine and proline metabolism, as well as glutamine and glutamate metabolism, were found in both CIN and non-CIN types (Figure 2), and those involving alanine, aspartate and glutamate metabolism, glyoxylate and dicarboxylate metabolism, histidine metabolism, and phenylalanine, tyrosine, and tryptophan biosynthesis were observed in the CIN type alone. NAD could be a predictor for GC tissue development in both CIN and non-CIN GC. Through gas chromatography-MS analysis, Song et al[15] revealed that the levels of several intermediate products of aerobic glycolytic pathways, such as fumaric and alpha-ketoglutaric acid, significantly increased in cancer tissues compared with healthy mucosa, suggesting that altered glucose metabolism is a vital parameter in distinguishing GC cells from healthy cells. Similarly, abnormal glucose metabolism has been observed by other researchers in GC tissue[16,23-25]. Signaling pathways are involved in glycolysis pathways with changes in hypoxia-inducible factor, insulin signaling pathway, and PI3K-Akt-mTOR levels[26].
Figure 2.
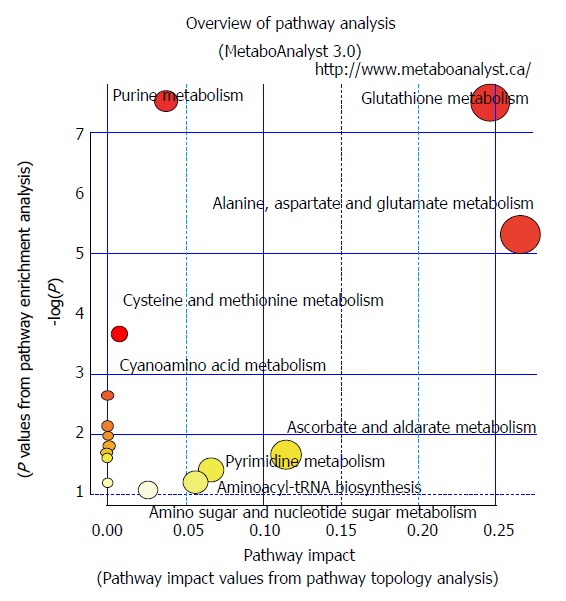
Pathways in chromosomal instability (CIN) and non-chromosomal instability gastric cancer. Pathways involving glutathione metabolism, aminoacyl-tRNA biosynthesis, arginine and proline metabolism, and glutamine and glutamate metabolism were found in CIN and non-CIN types. Pathways involving alanine, aspartate, and glutamate metabolism, glyoxylate and dicarboxylate metabolism, histidine metabolism, and phenylalanine, tyrosine, and tryptophan biosynthesis were observed in the CIN type but not the non-CIN type. CIN: Chromosomal instability.
A critical clinical implication of our GC metabolomics study is biomarker discovery. Sohn et al[27] demonstrated that patients with CIN GC had better overall survival than those with non-CIN GC. Metabolomics might offer an alternative way for stratification that could help to select more appropriate adjuvant chemotherapy. Shaukat et al[28] also demonstrated that CIN induction sensitizes GC to metabolic stress. Mild metabolic disruption that does not affect healthy cells can lead to high levels of oxidative stress and subsequent cell death in CIN GC cells because they are already managing elevated stress levels. Our data illustrated novel therapeutic possibilities regarding GC, as untargeted metabolomics screening identified key regulators that can exploit these changes that cause cell death, which may provide cancer-specific potential drug targets, particularly for advanced cancers exhibiting CIN. The interaction between CIN status and metabolic alteration is linked by aberrant DNA methylation, which could lead to CIN and transcriptional gene silencing of tumor suppressors or overexpression of oncogenes. Vitamin B has essential roles in carbon metabolism providing critical metabolites for DNA methylation, DNA repair, and nucleic acid synthesis[29], although they were not significantly changed in our study. Adjustment of vitamin B levels and genetic polymorphisms of related key enzymes in one carbon metabolism pathway might govern the bioavailability of metabolites and therefore cause changes in CIN phenotypes. Another aspect is the balance between the oxidized and reduced forms of NAD, which is a vital component of a redox state in a cell, and reflects both the metabolic activities and the health of cells[30]. NAD acts as a coenzyme in redox reactions, as a donor of ADP-ribose moieties in ADP-ribosylation reactions, as a precursor of the second messenger molecule cyclic ADP-ribose, and as a substrate for DNA ligases (Figure 3)[31,32].
Figure 3.
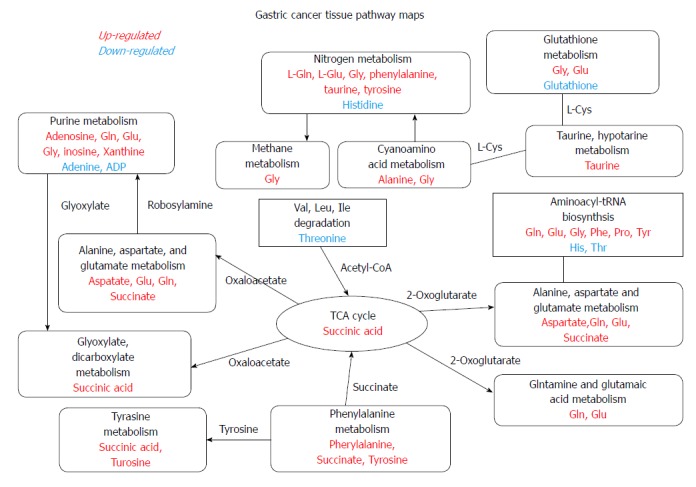
Metabolites in the chromosomal instability type and the non-chromosomal instability type. Upregulated (red) or down-regulated (blue) metabolites of pathways (black) involved in CIN and non-CIN types. CIN: Chromosomal instability.
One of the study limitations was a small number of participants undergoing an operation who were willing to contribute their tissue samples in each category, and therefore we did not correct for multiple comparisons. Metabolomics studies on GC have provided metabolite biomarkers that can differentiate GC patients from healthy controls[33]. We found that the metabolomic profiling of GC was directly correlated with the genetic CIN status but not the traditional Lauren’s classification system. The transomics approach in this study to analyze the genomics and metabolomics data inadvertently limited the cohort size of this study. Nevertheless, in this discovery phase, we demonstrated that CIN was associated with downstream biochemical alterations. Therefore, larger studies are required for validating this biomarker’s utility followed by its translation into a clinical setting. In future works, we would like to collect more patient survival data to understand what metabolites can affect cancer progression or patient prognosis.
In conclusion, we combined the classification method of gene molecules with a metabolomics method to discover metabolic information to accurately classify tumors. These findings on metabolomics profiling based on CIN status have translational potential for biomarker discovery and novel therapeutic development.
ARTICLE HIGHLIGHTS
Research background
Gastric cancer (GC) is one of the most common malignancies. GC can be histologically classified using the Lauren classification system, which divides GC into diffuse and intestinal subtypes. The Cancer Genome Atlas Research Group (TCGA) has developed molecular classification systems based on gene expression profiling. Metabolomics may offer practical solutions to the traditional methods for GC detection and treatment. We hypothesize that metabolic alternations reflect the chromosomal instability (CIN) or genomic stability status of GC and aim to study the comprehensive metabolomic profiles of GC and correlate them with its CIN status.
Research motivation
The numerous biomarkers discovered from metabolomic studies may play a noteworthy role in GC with regard to early-stage detection, diagnosis, prognosis, drug development, and chemosensitivity predictions, but the evidence of metabolomics’ association with CIN status remains lacking.
Research objectives
The aim of our study was to explore the correlation of metabolomics profiles of GC and its CIN status.
Research methods
Based on 409 oncogenes and tumor suppressor genes sequenced, 19 GC patients were classified as CIN and non-CIN type by TCGA. The aqueous metabolites of the GC tumor and its surrounding adjacent healthy tissues were identified through liquid chromatography-mass spectrometry.
Research results
GC tumors demonstrated significantly higher aspartic acid, citicoline, glutamic acid, oxidized glutathione, succinyladenosine, and uridine diphosphate-N-acetylglucosamine levels, but significantly lower butyrylcarnitine, glutathione hydroxyhexanoycarnitine, inosinic acid, isovalerylcarnitine, and threonine levels compared to the adjacent healthy tissues. CIN tumors contained significantly higher phosphocholine and uridine 5’-monophosphate levels but significantly lower beta-citryl-L-glutamic acid level than did non-CIN tumors. CIN GC tumors demonstrated additional altered pathways involving alanine, aspartate, and glutamate metabolism, glyoxylate and dicarboxylate metabolism, histidine metabolism, and phenylalanine, tyrosine, and tryptophan biosynthesis.
Research conclusions
Metabolomic profiles of GC tumors and the adjacent healthy tissue are distinct, and the CIN status is associated with downstream metabolic alterations in GC.
Research perspectives
The combination of classification method of gene molecules and a metabolomics method may reveal that metabolic information can be used to accurately classify tumors. These findings on metabolomics profiling based on CIN status have translational potential for biomarker discovery and novel therapeutic development.
ACKNOWLEDGMENTS
The authors express their thanks to the Metabolomics Core Laboratory, Healthy Aging Research Center, Chang Gung University and Clinical Metabolomics Core Laboratory, Chang Gung Memorial Hospital for performing the metabolomics analysis using liquid chromatography-mass spectrometry and/or NMR spectroscopy.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Taiwan
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): 0
Grade C (Good): C, C
Grade D (Fair): D
Grade E (Poor): 0
Institutional review board statement: All procedures in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Conflict-of-interest statement: The authors have no conflict of interest to declare.
Peer-review started: May 23, 2018
First decision: June 13, 2018
Article in press: July 16, 2018
P- Reviewer: Chen Z, De Re V, Kimura A S- Editor: Wang XJ L- Editor: Filipodia E- Editor: Huang Y
Contributor Information
Cheng-Kun Tsai, Clinical Metabolomics Core Lab, Chang Gung Memorial Hospital at Linkou and Chang Gung University, Taoyuan 333, Taiwan; Department of Medical Imaging and Intervention, Imaging Core Lab, Institute for Radiological Research, Chang Gung Memorial Hospital at Linkou and Chang Gung University, Taoyuan 333, Taiwan.
Ta-Sen Yeh, Department of Surgery, Chang Gung Memorial Hospital at Linkou and Chang Gung University, Taoyuan 333, Taiwan.
Ren-Chin Wu, Department of Pathology, Chang Gung Memorial Hospital at Linkou and Chang Gung University, Taoyuan 333, Taiwan.
Ying-Chieh Lai, Department of Medical Imaging and Intervention, Imaging Core Lab, Institute for Radiological Research, Chang Gung Memorial Hospital at Linkou and Chang Gung University, Taoyuan 333, Taiwan.
Meng-Han Chiang, Clinical Metabolomics Core Lab, Chang Gung Memorial Hospital at Linkou and Chang Gung University, Taoyuan 333, Taiwan; Department of Medical Imaging and Intervention, Imaging Core Lab, Institute for Radiological Research, Chang Gung Memorial Hospital at Linkou and Chang Gung University, Taoyuan 333, Taiwan.
Kuan-Ying Lu, Department of Medical Imaging and Intervention, Imaging Core Lab, Institute for Radiological Research, Chang Gung Memorial Hospital at Linkou and Chang Gung University, Taoyuan 333, Taiwan.
Cheng-Yu Hung, Department of Medical Imaging and Intervention, Imaging Core Lab, Institute for Radiological Research, Chang Gung Memorial Hospital at Linkou and Chang Gung University, Taoyuan 333, Taiwan.
Hung-Yao Ho, Clinical Metabolomics Core Lab, Chang Gung Memorial Hospital at Linkou and Chang Gung University, Taoyuan 333, Taiwan; Department of Medical Biotechnology and Laboratory Science, College of Medicine, Chang Gung University, Taoyuan 333, Taiwan.
Mei-Ling Cheng, Clinical Metabolomics Core Lab, Chang Gung Memorial Hospital at Linkou and Chang Gung University, Taoyuan 333, Taiwan; Department of Biomedical Science, College of Medicine, Chang Gung University, Taoyuan 333, Taiwan.
Gigin Lin, Clinical Metabolomics Core Lab, Chang Gung Memorial Hospital at Linkou and Chang Gung University, Taoyuan 333, Taiwan; Department of Medical Imaging and Intervention, Imaging Core Lab, Institute for Radiological Research, Chang Gung Memorial Hospital at Linkou and Chang Gung University, Taoyuan 333, Taiwan. giginlin@cgmh.org.tw.
References
- 1.Emadi-Baygi M, Nikpour P, Emadi-Andani E. SIX1 overexpression in diffuse-type and grade III gastric tumors: Features that are associated with poor prognosis. Adv Biomed Res. 2015;4:139. doi: 10.4103/2277-9175.161540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Quéro L, Guillerm S, Hennequin C. Neoadjuvant or adjuvant therapy for gastric cancer. World J Gastrointest Oncol. 2015;7:102–110. doi: 10.4251/wjgo.v7.i8.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 4.Compare D, Rocco A, Nardone G. Risk factors in gastric cancer. Eur Rev Med Pharmacol Sci. 2010;14:302–308. [PubMed] [Google Scholar]
- 5.Lauren P. The two histological main types of gastric carcinoma: diffuse and so-called intestinal-type carcinoma. An attempt at a histo-clinical classification. Acta Pathol Microbiol Scand. 1965;64:31–49. doi: 10.1111/apm.1965.64.1.31. [DOI] [PubMed] [Google Scholar]
- 6.Dicken BJ, Bigam DL, Cass C, Mackey JR, Joy AA, Hamilton SM. Gastric adenocarcinoma: review and considerations for future directions. Ann Surg. 2005;241:27–39. doi: 10.1097/01.sla.0000149300.28588.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grabsch HI, Tan P. Gastric cancer pathology and underlying molecular mechanisms. Dig Surg. 2013;30:150–158. doi: 10.1159/000350876. [DOI] [PubMed] [Google Scholar]
- 8.Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–209. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rochfort S. Metabolomics reviewed: a new “omics” platform technology for systems biology and implications for natural products research. J Nat Prod. 2005;68:1813–1820. doi: 10.1021/np050255w. [DOI] [PubMed] [Google Scholar]
- 10.Lim B, Kim JH, Kim M, Kim SY. Genomic and epigenomic heterogeneity in molecular subtypes of gastric cancer. World J Gastroenterol. 2016;22:1190–1201. doi: 10.3748/wjg.v22.i3.1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geigl JB, Obenauf AC, Schwarzbraun T, Speicher MR. Defining ‘chromosomal instability’. Trends Genet. 2008;24:64–69. doi: 10.1016/j.tig.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 12.Maleki SS, Röcken C. Chromosomal Instability in Gastric Cancer Biology. Neoplasia. 2017;19:412–420. doi: 10.1016/j.neo.2017.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jayavelu ND, Bar NS. Metabolomic studies of human gastric cancer: review. World J Gastroenterol. 2014;20:8092–8101. doi: 10.3748/wjg.v20.i25.8092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carvalho B, Sillars-Hardebol AH, Postma C, Mongera S, Terhaar Sive Droste J, Obulkasim A, van de Wiel M, van Criekinge W, Ylstra B, Fijneman RJ, et al. Colorectal adenoma to carcinoma progression is accompanied by changes in gene expression associated with ageing, chromosomal instability, and fatty acid metabolism. Cell Oncol (Dordr) 2012;35:53–63. doi: 10.1007/s13402-011-0065-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song H, Wang L, Liu HL, Wu XB, Wang HS, Liu ZH, Li Y, Diao DC, Chen HL, Peng JS. Tissue metabolomic fingerprinting reveals metabolic disorders associated with human gastric cancer morbidity. Oncol Rep. 2011;26:431–438. doi: 10.3892/or.2011.1302. [DOI] [PubMed] [Google Scholar]
- 16.Hur H, Paik MJ, Xuan Y, Nguyen DT, Ham IH, Yun J, Cho YK, Lee G, Han SU. Quantitative measurement of organic acids in tissues from gastric cancer patients indicates increased glucose metabolism in gastric cancer. PLoS One. 2014;9:e98581. doi: 10.1371/journal.pone.0098581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chan AW, Gill RS, Schiller D, Sawyer MB. Potential role of metabolomics in diagnosis and surveillance of gastric cancer. World J Gastroenterol. 2014;20:12874–12882. doi: 10.3748/wjg.v20.i36.12874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abbassi-Ghadi N, Kumar S, Huang J, Goldin R, Takats Z, Hanna GB. Metabolomic profiling of oesophago-gastric cancer: a systematic review. Eur J Cancer. 2013;49:3625–3637. doi: 10.1016/j.ejca.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 19.FOLCH J, LEES M, SLOANE STANLEY GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- 20.Want EJ, Wilson ID, Gika H, Theodoridis G, Plumb RS, Shockcor J, Holmes E, Nicholson JK. Global metabolic profiling procedures for urine using UPLC-MS. Nat Protoc. 2010;5:1005–1018. doi: 10.1038/nprot.2010.50. [DOI] [PubMed] [Google Scholar]
- 21.Fontana E, Smyth EC. Novel targets in the treatment of advanced gastric cancer: a perspective review. Ther Adv Med Oncol. 2016;8:113–125. doi: 10.1177/1758834015616935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rajagopalan H, Lengauer C. Aneuploidy and cancer. Nature. 2004;432:338–341. doi: 10.1038/nature03099. [DOI] [PubMed] [Google Scholar]
- 23.Wu H, Xue R, Tang Z, Deng C, Liu T, Zeng H, Sun Y, Shen X. Metabolomic investigation of gastric cancer tissue using gas chromatography/mass spectrometry. Anal Bioanal Chem. 2010;396:1385–1395. doi: 10.1007/s00216-009-3317-4. [DOI] [PubMed] [Google Scholar]
- 24.Hirayama A, Kami K, Sugimoto M, Sugawara M, Toki N, Onozuka H, Kinoshita T, Saito N, Ochiai A, Tomita M, et al. Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time-of-flight mass spectrometry. Cancer Res. 2009;69:4918–4925. doi: 10.1158/0008-5472.CAN-08-4806. [DOI] [PubMed] [Google Scholar]
- 25.Cai Z, Zhao JS, Li JJ, Peng DN, Wang XY, Chen TL, Qiu YP, Chen PP, Li WJ, Xu LY, et al. A combined proteomics and metabolomics profiling of gastric cardia cancer reveals characteristic dysregulations in glucose metabolism. Mol Cell Proteomics. 2010;9:2617–2628. doi: 10.1074/mcp.M110.000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuan LW, Yamashita H, Seto Y. Glucose metabolism in gastric cancer: The cutting-edge. World J Gastroenterol. 2016;22:2046–2059. doi: 10.3748/wjg.v22.i6.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sohn BH, Hwang JE, Jang HJ, Lee HS, Oh SC, Shim JJ, Lee KW, Kim EH, Yim SY, Lee SH, et al. Clinical Significance of Four Molecular Subtypes of Gastric Cancer Identified by The Cancer Genome Atlas Project. Clin Cancer Res. 2017 doi: 10.1158/1078-0432.CCR-16-2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shaukat Z, Liu D, Choo A, Hussain R, O’Keefe L, Richards R, Saint R, Gregory SL. Chromosomal instability causes sensitivity to metabolic stress. Oncogene. 2015;34:4044–4055. doi: 10.1038/onc.2014.344. [DOI] [PubMed] [Google Scholar]
- 29.Donkena KV, Yuan H, Young CY. Vitamin Bs, one carbon metabolism and prostate cancer. Mini Rev Med Chem. 2010;10:1385–1392. doi: 10.2174/138955710793564106. [DOI] [PubMed] [Google Scholar]
- 30.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 31.Smyth LM, Bobalova J, Mendoza MG, Lew C, Mutafova-Yambolieva VN. Release of beta-nicotinamide adenine dinucleotide upon stimulation of postganglionic nerve terminals in blood vessels and urinary bladder. J Biol Chem. 2004;279:48893–48903. doi: 10.1074/jbc.M407266200. [DOI] [PubMed] [Google Scholar]
- 32.Billington RA, Bruzzone S, De Flora A, Genazzani AA, Koch-Nolte F, Ziegler M, Zocchi E. Emerging functions of extracellular pyridine nucleotides. Mol Med. 2006;12:324–327. doi: 10.2119/2006-00075.Billington. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ni Y, Xie G, Jia W. Metabonomics of human colorectal cancer: new approaches for early diagnosis and biomarker discovery. J Proteome Res. 2014;13:3857–3870. doi: 10.1021/pr500443c. [DOI] [PubMed] [Google Scholar]