

Two of the constituent molecules in bis(3-carbamoylpyridin-1-ium) phosphite monohydrate, i.e. the phosphite anion and the water molecule, are situated on the symmetry plane. The molecules are held together by moderate N—H⋯O and O—H⋯N, and weak O—H⋯O and C—H⋯Ocarbonyl hydrogen bonds in which the primary and secondary amine and water H atoms are involved. The H atom directly bonded to the P atom avoids hydrogen bonding, as usual.
Keywords: crystal structure, hydrogen bonding, phosphite
Abstract
Two of the constituent molecules in the title structure, 2C6H7N2O+·HPO3 2−·H2O, i.e. the phosphite anion and the water molecule, are situated on a symmetry plane. The molecules are held together by moderate N—H⋯O and O—H⋯N, and weak O—H⋯O and C—H⋯Ocarbonyl hydrogen bonds in which the amide and secondary amine groups, and the water molecules are involved. The structural features are usual, among them the H atom bonded to the P atom avoids hydrogen bonding.
Chemical context
Nicotinamide (pyridine-2-carboxamide) is a biologically important molecule, being the active part of vitamin B3 and nicotinamide adenine dinucleotide (NAD) (e.g. Wald, 1991 ▸; Williamson et al., 1967 ▸).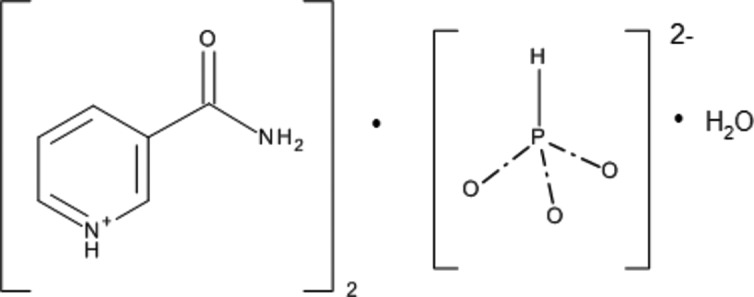
However, interest in the preparation of the title hydrated salt was called for with respect to an investigation of the configuration of the –NH2 group and its dependence on its environment.
It was hoped that 3-carbamoylpyridine (nicotinamide) would make a salt or a co-crystal with phosphorous acid, H3PO3. It is difficult to predict which of these two forms would be prefererred, because of a small difference of ΔpK a = pK a(base) − pK a(acid) (Childs et al., 2007 ▸). [The pK a values for 3-carbamoylpyridine and H3PO3 are 3.3 and 1.3 (first degree), respectively (CRC Handbook, 2009 ▸).]
Structural commentary
The title molecules are shown in Fig. 1 ▸. The resulting structure turned out to be a monohydrated salt. Table 1 ▸ lists the hydrogen bonds, which are shown in Fig. 2 ▸. The secondary amine hydrogen H1n1 is involved in the strongest hydrogen bond present in the structure (N1—H1n1⋯O3i). Its parameters indicate that this hydrogen bond is situated on the boundary between strong and moderate hydrogen bonds (Gilli & Gilli, 2009 ▸). The amide hydrogen H1n2 is donated to the water oxygen, while H2n2 is donated to atom O3 of the phosphite anion. Atom O2 is an acceptor of water hydrogen H1ow. Water hydrogen H2ow is donated to a pair of O3 atoms. The carbonyl oxygen O1 is an acceptor of two weak C—H⋯O hydrogen bonds, namely C3—H1c3⋯O1ii and C4—H1c4⋯O1iii. The water oxygen atom is also an acceptor of hydrogen H1c2 (see Table 1 ▸).
Figure 1.

The title molecule, with anisotropic atomic displacement ellipsoids shown at the 50% probability level (PLATON; Spek, 2009 ▸).
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C2—H1c2⋯Ow i | 0.95 | 2.64 | 3.5777 (14) | 168 |
| C3—H1c3⋯O1ii | 0.95 | 2.56 | 3.4790 (17) | 164 |
| C4—H1c4⋯O1iii | 0.95 | 2.56 | 3.1948 (13) | 125 |
| N1—H1n1⋯O3iv | 1.053 (15) | 1.455 (15) | 2.508 (3) | 178.1 (14) |
| N2—H1n2⋯Ow i | 0.849 (16) | 2.140 (16) | 2.9513 (13) | 159.8 (18) |
| N2—H2n2⋯O3v | 0.889 (18) | 1.955 (18) | 2.823 (3) | 165.2 (15) |
| Ow—H1ow⋯O2vi | 0.84 (2) | 1.82 (2) | 2.657 (4) | 172 (2) |
| Ow—H2ow⋯O3 | 0.96 (3) | 2.42 (2) | 3.263 (3) | 146.8 (9) |
| Ow—H2ow⋯O3vii | 0.96 (3) | 2.42 (2) | 3.263 (3) | 146.8 (9) |
Symmetry codes: (i)  ; (ii)
; (ii)  ; (iii)
; (iii)  ; (iv)
; (iv)  ; (v)
; (v)  ; (vi)
; (vi)  ; (vii)
; (vii)  .
.
Figure 2.

View of the title structure. C, H, N, O and P atoms are represented by gray, small gray, blue, red and violet circles, respectively. [Symmetry codes: (i) −x + 1, y, z; (ii) x, y − 1, z; (iii) x, y − 1, z; (iv) −x +  , −y + 1, z −
, −y + 1, z −  ; (v) −x + 2, y − 1, z; (vi) −x + , −y + 1, z + ; (vii) x − , −y + 1, z − ; (viii) x, y − 1, z − 1.] The hydrogen bonds are shown as yellow dashed lines (DIAMOND; Brandenburg & Putz, 2005 ▸).
; (v) −x + 2, y − 1, z; (vi) −x + , −y + 1, z + ; (vii) x − , −y + 1, z − ; (viii) x, y − 1, z − 1.] The hydrogen bonds are shown as yellow dashed lines (DIAMOND; Brandenburg & Putz, 2005 ▸).
Phosphite and fluorophosphonate, as well hydrogen phosphite and hydrogen fluorophosphonate, are similar molecules. Either molecule can be involved, not only in isostructural compounds, but even in mixed crystals (Fábry et al., 2012 ▸). Similarity regarding not only the shape of the molecules but also the avoidance both of P-bonded fluorines and hydrogens of involvement in strong or moderate hydrogen bonds (Matulková et al., 2017 ▸). The latter article shows a plot of the dependence of P—F distance on the longest P—O distance in flourophosphonate and hydrogen fluorophosphonate molecules. The P—F distance tends to be longer in [FPO3]2− than in [HFPO3]−. Fig. 3 ▸ shows a similar plot for the phosphites and hydrogen phosphites between both molecules despite the larger spread of P—H distances in phosphite molecules because of the lower accuracy of the H-atom determinations by X-ray diffraction experiments. The reason why the P—H bond tends to be longer follows from the conservation of the overall bond valence sum of the central P5+ or P3+ atom. It is worth pointing out that the tabulated value of the bond valence parameter for the P—H bond seems to yield too high values. For example, for the important values of the P—H distances, i.e. 1.28, 1.33 and 1.37 Å (cf. Fig. 3 ▸), the bond valences (Brese & O’Keeffe, 1991 ▸) are 1.42, 1.24 and 1.11, respectively. The P—H bond valence parameters are going to be checked as part of future work.
Figure 3.

The dependence of the longest P—O distance (Å) on the P—H distance (Å) in hydrogen phosphites (red circles); phosphites are represented by black squares and the title phosphite structure by a green triangle.
The C—NH2 group tends to be fairly planar for short C—N bonds (Fábry et al., 2014 ▸). In agreement with a short C—N bond length [C6—N2 = 1.3232 (18) Å] in the title structure, the best plane through C6/N2/H1n2/H2n2 reveals a maximum deviation of about 0.05 (2) Å for each hydrogen, while ξ2 = 12.6.
Supramolecular features
In the crystal, the most important graph-set motif (Etter et al., 1990 ▸) present is an  (10) ring motif, which is composed of atoms P1—O3⋯H2n2iv—N2iv—H1n2iv⋯Ow⋯H1n2vii—N2vii—H2n2vii⋯O3i (Table 1 ▸ and Fig. 2 ▸; symmetry codes are given in the figure cation). The phosphite anion and the water molecule are linked by Owater—H⋯Ophosphite hydrogen bonds, forming chains propagating along [001]. The cations are linked to these chains via N—H⋯O hydrogen bonds, forming layers parallel to the bc plane, as shown in Fig. 2 ▸. The layers are linked by C—H⋯O hydrogen bonds, resulting in the formation of a supramolecular three-dimensional structure.
(10) ring motif, which is composed of atoms P1—O3⋯H2n2iv—N2iv—H1n2iv⋯Ow⋯H1n2vii—N2vii—H2n2vii⋯O3i (Table 1 ▸ and Fig. 2 ▸; symmetry codes are given in the figure cation). The phosphite anion and the water molecule are linked by Owater—H⋯Ophosphite hydrogen bonds, forming chains propagating along [001]. The cations are linked to these chains via N—H⋯O hydrogen bonds, forming layers parallel to the bc plane, as shown in Fig. 2 ▸. The layers are linked by C—H⋯O hydrogen bonds, resulting in the formation of a supramolecular three-dimensional structure.
Database survey
The applied crystallographic databases were the Cambridge Crystallographic Database (Version 5.39, with updates to May 2018; Groom et al., 2016 ▸) and the Inorganic Crystal Structure Database (June 2018; ICSD, 2018 ▸). The search was carried out for all phosphites or hydrogen phosphites with a cation of one kind.
Synthesis and crystallization
The title structure was prepared by slow evaporation of a water solution (18 ml) of equimolar amounts of nicotinamide (1.49 g) and phosphorous acid (1 g). Colourless crystals were isolated after two months.
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 2 ▸. All the H atoms were discernible in the difference electron-density map. The aryl H atoms were constrained by the constraints C—H = 0.95 Å and U
iso(H) = 1.2U
eq(C). Water hydrogen H2ow was refined freely, while H1ow was restrained with a distance restraint of 0.84 Å with elasticity 0.02 Å (Müller, 2009 ▸), and with U
iso(H) = 1.5U
eq(O). The hydrogens of the primary amine N2 group and the secondary amine N1 group were constrained by U
iso(H) = 1.2U
eq(N). The P—H hydrogen was refined isotropically. Three reflections, i.e. 95 , 10,5, and 11,5,, were discarded from the refinement because |I(obs) − I(calc)|/σ(I) > 20.
, 10,5, and 11,5,, were discarded from the refinement because |I(obs) − I(calc)|/σ(I) > 20.
Table 2. Experimental details.
| Crystal data | |
| Chemical formula | 2C6H7N2O+·HPO3 2−·H2O |
| M r | 344.3 |
| Crystal system, space group | Orthorhombic, P m n21 |
| Temperature (K) | 95 |
| a, b, c (Å) | 22.9297 (4), 4.5910 (1), 7.0900 (1) |
| V (Å3) | 746.37 (2) |
| Z | 2 |
| Radiation type | Cu Kα |
| μ (mm−1) | 2.01 |
| Crystal size (mm) | 0.45 × 0.16 × 0.06 |
| Data collection | |
| Diffractometer | Rigaku OD SuperNova Dual source diffractometer with an AtlasS2 detector |
| Absorption correction | Multi-scan (CrysAlis PRO; Rigaku OD, 2017 ▸) |
| T min, T max | 0.599, 0.831 |
| No. of measured, independent and observed [I > 3σ(I)] reflections | 10796, 1592, 1588 |
| R int | 0.021 |
| (sin θ/λ)max (Å−1) | 0.630 |
| Refinement | |
| R[F > 3σ(F)], wR(F), S | 0.019, 0.054, 2.21 |
| No. of reflections | 1592 |
| No. of parameters | 181 |
| No. of restraints | 1 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 0.14, −0.15 |
| Absolute structure | Since the Flack parameter turned out to equal to 0.012(13) in the final stage of refinement it was set to 0. 726 Friedel pairs used in the refinement. |
| Absolute structure parameter | 0.0 |
Since the phosphite oxygens revealed large displacement ellipsoids, the anharmonic displacement parameters upto the fourth grade were included for atoms P1, O2 and O3. (The refinement with the harmonic approximation resulted in R obs = 0.0242, Rw obs = 0.0773, R all = 0.0243, Rw all = 0.0774 and S = 3.13, with number of parameters = 127. With application of anharmonic approximation, R obs = 0.0188, Rw obs = 0.0535, R all = 0.0188, Rw all = 0.0535 and S = 2.21, with number of parameters = 181. The respective values of the third- and fourth-order components of the displacement tensor are given in the CIF.)
Refinement with the assumption of the presence of inversion twinning resulted in a Flack parameter of 0.012 (13) (726 Friedel pairs used in the refinement). Therefore, the crystal was considered as single-domained in the final stage of the refiement, the results of which are presented here.
Supplementary Material
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S2056989018011192/eb2010sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989018011192/eb2010Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989018011192/eb2010Isup3.smi
Supporting information file. DOI: 10.1107/S2056989018011192/eb2010Isup4.cml
CCDC reference: 1860376
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
The author expresses the gratitude for the support of the Ministry of Education of the Czech Republic. Dr Michal Dušek from the Institute of Physics is thanked for careful data collection.
supplementary crystallographic information
Crystal data
| 2C6H7N2O+·HPO32−·H2O | F(000) = 360 |
| Mr = 344.3 | Dx = 1.532 Mg m−3 |
| Orthorhombic, Pmn21 | Cu Kα radiation, λ = 1.54184 Å |
| Hall symbol: P -2x;-2yac;2zac | Cell parameters from 9530 reflections |
| a = 22.9297 (4) Å | θ = 6.5–75.9° |
| b = 4.5910 (1) Å | µ = 2.01 mm−1 |
| c = 7.0900 (1) Å | T = 95 K |
| V = 746.37 (2) Å3 | Plate, colourless |
| Z = 2 | 0.45 × 0.16 × 0.06 mm |
Data collection
| Rigaku OD SuperNova Dual source diffractometer with an AtlasS2 detector | 1592 independent reflections |
| Radiation source: micro-focus sealed X-ray tube, SuperNova (Cu) X-ray Source | 1588 reflections with I > 3σ(I) |
| Mirror monochromator | Rint = 0.021 |
| Detector resolution: 5.2027 pixels mm-1 | θmax = 76.3°, θmin = 3.9° |
| ω/ scans | h = −28→28 |
| Absorption correction: multi-scan (CrysAlis PRO; Rigaku OD, 2017) | k = −5→5 |
| Tmin = 0.599, Tmax = 0.831 | l = −8→8 |
| 10796 measured reflections |
Refinement
| Refinement on F2 | Weighting scheme based on measured s.u.'s w = 1/(σ2(I) + 0.0004I2) |
| R[F > 3σ(F)] = 0.019 | (Δ/σ)max = 0.035 |
| wR(F) = 0.054 | Δρmax = 0.14 e Å−3 |
| S = 2.21 | Δρmin = −0.15 e Å−3 |
| 1592 reflections | Extinction correction: B-C type 1 Lorentzian isotropic (Becker & Coppens, 1974) |
| 181 parameters | Extinction coefficient: 910 (160) |
| 1 restraint | Absolute structure: Since the Flack parameter turned out to equal to 0.012(13) in the final stage of refinement it was set to 0. 726 Friedel pairs used in the refinement. |
| 22 constraints | Absolute structure parameter: 0.0 |
| H atoms treated by a mixture of independent and constrained refinement |
Special details
| Refinement. This part differs from the original article by Thanigaimani et al. (2006). It also differs from the refinement by Thanigaimani et al. (2006) by a different threshold for the consideration of the observed diffractions: F2 > 3sigma(F2) has been used as criterion for observed diffractions by JANA2006 which was used for the calculation of the corrected structural model.Three diffractions 9 5 -2, 10 5 -2, 11 5 -2 were discarded from the refinement because |I(obs)-I(calc)|/σ(I) > 20. Since the Flack parameter turned out to equal to 0.012 (13) in the final stage of refinement it was set to 0. 726 Friedel pairs used in the refinement. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| C1 | 0.84788 (5) | 0.8441 (2) | 0.47478 (18) | 0.0157 (3) | |
| C2 | 0.88464 (4) | 1.0560 (2) | 0.40131 (18) | 0.0159 (3) | |
| H1c2 | 0.916985 | 1.121048 | 0.473657 | 0.0191* | |
| N1 | 0.87528 (4) | 1.1699 (2) | 0.23035 (17) | 0.0168 (2) | |
| H1n1 | 0.9056 (7) | 1.320 (3) | 0.175 (2) | 0.0202* | |
| C3 | 0.82992 (5) | 1.0852 (2) | 0.1232 (2) | 0.0169 (3) | |
| H1c3 | 0.824169 | 1.170176 | 0.002456 | 0.0202* | |
| C4 | 0.79162 (5) | 0.8751 (2) | 0.1879 (2) | 0.0178 (3) | |
| H1c4 | 0.759552 | 0.814668 | 0.112553 | 0.0213* | |
| C5 | 0.80087 (4) | 0.7536 (2) | 0.36522 (18) | 0.0168 (3) | |
| H1c5 | 0.77504 | 0.608615 | 0.411623 | 0.0201* | |
| C6 | 0.85727 (5) | 0.7074 (3) | 0.66613 (19) | 0.0173 (3) | |
| O1 | 0.82199 (4) | 0.52392 (19) | 0.72395 (16) | 0.0271 (2) | |
| N2 | 0.90344 (4) | 0.7930 (2) | 0.76358 (18) | 0.0202 (3) | |
| H1n2 | 0.9281 (7) | 0.917 (3) | 0.724 (3) | 0.0243* | |
| H2n2 | 0.9105 (7) | 0.698 (4) | 0.871 (3) | 0.0243* | |
| P1 | 0.5 | 0.3758 (3) | 0.7085 (2) | 0.0181 (6) | |
| H1p1 | 0.5 | 0.092 (5) | 0.698 (4) | 0.034 (6)* | |
| O2 | 0.5 | 0.4732 (8) | 0.9100 (6) | 0.0287 (14) | |
| O3 | 0.55414 (11) | 0.4668 (5) | 0.5974 (3) | 0.0350 (8) | |
| Ow | 0.5 | 0.7861 (2) | 0.22531 (19) | 0.0219 (3) | |
| H1ow | 0.5 | 0.672 (5) | 0.132 (3) | 0.0328* | |
| H2ow | 0.5 | 0.658 (6) | 0.332 (4) | 0.0328* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| C1 | 0.0143 (4) | 0.0170 (5) | 0.0159 (5) | 0.0034 (3) | 0.0021 (4) | −0.0004 (4) |
| C2 | 0.0157 (4) | 0.0167 (5) | 0.0152 (5) | 0.0009 (4) | −0.0018 (4) | −0.0008 (4) |
| N1 | 0.0174 (4) | 0.0158 (4) | 0.0172 (4) | 0.0004 (3) | −0.0006 (4) | 0.0018 (4) |
| C3 | 0.0183 (4) | 0.0170 (5) | 0.0153 (5) | 0.0020 (4) | −0.0026 (4) | 0.0000 (4) |
| C4 | 0.0163 (4) | 0.0189 (5) | 0.0182 (5) | 0.0008 (3) | −0.0032 (4) | −0.0018 (4) |
| C5 | 0.0142 (4) | 0.0177 (5) | 0.0184 (5) | −0.0001 (4) | 0.0019 (3) | −0.0001 (4) |
| C6 | 0.0176 (5) | 0.0183 (5) | 0.0161 (5) | 0.0013 (4) | 0.0026 (3) | 0.0014 (4) |
| O1 | 0.0275 (4) | 0.0331 (4) | 0.0207 (4) | −0.0115 (3) | −0.0015 (4) | 0.0082 (4) |
| N2 | 0.0179 (4) | 0.0265 (5) | 0.0162 (4) | −0.0015 (4) | −0.0011 (3) | 0.0072 (4) |
| P1 | 0.0192 (11) | 0.0147 (7) | 0.0205 (12) | 0 | 0 | −0.0018 (8) |
| O2 | 0.038 (3) | 0.040 (2) | 0.008 (2) | 0 | 0 | −0.0080 (19) |
| O3 | 0.0349 (14) | 0.0382 (15) | 0.0318 (15) | −0.0234 (11) | 0.0241 (13) | −0.0182 (12) |
| Ow | 0.0283 (5) | 0.0185 (5) | 0.0188 (5) | 0 | 0 | −0.0010 (5) |
Geometric parameters (Å, º)
| C1—C2 | 1.3884 (15) | C5—H1c5 | 0.95 |
| C1—C5 | 1.3920 (16) | C6—O1 | 1.2379 (15) |
| C1—C6 | 1.5103 (18) | C6—N2 | 1.3237 (16) |
| C2—H1c2 | 0.95 | N2—H1n2 | 0.849 (16) |
| C2—N1 | 1.3375 (17) | N2—H2n2 | 0.889 (18) |
| N1—H1n1 | 1.053 (15) | H1n2—H2n2 | 1.50 (2) |
| N1—C3 | 1.3455 (16) | P1—H1p1 | 1.30 (3) |
| H1n1—O3i | 1.455 (15) | P1—O2 | 1.497 (4) |
| C3—H1c3 | 0.9501 | P1—O3 | 1.528 (3) |
| C3—C4 | 1.3827 (15) | P1—O3ii | 1.528 (3) |
| C4—H1c4 | 0.95 | Ow—H1ow | 0.84 (2) |
| C4—C5 | 1.3917 (18) | Ow—H2ow | 0.96 (3) |
| C2—C1—C5 | 118.02 (11) | C1—C5—H1c5 | 119.91 |
| C2—C1—C6 | 122.79 (10) | C4—C5—H1c5 | 119.91 |
| C5—C1—C6 | 119.19 (10) | C1—C6—O1 | 119.15 (11) |
| C1—C2—H1c2 | 119.45 | C1—C6—N2 | 117.36 (10) |
| C1—C2—N1 | 121.10 (10) | O1—C6—N2 | 123.49 (13) |
| H1c2—C2—N1 | 119.45 | C6—N2—H1n2 | 124.0 (12) |
| C2—N1—H1n1 | 119.1 (9) | C6—N2—H2n2 | 116.4 (11) |
| C2—N1—C3 | 121.51 (10) | H1n2—N2—H2n2 | 119.2 (16) |
| H1n1—N1—C3 | 119.3 (9) | H1p1—P1—O2 | 110.6 (13) |
| N1—H1n1—O3i | 178.1 (14) | H1p1—P1—O3 | 104.1 (7) |
| N1—C3—H1c3 | 119.82 | H1p1—P1—O3ii | 104.1 (7) |
| N1—C3—C4 | 120.37 (12) | O2—P1—O3 | 114.21 (12) |
| H1c3—C3—C4 | 119.82 | O2—P1—O3ii | 114.21 (12) |
| C3—C4—H1c4 | 120.58 | O3—P1—O3ii | 108.64 (15) |
| C3—C4—C5 | 118.82 (11) | H1n1iii—O3—P1 | 120.3 (6) |
| H1c4—C4—C5 | 120.59 | H1ow—Ow—H2ow | 104 (2) |
| C1—C5—C4 | 120.18 (10) |
Symmetry codes: (i) −x+3/2, −y+2, z−1/2; (ii) −x+1, y, z; (iii) −x+3/2, −y+2, z+1/2.
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C2—H1c2···Owiii | 0.95 | 2.64 | 3.5777 (14) | 167.77 |
| C3—H1c3···O1iv | 0.95 | 2.56 | 3.4790 (17) | 163.62 |
| C4—H1c4···O1v | 0.95 | 2.56 | 3.1948 (13) | 124.74 |
| N1—H1n1···O3i | 1.053 (15) | 1.455 (15) | 2.508 (3) | 178.1 (14) |
| N2—H1n2···Owiii | 0.849 (16) | 2.140 (16) | 2.9513 (13) | 159.8 (18) |
| N2—H2n2···O3vi | 0.889 (18) | 1.955 (18) | 2.823 (3) | 165.2 (15) |
| Ow—H1ow···O2vii | 0.84 (2) | 1.82 (2) | 2.657 (4) | 172 (2) |
| Ow—H2ow···O3 | 0.96 (3) | 2.42 (2) | 3.263 (3) | 146.8 (9) |
| Ow—H2ow···O3ii | 0.96 (3) | 2.42 (2) | 3.263 (3) | 146.8 (9) |
Symmetry codes: (i) −x+3/2, −y+2, z−1/2; (ii) −x+1, y, z; (iii) −x+3/2, −y+2, z+1/2; (iv) x, y+1, z−1; (v) −x+3/2, −y+1, z−1/2; (vi) −x+3/2, −y+1, z+1/2; (vii) x, y, z−1.
Funding Statement
This work was funded by Ministry of Education of the Czech Republic grant NPU I - LO1603.
References
- Becker, P. J. & Coppens, P. (1974). Acta Cryst. A30, 129–147.
- Brandenburg, K. & Putz, H. (2005). DIAMOND. Crystal Impact GbR, Bonn, Germany.
- Brese, N. E. & O’Keeffe, M. (1991). Acta Cryst. B47, 192–197.
- Burla, M. C., Caliandro, R., Carrozzini, B., Cascarano, G. L., Cuocci, C., Giacovazzo, C., Mallamo, M., Mazzone, A. & Polidori, G. (2015). J. Appl. Cryst. 48, 306–309.
- Childs, S. L., Stahly, G. P. & Park, A. (2007). Mol. Pharm. 4, 323–338. [DOI] [PubMed]
- CRC Handbook (2009). CRC Handbook of Chemistry and Physics, 90th ed., edited by D. R. Lidl, pp. 8–40 and 8–45. Boca Raton, London, New York: CRC Press.
- Etter, M. C., MacDonald, J. C. & Bernstein, J. (1990). Acta Cryst. B46, 256–262. [DOI] [PubMed]
- Fábry, J., Dušek, M., Vaněk, P., Rafalovskyi, I., Hlinka, J. & Urban, J. (2014). Acta Cryst. C70, 1153–1160. [DOI] [PubMed]
- Fábry, J., Fridrichová, M., Dušek, M., Fejfarová, K. & Krupková, R. (2012). Acta Cryst. C68, o76–o83. [DOI] [PubMed]
- Gilli, G. & Gilli, P. (2009). The Nature of the Hydrogen Bond, p. 61. New York: Oxford University Press Inc.
- Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. (2016). Acta Cryst. B72, 171–179. [DOI] [PMC free article] [PubMed]
- ICSD (2018). Inorganic Crystal Structure Database. FIZ-Karlsruhe, Germany. http://www.fiz-karlsruhe.de/fiz/products/icsd/welcome.html.
- Matulková, I., Fábry, J., Němec, I., Císařová, I. & Vaněk, P. (2017). Acta Cryst. B73, 1114–1124.
- Müller, P. (2009). Crystallogr. Rev. 15, 57–83.
- Petříček, V., Dušek, M. & Palatinus, L. (2014). Z. Kristallogr. 229, 345–352.
- Rigaku OD (2017). CrysAlis PRO. Rigaku Oxford Diffraction, Yarnton, Oxfordshire, England.
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Wald, N. (1991). Lancet, 338, 131–137.
- Williamson, D. H., Lund, P. & Krebs, H. A. (1967). Biochem J. 103, 514–527. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S2056989018011192/eb2010sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989018011192/eb2010Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989018011192/eb2010Isup3.smi
Supporting information file. DOI: 10.1107/S2056989018011192/eb2010Isup4.cml
CCDC reference: 1860376
Additional supporting information: crystallographic information; 3D view; checkCIF report