

Abstract
Tardive dyskinesia (TD) risk with D2/serotonin receptor antagonists or D2 receptor partial agonists (second‐generation antipsychotics, SGAs) is considered significantly lower than with D2 antagonists (first‐generation antipsychotics, FGAs). As some reports questioned this notion, we meta‐analyzed randomized controlled studies (RCTs) to estimate the risk ratio (RR) and annualized rate ratio (RaR) of TD comparing SGAs vs. FGAs and SGAs vs. SGAs. Additionally, we calculated raw and annualized pooled TD rates for each antipsychotic. Data from 57 head‐to‐head RCTs, including 32 FGA and 86 SGA arms, were meta‐analyzed, yielding 32 FGA‐SGA pairs and 35 SGA‐SGA pairs. The annualized TD incidence across FGA arms was 6.5% (95% CI: 5.3‐7.8%) vs. 2.6% (95% CI: 2.0‐3.1%) across SGA arms. TD risk and annualized rates were lower with SGAs vs. FGAs (RR=0.47, 95% CI: 0.39‐0.57, p<0.0001, k=28; RaR=0.35, 95% CI: 0.28‐0.45, p<0.0001, number‐needed‐to‐treat, NNT=20). Meta‐regression showed no FGA dose effect on FGA‐SGA comparisons (Z=−1.03, p=0.30). FGA‐SGA TD RaRs differed by SGA comparator (Q=21.8, df=7, p=0.003), with a significant advantage of olanzapine and aripiprazole over other non‐clozapine SGAs in exploratory pairwise comparisons. SGA‐SGA comparisons confirmed the olanzapine advantage vs. non‐clozapine SGAs (RaR=0.66, 95% CI: 0.49‐0.88, p=0.006, k=17, NNT=100). This meta‐analysis confirms a clinically meaningfully lower TD risk with SGAs vs. FGAs, which is not driven by high dose FGA comparators, and documents significant differences with respect to this risk between individual SGAs.
Keywords: Tardive dyskinesia, first‐generation antipsychotics, second‐generation antipsychotics, randomized controlled studies, schizophrenia, meta‐analysis, annualized incidence, clozapine, aripiprazole
Can tardive dyskinesia (TD), a condition of potentially irreversible abnormal involuntary movements associated to treatment with D2 receptor antagonists (first‐generation antipsychotics, FGAs), and producing a significant impairment of functioning and quality of life1, 2, be considered relatively irrelevant for treatment with second‐generation antipsychotics (SGAs)?
Based on studies conducted until 2004, the annual TD incidence during SGA treatment was estimated as 0.8% in non‐elderly adults3, one fifth of the rate (5.4%) with FGAs. Surprisingly, however, equal rates of TD during SGA or select FGA treatment were reported in two large randomized controlled trials (RCTs) in schizophrenia, the UK‐based Cost Utility of the Latest Antipsychotic Drugs in Schizophrenia Study (CUtLASS‐1)4 and the US‐based Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study5. As a pre‐emptive risk reduction, the CATIE study5 did not include subjects with a history of TD into the FGA arms, but only in the SGA arms, limiting the interpretation of reported TD rates. Moreover, most meta‐analyses of SGA treatment do not comment on TD risk, as most RCTs are shorter than 3 months (the minimum duration required to diagnose TD6, 7 and/or report continuous outcome measures for dyskinesia), being not suited to identify TD cases8, 9, 10. Thus, information on incident TD during RCTs is very scarce.
We recently summarized prevalence rates of TD11, finding that 20% of subjects with current SGA treatment presented with at least mild screening‐based probable TD. This observation contrasts with the clinical perception of vanishing TD during SGA treatment, possibly due to the overrating of mild TD cases detected only with screening, but not perceived as clinically meaningful. Importantly, however, prevalence rates are inappropriate indicators of treatment‐associated risk for TD, as the antipsychotic given at the time of TD development may not be the one prescribed at the time of TD assessment.
To determine the incidence and the relative risk of treatment‐emergent TD with FGA or SGA treatment, we searched for RCTs with a duration of ≥3 months comparing ≥2 antipsychotics and reporting incident TD cases. We aimed to describe TD risk by: a) class‐wise comparisons of pooled FGAs to each specific SGA, b) drug‐wise comparisons of a specific FGA to one or more specific SGAs, and c) comparisons between individual SGAs.
Based on our earlier study3, we hypothesized that the incidence and the relative TD risk would be lower with SGAs vs. FGAs. Moreover, based on the association between TD incidence and FGA dose12, we hypothesized that studies using high dose FGA comparators would drive the estimated risk reduction of SGAs vs. FGAs. Lastly, based on the association between TD incidence and early parkinsonian side effects12, we hypothesized that SGAs with a higher propensity for extrapyramidal side effects (EPS) would be associated to a higher risk for TD vs. SGAs with lower EPS potential.
METHODS
Literature search
Two authors (CC, MC) independently conducted a PubMed/Web of Science search without language or time restriction for comparative randomized antipsychotic studies (last search update: January 31, 2018). The following terms were used in the advanced search option: all fields (= any field): (antipsychotic*) AND (amisulprid* OR aripiprazol* OR asenapin* OR clozapine OR olanzapin* OR paliperidon* OR quetiapin* OR risperidon* OR sertindole OR ziprasidon* OR zotepin* OR iloperidon* OR cariprazin* OR brexpiprazol* OR lurasidone OR blonanserin OR melperone) AND (randomized controlled trial); publication type: NOT review.
Additionally, we hand‐searched references from Cochrane meta‐analyses on SGAs. Whenever data needed for the meta‐analysis were missing, we repeatedly contacted the authors for additional information and for access to data repositories for unpublished data.
Study inclusion criteria
We included all head‐to‐head comparisons of one of the above listed antipsychotics in any (oral or i.m.) form of administration to any FGA or other SGA without restriction on age or gender of participants. Requiring that at least one arm consisted of an SGA, we ensured that any FGA comparator was studied at the same time as SGAs were available, avoiding potential biases due to time effects regarding different patient populations and dosing schemas in trials before availability of SGAs.
Included studies had to provide information on the rate of probable treatment‐emergent TD in subjects free of TD at study baseline. The diagnosis of probable TD could be clinical or scale‐based, as long as diagnostic criteria were clearly defined and identical for all treatment arms. Fixed and flexible dose studies were eligible, as long as baseline randomization was present. Any psychiatric or medical diagnosis was allowed, except for movement disorders. Hence, antipsychotic trials in schizophrenia patients with a serious concomitant medical illness as an inclusion criterion were not excluded. Studies allowing concomitant or prophylactic anticholinergic medication were similarly included, and anticholinergic medication was assessed as moderator. Trials that allowed switching of treatments between groups were excluded. For trials which had a crossover design, only results from the first randomization period were considered, to avoid carry‐over effects.
A minimum trial duration of three months was considered for the identification of cases of probable dyskinesia. However, longer observation periods are more appropriate, and the duration of exposure was explored as moderator of TD incidence. Trials in which treatment consisted of concomitant use of ≥2 antipsychotics per individual were excluded, as we aimed to differentiate effects of specific antipsychotics. We did not exclude randomized, open‐label studies, but excluded these in sensitivity analyses to address the lack of blinding, which has been shown to be a substantial source of bias13.
Data extraction
Two authors (MC, CC) independently checked eligibility and extracted data. Any disagreement was resolved by discussion/consensus.
In addition to antipsychotic‐specific and class‐wise TD rates, we extracted: chlorpromazine equivalent dose for FGAs and olanzapine equivalent dose for SGAs (using the methods described by Leucht et al14, where 300 mg of chlorpromazine equals 10 mg of olanzapine according to the daily dose method), publication year, study design, geographic region, patient gender, age, ethnicity, clinical diagnoses, illness duration, disease severity, comorbid parkinsonism, TD rating scale and scores, diagnostic TD criteria. Data on TD severity were provided in a minority of studies only. Whenever possible, rates of persistent TD were used, as this measure is clinically more meaningful and less prone to variability than probable TD.
Statistical analysis
We conducted two parallel sets of pairwise, head‐to‐head analyses: FGA‐SGA across‐class comparisons and SGA‐SGA within‐class comparisons. In both sets of analyses, the effect size calculation was based on the pairing of treatment arms as in the original RCT to maintain control of confounding variables. Thus, we calculated an effect size for each available head‐to‐head combination. From all studies we extracted the number of TD cases per treatment arm (N = intent‐to‐treat, ITT) and the duration of antipsychotic exposure within this group. Then, we calculated the raw TD incidence risk, as the ratio of TD cases per total exposed subject number, and the annualized TD incidence risk, as cases per exposed subject number per time of antipsychotic exposure (i.e., person years), each with their respective 95% confidence intervals (CIs). Numbers‐needed‐to‐treat (NNTs) reflecting reduced annualized risk for TD were calculated dividing 1 by the rate difference.
We next used subgroup comparisons and meta‐regression to explore the relative risk change by antipsychotic class and for specific moderators: mean age, male gender percentage, Caucasian ethnicity percentage, dosing (dichotomizing mean/median study dose – i.e., <500 mg vs. ≥500 mg chlorpromazine equivalent, and <3 mg vs. ≥3 mg haloperidol in studies using haloperidol – or dichotomizing the maximum allowed haloperidol dose range: <10 mg vs. ≥10 mg), illness duration and stage, disease severity, comorbid parkinsonism, information on prior FGA exposure, publication year, study design, duration and sponsorship, data source, case definition, geographic region.
A multivariable analysis was performed, including significant moderators which had been identified in univariate analysis. Since TD risk is cumulative and higher the longer patients are followed, we conducted these moderator analyses only for annualized rate ratios (RaRs) that correct for any differences in observation time. All analyses used a random effects model and were two‐sided, with alpha=0.05. Data were analyzed with Comprehensive Meta‐Analysis Version 3.
RESULTS
Search results
Of 3,438 hits in PubMed and Web of Science, 273 full‐text articles were screened, resulting in 57 articles that fulfilled all inclusion criteria (Figure 1). Notably, a lack of TD reporting resulted in the exclusion of 75 studies of eligible cohorts.
Figure 1.

PRISMA flow chart. TD – tardive dyskinesia, RCTs – randomized controlled trials
Sample characteristics
FGA‐SGA studies
Thirty‐two randomized studies comparing FGAs to SGAs and providing data on TD incidence in 10,706 subjects were included5, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45. Twenty‐two of them were double‐blind studies5, 15, 17, 19, 21, 22, 23, 24, 25, 27, 28, 29, 31, 33, 36, 38, 39, 40, 41, 42, 43, 44 and ten were randomized open studies16, 18, 20, 26, 30, 32, 34, 37, 39, 45 (partly with blinded ratings). Haloperidol was the FGA comparator in the majority of studies. All but one study43 included subjects with schizophrenia‐spectrum disorders, and one third of the studies were industry independent5, 22, 23, 25, 28, 30, 34, 35, 36, 39, 42, 45.
Overall, the studies included 65.6% males, 63.2% Caucasians, with a mean age of 37.7±12.4 years and a mean illness duration of 13.2±12.6 years. The mean chlorpromazine equivalent dose was 423.9±252.4 mg/day (range: 69‐733.5 mg/day) for FGA treatment, and 522.6±199.3 mg/day (range: 118‐905.7 mg/day) for SGA treatment14. The mean PANSS total (from the fifteen studies reporting this measure5, 21, 22, 23, 24, 27, 28, 29, 30, 33, 34, 36, 38, 40, 42) was 76.3±20.2 at baseline. The median study duration (by design) was one year (interquartile range, IQR: 0.44‐2.0). The median observed follow‐up lasted 108 person years (IQR: 34.5‐254.3) for SGAs and 73.1 person years (IQR: 25.0‐117.6) for FGAs.
Few studies included only subjects with first‐episode or early‐phase schizophrenia. The reported mean illness duration was 14 years (IQR: 10.6‐17.1, k=21) for patients on SGAs and 13.7 years (IQR: 10.2‐17.4, k=21) for patients on FGAs. Data on prior FGA exposure were very sparse; sixteen studies5, 16, 24, 27, 29, 30, 32, 34, 35, 36, 37, 39, 41, 42, 45, 46 reported that included subjects had prior FGA exposure. The mean percentage of patients with pre‐baseline FGA exposure was 76.4±30.5% (IQR: 40.0‐100%, k=12).
The majority (81.3%) of FGA‐SGA studies used standardized screening instruments to detect and report dyskinesia. The Abnormal Involuntary Movement Scale (AIMS) was mostly used in conjunction with Schooler‐Kane criteria for probable TD. Four studies5, 21, 23, 32 reported both probable and persistent TD, and five studies15, 22, 24, 38, 42 reported persistent TD only. Systematic screening was also performed using the Extrapyramidal Symptom Rating Scale (ESRS)19, 23, 24, 41, 47, the St. Hans Rating Scale30 and the Dyskinesia Rating Scale25. Only six studies assessed TD based on clinical reporting16, 17, 26, 28, 39, 45.
SGA‐SGA studies
Twenty‐three randomized studies comparing one SGA to another SGA and providing data on TD incidence in 9,153 subjects were included in the analysis (comparators: olanzapine31, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, clozapine60, 61, 62, or risperidone63, 64, 65, 66, 67, 68).
SGA‐SGA comparisons also included treatment arms from FGA‐SGA studies with randomization into multiple SGA arms and with TD data reporting by specific antipsychotic, thus adding data on SGA treatment from five studies listed also above5, 30, 31, 34, 41.
Based on the frequency of included studies and their use of specific antipsychotics (but not a priori), the SGA‐SGA studies were grouped into the following three subsets for analyses: a) 19 comparisons with olanzapine as one comparator; b) three comparisons with clozapine as one comparator; c) six comparisons with risperidone or paliperidone (which is metabolized from risperidone) as one comparator. This latter group excluded olanzapine as the other comparator, as these head‐to‐head comparisons had been included in the first subset. We used sensitivity analyses to separate the effect of risperidone‐olanzapine head‐to‐head comparisons in the studies of the first and third group.
The majority of studies included adults with schizophrenia spectrum disorders; two studies included subjects with bipolar disorder57, 64; two subjects with acute psychosis51, 52; one patients with first‐episode psychosis48; and one patients with pediatric schizophrenia or schizoaffective disorder22.
Overall, these studies included 62.2% males, 67.0% Caucasian, with a mean age of 37.0±10.7 years and a mean illness duration of 11.8±6.8 years. Four studies included only subjects with first‐episode or early‐phase schizophrenia22, 30, 48, 51. Data on prior FGA exposure were reported only in six studies (being 0‐33% in three studies30, 47, 51, and >50% in two5, 50).
SGA doses converted into mean olanzapine equivalent doses14 were: a) for studies with olanzapine, 13.5±3.4 mg/day (range: 4.8‐20.1 mg/day) for olanzapine and 10.6±2.9 mg/day (range: 6.8‐13.6 mg/day) for the comparators; b) for studies with clozapine, 10.7 mg/day for clozapine and 21.0±4.6 mg/day (range: 19.0‐23.4 mg/day) for the comparators; c) for studies with risperidone or paliperidone, 12.1±3.2 mg/day (range: 8.6‐16.0 mg/day) for risperidone/paliperidone and 12.3±2.7 mg/day (range: 8.6‐15.0 mg/day) for the comparators. The mean PANSS total (from studies reporting this measure22, 47, 48, 49, 52, 53, 54, 55, 56, 60, 61, 62, 64, 65, 66, 67) was 79.5±16.6 at baseline.
In contrast to FGA‐SGA studies, the use of standardized TD screening instruments was slightly lower in SGA‐SGA studies (64.3%), with a third of studies relying on clinical or patient reporting48, 50, 52, 54, 58, 63, 67, 68. The majority of studies reported probable TD, except for CATIE5.
Full sample analyses: mean TD incidence in randomized studies
The estimated weighted mean incidence of TD across all FGA treatment groups was 6.5% (95% CI: 4.6‐9.0%) for 3,763 subjects in 32 treatment arms. Similarly, the annualized incidence was 6.5% (95% CI: 5.3‐7.8%; see Table 1 for specific FGAs).
Table 1.
Incidence of tardive dyskinesia (TD) for specific antipsychotics
| Mean raw TD incidence | Mean annualized TD incidence | |||||
|---|---|---|---|---|---|---|
| Antipsychotic | N. studies/treatment arms | N. subjects | % | 95% CI | % | 95% CI |
| FGAs | ||||||
| Perphenazine | 1 | 853 | 3.3 | 0.5‐17.4 | 3.7 | 0.1‐6.7 |
| Molindone | 1 | 20 | 2.4 | 0.1‐38.5 | 4.2 | −0.8 to 16.5 |
| Haloperidol | 22 | 2,975 | 6.6 | 4.5‐9.6 | 7.5 | 5.9‐9.2 |
| Chlorpromazine | 1 | 80 | 21.3 | 4.4‐60.1 | 11.2 | 4.8‐17.7 |
| Fluphenazine | 1 | 28 | 3.6 | 3.0‐32.8 | 12.5 | 12.3‐37.3 |
| SGAs | ||||||
| Aripiprazole | 3 | 1,215 | 0.9 | 0.3‐3.1 | 1.7 | −0.8 to 4.1 |
| Amisulpride | 3 | 558 | 1.5 | 0.4‐5.4 | 2.4 | −0.4 to 5.2 |
| Asenapine | 4 | 1,472 | 1.2 | 0.5‐3.3 | 2.4 | −0.1 to 4.8 |
| Risperidone | 20 | 853 | 4.2 | 2.6‐6.6 | 2.4 | 1.2‐3.5 |
| Quetiapine | 6 | 221 | 2.8 | 0.2‐1.1 | 2.5 | 0.2‐4.8 |
| Olanzapine | 29 | 5,686 | 2.7 | 1.9‐4.0 | 2.9 | 1.8‐3.9 |
| Ziprasidone | 7 | 918 | 3.5 | 1.6‐7.5 | 3.5 | 1.3‐5.7 |
| Clozapine | 6 | 348 | 8.2 | 3.9‐16.6 | 4.2 | 1.7‐6.7 |
| Lurasidone | 1 | 427 | 2.6 | 0.5‐13.1 | 4.8 | −0.2 to 9.3 |
The TD rates for the individual medications are not directly comparable, as they do not originate from randomized trials in which those medications were compared head‐to‐head, but are pooled irrespective of study comparators. FGAs – first‐generation antipsychotics, SGAs – second‐generation antipsychotics
The estimated weighted mean incidence of TD across all SGA treatment groups was 3.0% (95% CI: 2.4‐3.8%) for 15,092 subjects in 86 treatment arms. The annualized incidence was 2.6% (95% CI: 2.0‐3.1%; see Table 1 for specific SGAs).
As randomization with regard to TD was unbalanced in the CATIE study (subjects with a history of TD were barred from randomization to FGA treatment), a sensitivity analysis was performed after excluding the CATIE data, yielding a weighted mean annualized incidence of TD for FGAs of 6.8% (95% CI: 5.5‐8.1%) and for SGAs of 2.6% (95% CI: 2.1‐3.2%).
Further sensitivity analyses corroborated the range of observations above, but also showed slightly lower rates for SGAs after exclusion of clozapine studies (weighted mean annualized incidence of 2.4%, 95% CI: 1.9‐3.0).
TD incidence in studies using only clinical reporting (but not systematic screening) was 3.8% (95% CI: 2.1‐6.7%) for FGA arms, and 0.9% (95% CI: 0.3‐2.4%) for SGA arms.
FGA‐SGA sample analyses: TD risk and moderators of TD risk differences
Of the 32 FGA‐SGA included studies, only 28 contributed analytically to the comparative TD risk analysis, as meta‐analytic risk calculations exclude studies with zero events in both treatment arms.
Primary analysis
The estimated TD risk ratio (RR) was significantly lower with SGAs relative to FGAs (RR=0.47, 95% CI: 0.39‐0.57, p<0.0001, k=28). Similarly, the estimated TD RaR, reflecting the annualized incidence, was significantly lower in SGAs relative to FGAs (RaR=0.35, 95% CI: 0.28‐0.45, p<0.0001, k=28; NNT=20, 95% CI: 15‐31) (Figure 2).
Figure 2.
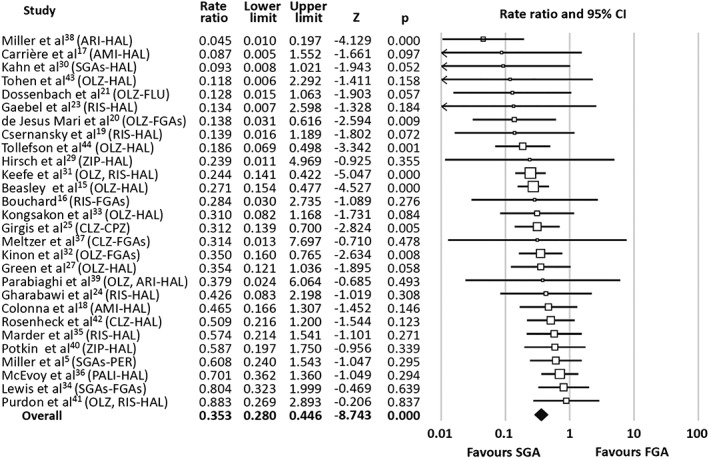
Forest plot of tardive dyskinesia rate ratios of randomized studies comparing first‐generation antipsychotics (FGAs) to second generation antipsychotics (SGAs). ARI – aripiprazole, AMI – amisulpride, CLZ – clozapine, CPZ – chlorpromazine, HAL – haloperidol, OLZ – olanzapine, PALI – paliperidone, PER – perphenazine, RIS – risperidone, ZIP – ziprasidone
The estimated TD RR after exclusion of CATIE data was 0.35 (95% CI: 0.27‐0.44, p<0.0001, k=27). The TD RaR after CATIE exclusion was 0.34 (95% CI: 0.27‐0.44, p<0.0001, k=27).
Moderator analyses of TD RaRs
The mean TD RaR varied significantly by SGA comparator (Q=21.8, df=7, p=0.003). The advantage of SGAs vs. FGAs was most prominent with aripiprazole and smallest with quetiapine (Table 2). Exploratory pairwise comparisons showed significantly lower RaRs with aripiprazole relative to all other SGAs. Moreover, RaRs with olanzapine were also significantly lower relative to risperidone and quetiapine (Table 3).
Table 2.
Mean annualized tardive dyskinesia (TD) rate ratios in SGAs vs. FGAs
| SGA | N. treatment arms | N. subjects | Mean TD rate ratio vs. FGA (95% CI) | p |
|---|---|---|---|---|
| Amisulpride | 3 | 558 | 0.37 (0.15‐0.91) | 0.032 |
| Aripiprazole | 1 | 1,215 | 0.045 (0.01‐0.19) | 0.000 |
| Clozapine | 2 | 405 | 0.39 (0.22‐0.70) | 0.001 |
| Olanzapine | 12 | 5,624 | 0.25 (0.19‐0.34) | 0.000 |
| Paliperidone | 1 | 145 | 0.70 (0.35‐1.36) | 0.294 |
| Quetiapine | 2 | 786 | 0.94 (0.35‐0.91) | 0.915 |
| Risperidone | 8 | 2,479 | 0.38 (0.25‐0.58) | 0.000 |
| Ziprasidone | 4 | 887 | 0.57 (0.26‐1.27) | 0.169 |
FGAs – first‐generation antipsychotics, SGAs – second‐generation antipsychotics
Table 3.
Moderating effects of the comparator SGA on annualized rate ratios of treatment emergent tardive dyskinesia in studies comparing FGAs vs. SGAs
| SGA comparator in FGA study | Amisulpride | Clozapine | Olanzapine | Quetiapine | Risperidone | Ziprasidone |
|---|---|---|---|---|---|---|
| Aripiprazole | Q=5.75, p=0.017 | Q=7.14, p=0.008 | Q=5.04, p=0.025 | Q=4.24, p=0.039 | Q=11.27, p=0.004 | Q=8.84, p=0.003 |
| Amisulpride | ‐ | Q=0.07, p=0.94 | Q=0.65, p=0.42 | Q=1.84, p=0.17 | Q=2.84, p=0.28 | Q=0.49, p=0.49 |
| Clozapine | ‐ | Q=1.72, p=0.19 | Q=2.72, p=0.13 | Q=2.52, p=0.28 | Q=0.58, p=0.45 | |
| Olanzapine | ‐ | Q=6.12, p=0.013 | Q=8.5, p=0.015 | Q=3.6, p=0.06 | ||
| Quetiapine | ‐ | Q=4.15, p=0.15 | Q=0.608, p=0.43 | |||
| Risperidone | ‐ | Q=2.58, p=0.27 |
Significant effects are highlighted in bold prints. FGAs – first‐generation antipsychotics, SGAs – second‐generation antipsychotics
The mean TD RaR did not vary significantly by FGA comparator (Q=0.23, df=1, p=0.63). The advantage of SGAs over FGAs persisted after exclusion of all studies in which haloperidol was used as the FGA comparator (RaR=0.39, 95% CI: 0.25‐0.61, p<0.0001, k=8). In those studies with haloperidol as the comparator, similar RaRs were obtained (RaR=0.34, 95% CI: 0.26‐0.45, p<0.0001, k=20). In the studies with specified, non‐haloperidol FGAs5, 21, 25, 34, a somewhat weaker, but still significant advantage of SGAs over FGAs emerged (RaR=0.47, 95% CI: 0.27‐0.82, p=0.007, k=4). The comparison with all other studies (i.e., those that included haloperidol as the only, or as one possible FGA) was non‐significant (Q=1.27, df=1, p=0.26).
TD RaRs were similar in first episode/early‐phase psychosis cohorts22, 23, 24, 25, 27, 30 vs. the remaining unrestricted cohorts (Q=0.04, df=1, p=0.85). Prior FGA exposure was not consistently reported, and no cohort explicitly included only subjects without prior FGA exposure.
TD RaRs varied significantly by sponsorship (Q=10.0, df=1, p=0.003). The reduction in TD incidence associated with SGA treatment was accentuated in industry‐sponsored relative to academic studies, but persisted independently in both of them (academic studies: RaR=0.59, 95% CI: 0.39‐0.87, p=0.008, k=11; industry‐sponsored studies: RaR=0.28, 95% CI: 0.21‐0.35, p<0.001, k=20).
A mixed regression model including SGA comparator and sponsorship (Q=20.74, df=6, p=0.002) confirmed the independent effect of SGA comparator (Q=10.78, df=5, p=0.05), while no significant effect of sponsorship was found (Z=−1.2, df=1, p=0.23).
Moderator and meta‐regression analyses were non‐significant for age, gender, illness duration (both as years of disease duration and categorically as first episode/early‐phase psychosis vs. other cohorts), disease severity, study region, study duration, anticholinergic use, study design (open‐label extension vs. blinded trial), year of study start, and case definition. There was no effect of FGA dose on TD rate ratios, neither when data were dichotomized (below vs. above 500 mg chlorpromazine equivalents, Q=0.19, df=1, p=0.66), nor when mean FGA dose in chlorpromazine equivalents was used in a meta‐regression model (Z=−1.03, p=0.30). Similarly, there was no effect of the FGA‐SGA ratio on TD RaRs (Z=1.56; p=0.12).
Data on TD severity were provided in a minority of studies (31.3%). TD severity was described in very heterogeneous formats, which did not allow for the use of this variable in the meta‐analysis. Discontinuation due to TD was reported rarely, but slightly more frequently in FGAs (N=1 in FGA vs. N=0 in SGA21; 2.7% in FGA vs. 0.7% with paliperidone36). Greater severity of TD with FGAs was reported repeatedly5, 23, 32, 35, 38. Reports of severe cases were rare in general, and found only in FGA‐treated subjects in four studies27, 38, 40, 41.
SGA‐SGA sample analyses: TD risk and moderators of TD RaR differences
Olanzapine vs. all other non‐clozapine SGAs
Of the 28 SGA‐SGA included studies, the most frequently studied SGA was olanzapine, which was a SGA comparator in 17 studies against other non‐clozapine SGAs. As CATIE5 and EUFEST30 randomized patients also to SGA treatment arms, 19 comparative olanzapine/non‐clozapine SGA studies were available. In these 19 studies, comparators were risperidone (k=9), asenapine (k=5), quetiapine (k=4), ziprasidone (k=3), amisulpiride (k=1) and aripiprazole (k=1). The mean dose in the olanzapine treatment arms was 13.5±3.4 mg/day, being 10.6±2.9 mg/day in the comparators, recalculated via mean olanzapine equivalent for all14. Three studies22, 52, 68 reported no cases of TD in all arms and did thus not contribute to risk calculations, as the risk of non‐occurrence is formally inestimable.
The estimated TD risk ratio was significantly lower with olanzapine relative to other non‐clozapine SGAs (RR=0.67, 95% CI: 0.50‐0.90, p=0.008, k=17; NNT=100, 95% CI: 63‐250). Similarly, the estimated TD RaR was significantly lower with olanzapine relative to non‐clozapine SGAs (RaR=0.66, 95% CI: 0.49‐0.88, p=0.006, k=17).
Moderator analyses of TD RaRs were non‐significant, including age, gender, study region, olanzapine equivalent dose, anticholinergic use, percentage of subjects with prior FGA exposure, year of study start, illness duration, illness stage (first‐episode yes/no), sponsorship, and comparator SGA study design.
Treatment‐related TD RaRs were significantly lower with olanzapine relative to risperidone (RaR=0.57, 95% CI: 0.37‐0.89, p=0.015, k=6), which represented the largest comparator subgroup. No other differences were found for the other SGAs when analyzed in exploratory pairwise comparisons.
To address the possibility that the head‐to‐head studies comparing olanzapine vs. risperidone were driving the favorable TD RaRs of olanzapine vs. non‐clozapine SGAs, we excluded those studies in a sensitivity analysis. The TD RaR remained significantly lower with olanzapine relative to all other non‐clozapine and non‐risperidone SGAs (RaR=0.68, 95% CI: 0.46‐0.99, p=0.047, k=12).
Clozapine vs. non‐clozapine SGAs
There were only three studies comparing clozapine against another SGA, with two of them vs. olanzapine61, 62, and one vs. mixed SGAs60, where olanzapine was used in 50% of subjects. These studies included only subjects with a schizophrenia spectrum disorder diagnosis. Subjects were 71% male, 77% Caucasian, with a mean age of 38.7±1.1 years, a mean illness duration of 17.3±5.1 years, and mean doses of 318.0±17.1 mg/day for clozapine and 21.0±2.2 mg/day for olanzapine.
There was no difference regarding probable treatment‐emergent TD between clozapine and non‐clozapine SGAs (RR=1.07, 95% CI: 0.49‐2.34, p=0.86, k=3; RaR=1.10, 95% CI: 0.66‐1.90, p=0.71, k=3).
Risperidone/paliperidone vs. non‐olanzapine SGAs
There were six studies comparing risperidone or paliperidone against a different non‐olanzapine/non‐clozapine SGA, four of which used long‐acting injectables (LAIs) (risperidone‐LAI = 3, paliperidone‐LAI = 1), and additional treatment arms from CATIE were also included in this subgroup comparison. The comparators were aripiprazole (k=2), lurasidone (k=1), quetiapine (k=1), ziprasidone (k=1) and mixed SGAs (k=1).
Five of the studies included subjects with a schizophrenia spectrum disorder diagnosis, and one was performed in bipolar disorder patients64. Subjects were 56% male, 51% Caucasian, with a mean age of 36.8±5.7 years, and a mean illness duration of 10.9±3.3 years.
There was no difference in RRs or RaRs of probable treatment‐emergent TD between risperidone/paliperidone treatment arms and SGA comparators (RR=0.88; 95% CI: 0.50‐1.56, p=0.66, k=7; RaR=0.94, 95% CI: 0.65‐1.35, p=0.72, k=7).
To address the influence of risperidone‐olanzapine head‐to‐head comparisons that were grouped primarily into the first analysis, we added these studies in a sensitivity analysis to the third set analyses. Doing so, there was no difference in RaRs for probable treatment‐emergent TD between risperidone/paliperidone treatment arms and all other SGA comparators including olanzapine (RaR=1.24, 95% CI: 0.91‐1.60, p=0.20, k=12).
DISCUSSION
Class‐wise TD risk
This meta‐analysis on treatment‐emergent TD during comparative randomized controlled trials shows an overall low, but still clinically relevant incidence of TD (annualized incidence: FGAs=6.5%; SGAs=2.6%). However, these rates are based only on probable and not persistent TD and may be inflated due to rating scale based assessments that do not take into consideration severity or impact. Conversely, the raw TD incidence rates based on clinical observation (FGAs=3.8%, SGAs=0.9%) have clinical face validity, but may be underestimates.
We confirmed our hypothesis of lower incidence rates of TD during SGA vs. FGA treatment, with a relative risk and annualized rate reduction to one third of the FGA rate and a NNT=20. Interestingly, the rate of clinically reported, but not screening‐based TD, which may reflect the more severe and clinically relevant cases, matches earlier incidence reports of TD risk during clinical FGA use and during early clinical trials with SGA treatment3.
The significant TD risk reduction with SGAs found in this meta‐analysis contrasts to the findings of the UK‐based CUtLASS‐1 study4 and the US‐based CATIE study5, which both conveyed the impression that the TD risk of FGAs and SGAs did not differ. While our dataset consisted predominantly of studies that used haloperidol as the FGA comparator, our analyses exploring the role of haloperidol as the comparator showed that it did not drive the FGA‐SGA class difference. TD RaRs in studies with haloperidol as the FGA comparator did not differ from those that used pooled groups of mixed FGAs (including CATIE and CUtLASS); and both subgroups of FGA comparators were independently associated with significantly greater TD risk than the respective SGA comparators.
Notably, CATIE and CUtLASS had methodological limitations regarding the specific question of EPS, as they were designed to assess antipsychotic efficacy. Contrasting to the remaining body of studies included in this meta‐analysis, the FGA arms of the CATIE study5 did not include subjects with a history of TD, as these subjects are at increased risk of TD upon re‐exposure to FGAs. Similarly, contrasting to the remaining body of studies included in this meta‐analysis, the FGA arm of the CUtLASS‐1 study4 was dominated by the use of sulpiride (58% of the FGA group). This antipsychotic has been categorized as atypical based on its high dissociation constant at the D2 receptor69, consistent with its exceptionally low EPS risk70. Interestingly, our results are close to the findings of earlier large incidence studies in clinical cohorts that demonstrated a significant risk reduction for SGAs vs. FGAs of 0.5171 and of 0.55 after adjustment for lifetime antipsychotic exposure72.
Due to the relatively low absolute annual risk of TD even during FGA treatment, the NNT to achieve a risk reduction for TD of 20 may appear too high to warrant an influence on clinical antipsychotic choice. On the other hand, the annual risk underestimates the individual lifetime risk in chronic mental illness, where antipsychotic exposure times range around six years in 40 year olds72, being closer to 15 years in people with schizophrenia spectrum disorders5. As the individual risk to develop TD is cumulative, at least during the first five years, the risk reduction should also be seen as a cumulative gain throughout the expected treatment period in each individual.
Contrary to earlier suggestions, the advantage of SGAs over FGAs was not driven by high dose FGA studies, as dose was excluded as a systematic confound of the FGA‐SGA risk comparisons. The potential dose effect was consistently ruled out via different approaches: a) mean/median study dose; b) <500 mg vs. ≥500 mg chlorpromazine equivalent and <3 mg vs. ≥3 mg haloperidol; and c) <10 mg vs. ≥10 mg haloperidol. Importantly, this finding does contradict the repeated observation that the individual risk for TD increases with higher FGA doses and clinically relevant EPS12. Other risk factors, which are established for individual TD risk, such as age and gender, also had no influence on the comparison of TD risks by antipsychotic class, suggesting that antipsychotic dose, age and gender generalize as risk factors across different antipsychotics.
Consistent with our hypothesis, however, moderator analyses showed that TD risk reduction with SGAs differed with specific SGA comparator. The relative risk reduction in comparisons vs. FGAs was most pronounced in studies with aripiprazole or olanzapine as SGA comparator. However, this information has to be interpreted cautiously, as TD RRs and RaRs vs. FGAs were accentuated in industry‐sponsored studies. The majority of olanzapine studies was industry sponsored and, although the mixed regression model formally confirmed the independent effect of SGA comparator, while no effect of sponsorship was found, no definite statement on this interaction can be made due to the effect of factor collinearity between olanzapine treatment and industry sponsorship.
Conversely, why would industry‐sponsored trials with predominantly blinded ratings provide more favourable TD rates for SGAs? The industry‐sponsored studies differed regarding the following factors: a) a higher number of subjects in FGA treatment arms (but the number of subjects per treatment arm is controlled by weighting in meta‐analyses); b) higher dose FGA comparator studies; c) higher mean age; d) fewer early psychosis cohorts; and e) more clinical reporting‐based TD assessment, which likely disfavours treatments with more severe TD expression, such as FGAs. Nevertheless, none of these factors was a significant moderator of RaRs in this meta‐analysis.
Within‐SGA class comparisons
Consistent with the TD risk reduction of olanzapine and aripiprazole relative to FGAs, SGA‐SGA comparisons seemed to confirm that agents with the lowest acute EPS risk73, i.e., clozapine and olanzapine, also have the lowest TD risk. However, TD rate differences within the SGA class, based on an NNT of 100 for olanzapine vs. other non‐clozapine SGAs, were rather subtle, and quetiapine – that meta‐analytically has very similar acute EPS risk to olanzapine and aripiprazole73 – had the highest individual TD risk among SGA comparator trials.
Contrasting to the FGA‐SGA comparisons, no effect of study sponsor was present in the SGA‐SGA analyses. However, most of these studies were industry‐sponsored, thus reducing the ability to assess the impact of this factor. Nevertheless, the NNT of 100 for the advantage of olanzapine underscores the need to consider other adverse effects, particularly cardiometabolic risk, when making SGA choices.
Interestingly, and consistent with an earlier clinical TD incidence study72, the TD risk of clozapine and olanzapine was similar in the three relevant studies. However, clozapine was relatively under‐dosed in those studies. Furthermore, there were no studies of clozapine vs. non‐olanzapine SGAs, a shortcoming that is all the more unfortunate, as olanzapine and clozapine share the major disadvantage of very significant weight gain. Since aripiprazole also seemed to have a particularly lower TD risk vs. FGAs, comparing a D2 partial agonist to clozapine for TD risk would be valuable.
In contrast to our initial hypothesis that SGAs with higher EPS potential, such as risperidone, would also have a higher TD risk, there were no notable differences of TD risks between non‐olanzapine, non‐clozapine SGAs, including risperidone/paliperidone in particular, but analyzable data were limited.
Limitations
Multiple limitations have to be taken into consideration when interpreting these results.
First, despite attempts to access this information, 75 randomized studies with an appropriate duration to provide information on TD risk could not be included in the analysis, as they did not provide any information on TD (and we were unable to obtain such information from the authors). This number of studies signifies a tremendous loss of information. Importantly, 32 of these studies reported on systematic screening in their methods, but did not provide information on TD cases. Instead, reporting of continuous sum scores of various dyskinesia‐rating scales is common, but not informative, as has been discussed before10, 11.
Second, meta‐analyses cannot estimate the risk of non‐occurrence. Thus, studies that did not observe any cases of TD in either arm did not contribute to risk calculations. They contribute to RARs though, thus their effect is reflected in the NNTs. On the one hand, this is an unfortunate shortcoming of the methodology; on the other hand, the data quality of large studies in adults with chronic mental illness without a single case of probable incident TD may also be questionable. Altogether, five studies including four treatment arm pairs for FGA‐SGA comparisons22, 26, 28, 45 and two treatment arm pairs for SGA‐SGA comparisons22, 52 did not contribute to the meta‐analytic risk estimates. Subsequently, the estimated raw TD rate is likely slightly lower under controlled study conditions, and similarly the observed differences may also be minimally lower.
Third, as the minimal exposure duration before TD can be diagnosed is >3 months6, 7, this time frame was chosen during title/abstract screening for eligible studies. However, within the time bin of studies lasting <6 months, only one (4‐month) study formally reported treatment‐emergent dyskinesia. We, thus, refrained from systematically requesting data on TD from studies lasting <6 months (unless a formal screening for dyskinesia was part of the study design). For all studies lasting ≥6 months, data were requested (even if the study design did not include formal TD screening). It is often argued that shorter observation periods are contaminated by high rates of withdrawal dyskinesia. While this argument can only be tested on a case‐by‐case evaluation of emerging dyskinesia cases, the observation of significantly increasing hyperkinesias during the early treatment phase with haloperidol, but not with risperidone74, argues against this notion. The median study duration was one year, but longer observation periods would be desirable.
Fourth, most studies reported probable mild TD, likely providing an overestimate of clinically relevant cases. While this is a cautious, safety‐oriented and well‐recognized strategy, it would be important to learn about the rates of persistent TD along with scale‐based severity measures. On the other, conservative end of the sensitivity spectrum, some studies, and increasingly more in SGA‐SGA comparator trials, only provide rates of clinically reported TD cases, not relying on rating scale information. Our moderator analysis did not identify a systematic influence of screening techniques on RRs or RaRs, but differences in primary data acquisition need to be considered when interpreting the results.
Fifth, the prevalence of TD in FGA‐naïve cohorts treated with SGAs has been reported to be lower than in subjects with a history of FGA exposure11. There was, however, not a single head‐to‐head RCT to test the effect of prior FGA exposure on TD rates in the FGA‐SGA comparison, and relatively sparse information on this issues in the SGA‐SGA comparisons.
Sixth, as the absolute TD risk gets lower (in SGA studies in general, relative to all FGA studies), the influence of potential co‐factors, such as co‐treatment with other potentially TD‐causing medications (e.g., metoclopramide75, 76, flunarizine77, or antidepressants78) may represent a confound that has not been addressed in the primary sources.
EPS have been characterized as the most important factor predicting non‐adherence during FGA treatment79. By contrast, akathisia, parkinsonism and dyskinesia failed to predict adherence during the EUFEST and CATIE studies80, but predicted adherence in another large study including more than 2,000 patients treated with SGAs or FGAs81. Considering the high incidence of clinically meaningful weight gain during SGA treatment82, EPS partly lost their weight in clinical decision‐making. Nevertheless, TD remains a clinical problem in movement disorders clinics: SGA‐related tardive syndromes lately outnumbered FGA‐related TD cases in a movement disorders clinic83, likely due to increasing on‐label, as well as off‐label use of SGAs.
CONCLUSIONS
In conclusion, the historic notion of an SGA advantage with regard to TD risk, which had been challenged by CATIE and CUtLASS, has been confirmed.
Importantly, the quality of data acquisition and reporting of TD needs to return to earlier standards in future comparative studies to understand whether or not SGAs differ among one another with regard to TD risk. Risk estimates should span incidence, severity and persistence as well as impact on functioning and quality of life. Ideally, patient‐level data repositories should help to accurately estimate rare side effects in antipsychotic studies.
ACKNOWLEDGEMENTS
The authors thank C. Arango, L. Citrome, M.R.K. Dossenbach, R. Rosenheck, S.W. Lewis, K.P. Hayhurst, P. Tyrer, S. Marder, D. Naber, S.G. Potkin, A. Parabiaghi, S.E. Purdon, A.I. Green, R.S. Keefe, R. Kongsakon, G. Gründer, B.J. Kinon, M.J. Cuesta, E. Johnsen, and the Lilly data source management team for providing unpublished data, and for clarifying published data.
REFERENCES
- 1. Yassa R, Jones BD. Complications of tardive dyskinesia: a review. Psychosomatics 1985;26:305‐7. [DOI] [PubMed] [Google Scholar]
- 2. Strassnig M, Rosenfeld A, Harvey PD. Tardive dyskinesia: motor system impairments, cognition and everyday functioning. CNS Spectr 2017;7:1‐8. [DOI] [PubMed] [Google Scholar]
- 3. Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second‐generation antipsychotics: a systematic review of 1‐year studies. Am J Psychiatry 2004;161:414‐25. [DOI] [PubMed] [Google Scholar]
- 4. Peluso MJ, Lewis SW, Barnes TR et al. Extrapyramidal motor side‐effects of first‐ and second‐generation antipsychotic drugs. Br J Psychiatry 2012;200:387‐92. [DOI] [PubMed] [Google Scholar]
- 5. Miller DD, Caroff SN, Davis SM et al. Extrapyramidal side‐effects of antipsychotics in a randomised trial. Br J Psychiatry 2008;193:279‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schooler NR, Kane JM. Research diagnoses for tardive dyskinesia. Arch Gen Psychiatry 1982;39:486‐7. [DOI] [PubMed] [Google Scholar]
- 7. American Psychiatric Association . Diagnostic and statistical manual of mental disorders, 5th ed. Arlington: American Psychiatric Publishing, 2013. [Google Scholar]
- 8. Komossa K, Rummel‐Kluge C, Hunger H et al. Olanzapine versus other atypical antipsychotics for schizophrenia. Cochrane Database Syst Rev 2010;3:CD006654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Komossa K, Rummel‐Kluge C, Schmid F et al. Quetiapine versus other atypical antipsychotics for schizophrenia. Cochrane Database Syst Rev 2010;1:CD006625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Haddad PM, Das A, Keyhani S et al. Antipsychotic drugs and extrapyramidal side effects in first episode psychosis: a systematic review of head‐head comparisons. J Psychopharmacol 2012;26:15‐26. [DOI] [PubMed] [Google Scholar]
- 11. Carbon M, Hsieh CH, Kane JM et al. Tardive dyskinesia prevalence in the period of second‐generation antipsychotic use: a meta‐analysis. J Clin Psychiatry 2017;78:e264‐78. [DOI] [PubMed] [Google Scholar]
- 12. Jankelowitz SK. Treatment of neurolept‐induced tardive dyskinesia. Neuropsychiatr Dis Treat 2013;9:1371‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Leucht S, Kissling W, Davis JM. How to read and understand and use systematic reviews and meta‐analyses. Acta Psychiatr Scand 2009;119:443‐50. [DOI] [PubMed] [Google Scholar]
- 14. Leucht S, Samara M, Heres S et al. Dose equivalents for antipsychotic drugs: the DDD method. Schizophr Bull 2016;42(Suppl. 1):S90‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Beasley CM, Dellva MA, Tamura RN et al. Randomised double‐blind comparison of the incidence of tardive dyskinesia in patients with schizophrenia during long‐term treatment with olanzapine or haloperidol. Br J Psychiatry 1999;174:23‐30. [DOI] [PubMed] [Google Scholar]
- 16. Bouchard RH. Longitudinal comparative study of risperidone vs classical neuroleptics in the treatment of schizophrenia: 24 months of observation. Encéphale 2002;28:S31‐2. [PubMed] [Google Scholar]
- 17. Carrière P, Bonhomme D, Lempérière T. Amisulpride has a superior benefit/risk profile to haloperidol in schizophrenia: results of a multicentre, double‐blind study (the Amisulpride Study Group). Eur Psychiatry 2000;15:321‐9. [DOI] [PubMed] [Google Scholar]
- 18. Colonna L, Saleem P, Dondey‐Nouvel L et al. Long‐term safety and efficacy of amisulpride in subchronic or chronic schizophrenia. Amisulpride Study Group. Int Clin Psychopharmacol 2000;15:13‐22. [DOI] [PubMed] [Google Scholar]
- 19. Csernansky JG, Mahmoud R, Brenner R et al. A comparison of risperidone and haloperidol for the prevention of relapse in patients with schizophrenia. N Engl J Med 2002;346:16‐22. [DOI] [PubMed] [Google Scholar]
- 20. de Jesus Mari J, Lima MS, Costa AN et al. The prevalence of tardive dyskinesia after a nine month naturalistic randomized trial comparing olanzapine with conventional treatment for schizophrenia and related disorders. Eur Arch Psychiatry Clin Neurosci 2004;254:356‐61. [DOI] [PubMed] [Google Scholar]
- 21. Dossenbach MR, Folnegovic‐Smalc V, Hotujac L et al. Double‐blind, randomized comparison of olanzapine versus fluphenazine in the long‐term treatment of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 2004;28:311‐8. [DOI] [PubMed] [Google Scholar]
- 22. Findling RL, Johnson JL, McClellan J et al. Double‐blind maintenance safety and effectiveness findings from the Treatment of Early‐Onset Schizophrenia Spectrum (TEOSS) study. J Am Acad Child Adolesc Psychiatry 2010;49:583‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gaebel W, Riesbeck M, Wolwer W et al. Maintenance treatment with risperidone or low‐dose haloperidol in first‐episode schizophrenia: 1‐year results of a randomized controlled trial within the German Research Network on Schizophrenia. J Clin Psychiatry 2007;68:1763‐74. [DOI] [PubMed] [Google Scholar]
- 24. Gharabawi GM, Bossie CA, Zhu Y. New‐onset tardive dyskinesia in patients with first‐episode psychosis receiving risperidone or haloperidol. Am J Psychiatry 2006;163:938‐9. [DOI] [PubMed] [Google Scholar]
- 25. Girgis RR, Phillips MR, Li X et al. Clozapine v. chlorpromazine in treatment‐naive, first‐episode schizophrenia: 9‐year outcomes of a randomised clinical trial. Br J Psychiatry 2011;199:281‐8. [DOI] [PubMed] [Google Scholar]
- 26. Glick ID, Marder SR. Long‐term maintenance therapy with quetiapine versus haloperidol decanoate in patients with schizophrenia or schizoaffective disorder. J Clin Psychiatry 2005;66:638‐41. [DOI] [PubMed] [Google Scholar]
- 27. Green AI, Lieberman JA, Hamer RM et al. Olanzapine and haloperidol in first episode psychosis: two‐year data. Schizophr Res 2006;86:234‐43. [DOI] [PubMed] [Google Scholar]
- 28. Grunder G, Heinze M, Cordes J et al. Effects of first‐generation antipsychotics versus second‐generation antipsychotics on quality of life in schizophrenia: a double‐blind, randomised study. Lancet Psychiatry 2016;3:717‐29. [DOI] [PubMed] [Google Scholar]
- 29. Hirsch SR, Kissling W, Bauml J et al. A 28‐week comparison of ziprasidone and haloperidol in outpatients with stable schizophrenia. J Clin Psychiatry 2002;63:516‐23. [DOI] [PubMed] [Google Scholar]
- 30. Kahn RS, Fleischhacker WW, Boter H et al. Effectiveness of antipsychotic drugs in first‐episode schizophrenia and schizophreniform disorder: an open randomised clinical trial. Lancet 2008;371:1085‐97. [DOI] [PubMed] [Google Scholar]
- 31. Keefe RS, Young CA, Rock SL et al. One‐year double‐blind study of the neurocognitive efficacy of olanzapine, risperidone, and haloperidol in schizophrenia. Schizophr Res 2006;81:1‐15. [DOI] [PubMed] [Google Scholar]
- 32. Kinon BJ, Kollack‐Walker S, Jeste D et al. Incidence of tardive dyskinesia in older adult patients treated with olanzapine or conventional antipsychotics. J Geriatr Psychiatry Neurol 2015;28:67‐79. [DOI] [PubMed] [Google Scholar]
- 33. Kongsakon R, Trinidad‐Onate P, Chaudhry HR et al. Asian outpatients with schizophrenia: a double‐blind randomized comparison of quality of life and clinical outcomes for patients treated with olanzapine or haloperidol. J Med Assoc Thai 2006;89:1157‐70. [PubMed] [Google Scholar]
- 34. Lewis SW, Davies L, Jones PB et al. Randomised controlled trials of conventional antipsychotic versus new atypical drugs, and new atypical drugs versus clozapine, in people with schizophrenia responding poorly to, or intolerant of, current drug treatment. Health Technol Assess 2006;10:1‐165. [DOI] [PubMed] [Google Scholar]
- 35. Marder SR, Glynn SM, Wirshing WC et al. Maintenance treatment of schizophrenia with risperidone or haloperidol: 2‐year outcomes. Am J Psychiatry 2003;160:1405‐12. [DOI] [PubMed] [Google Scholar]
- 36. McEvoy JP, Byerly M, Hamer RM et al. Effectiveness of paliperidone palmitate vs haloperidol decanoate for maintenance treatment of schizophrenia: a randomized clinical trial. JAMA 2014;311:1978‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Meltzer HY, Bobo WV, Lee MA et al. A randomized trial comparing clozapine and typical neuroleptic drugs in non‐treatment‐resistant schizophrenia. Psychiatry Res 2010;177:286‐93. [DOI] [PubMed] [Google Scholar]
- 38. Miller DD, Eudicone JM, Pikalov A et al. Comparative assessment of the incidence and severity of tardive dyskinesia in patients receiving aripiprazole or haloperidol for the treatment of schizophrenia: a post hoc analysis. J Clin Psychiatry 2007;68:1901‐6. [DOI] [PubMed] [Google Scholar]
- 39. Parabiaghi A, Tettamanti M, D'Avanzo B et al. Metabolic syndrome and drug discontinuation in schizophrenia: a randomized trial comparing aripiprazole olanzapine and haloperidol. Acta Psychiatr Scand 2016;133:63‐75. [DOI] [PubMed] [Google Scholar]
- 40. Potkin SG, Weiden PJ, Loebel AD et al. Remission in schizophrenia: 196‐week, double‐blind treatment with ziprasidone vs. haloperidol. Int J Neuropsychopharmacol 2009;12:1233‐48. [DOI] [PubMed] [Google Scholar]
- 41. Purdon SE, Jones BD, Stip E et al. Neuropsychological change in early phase schizophrenia during 12 months of treatment with olanzapine, risperidone, or haloperidol. The Canadian Collaborative Group for research in schizophrenia. Arch Gen Psychiatry 2000;57:249‐58. [DOI] [PubMed] [Google Scholar]
- 42. Rosenheck R, Cramer J, Xu W et al. A comparison of clozapine and haloperidol in hospitalized patients with refractory schizophrenia. Department of Veterans Affairs Cooperative Study Group on Clozapine in Refractory Schizophrenia. N Engl J Med 1997;337:809‐15. [DOI] [PubMed] [Google Scholar]
- 43. Tohen M, Baker RW, Altshuler LL et al. Olanzapine versus divalproex in the treatment of acute mania. Am J Psychiatry 2002;159:1011‐7. [DOI] [PubMed] [Google Scholar]
- 44. Tollefson GD, Beasley CM Jr, Tran PV et al. Olanzapine versus haloperidol in the treatment of schizophrenia and schizoaffective and schizophreniform disorders: results of an international collaborative trial. Am J Psychiatry 1997;154:457‐65. [DOI] [PubMed] [Google Scholar]
- 45. Tyrer P, Oliver‐Africano PC, Ahmed Z et al. Risperidone, haloperidol, and placebo in the treatment of aggressive challenging behaviour in patients with intellectual disability: a randomised controlled trial. Lancet 2008;371:57‐63. [DOI] [PubMed] [Google Scholar]
- 46. Tollefson GD, Beasley CM Jr, Tamura RN et al. Blind, controlled, long‐term study of the comparative incidence of treatment‐emergent tardive dyskinesia with olanzapine or haloperidol. Am J Psychiatry 1997;154:1248‐54. [DOI] [PubMed] [Google Scholar]
- 47. Buchanan RW, Panagides J, Zhao J et al. Asenapine versus olanzapine in people with persistent negative symptoms of schizophrenia. J Clin Psychopharmacol 2012;32:36‐45. [DOI] [PubMed] [Google Scholar]
- 48. Arango C, Robles O, Parellada M et al. Olanzapine compared to quetiapine in adolescents with a first psychotic episode. Eur Child Adolesc Psychiatry 2009;18:418‐28. [DOI] [PubMed] [Google Scholar]
- 49. Breier A, Berg PH, Thakore JH et al. Olanzapine versus ziprasidone: results of a 28‐week double‐blind study in patients with schizophrenia. Am J Psychiatry 2005;162:1879‐87. [DOI] [PubMed] [Google Scholar]
- 50. Ciudad A, Alvarez E, Bousono M et al. Safety and tolerability of olanzapine versus risperidone: a one‐year randomized study in outpatients with schizophrenia with prominent negative symptoms. Actas Esp Psiquiatr 2007;35:105‐14. [PubMed] [Google Scholar]
- 51. Cuesta MJ, Garcia de Jalon E, Campos MS et al. Cognitive effectiveness of olanzapine and risperidone in first‐episode psychosis. Br J Psychiatry 2009;194:439‐45. [DOI] [PubMed] [Google Scholar]
- 52. Johnsen E, Jorgensen HA, Kroken RA et al. Neurocognitive effectiveness of quetiapine, olanzapine, risperidone, and ziprasidone: a pragmatic, randomized trial. Eur Psychiatry 2013;28:174‐84. [DOI] [PubMed] [Google Scholar]
- 53. Kane JM, Osuntokun O, Kryzhanovskaya LA et al. A 28‐week, randomized, double‐blind study of olanzapine versus aripiprazole in the treatment of schizophrenia. J Clin Psychiatry 2009;70:572‐81. [DOI] [PubMed] [Google Scholar]
- 54. Keks NA, Ingham M, Khan A et al. Long‐acting injectable risperidone v. olanzapine tablets for schizophrenia or schizoaffective disorder. Randomised, controlled, open‐label study. Br J Psychiatry 2007;191:131‐9. [DOI] [PubMed] [Google Scholar]
- 55. Kinon BJ, Lipkovich I, Edwards SB et al. A 24‐week randomized study of olanzapine versus ziprasidone in the treatment of schizophrenia or schizoaffective disorder in patients with prominent depressive symptoms. J Clin Psychopharmacol 2006;26:157‐62. [DOI] [PubMed] [Google Scholar]
- 56. Kinon BJ, Noordsy DL, Liu‐Seifert H et al. Randomized, double‐blind 6‐month comparison of olanzapine and quetiapine in patients with schizophrenia or schizoaffective disorder with prominent negative symptoms and poor functioning. J Clin Psychopharmacol 2006;26:453‐61. [DOI] [PubMed] [Google Scholar]
- 57. McIntyre RS, Cohen M, Zhao J et al. Asenapine for long‐term treatment of bipolar disorder: a double‐blind 40‐week extension study. J Affect Disord 2010;126:358‐65. [DOI] [PubMed] [Google Scholar]
- 58. Schoemaker J, Naber D, Vrijland P et al. Long‐term assessment of asenapine vs. olanzapine in patients with schizophrenia or schizoaffective disorder. Pharmacopsychiatry 2010;43:138‐46. [DOI] [PubMed] [Google Scholar]
- 59. Tran PV, Hamilton SH, Kuntz AJ et al. Double‐blind comparison of olanzapine versus risperidone in the treatment of schizophrenia and other psychotic disorders. J Clin Psychopharmacol 1997;17:407‐18. [DOI] [PubMed] [Google Scholar]
- 60. Lewis SW, Barnes TR, Davies L et al. Randomized controlled trial of effect of prescription of clozapine versus other second‐generation antipsychotic drugs in resistant schizophrenia. Schizophr Bull 2006;32:715‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. McEvoy JP, Lieberman JA, Stroup TS et al. Effectiveness of clozapine versus olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia who did not respond to prior atypical antipsychotic treatment. Am J Psychiatry 2006;163:600‐10. [DOI] [PubMed] [Google Scholar]
- 62. Tollefson GD, Birkett MA, Kiesler GM et al. Double‐blind comparison of olanzapine versus clozapine in schizophrenic patients clinically eligible for treatment with clozapine. Biol Psychiatry 2001;49:52‐63. [DOI] [PubMed] [Google Scholar]
- 63. Addington DE, Labelle A, Kulkarni J et al. A comparison of ziprasidone and risperidone in the long‐term treatment of schizophrenia: a 44‐week, double‐blind, continuation study. Can J Psychiatry 2009;54:46‐54. [DOI] [PubMed] [Google Scholar]
- 64. Chengappa KNR, Turkin SR, Schlicht PJ et al. A pilot, 15‐month, randomised effectiveness trial of risperidone long‐acting injection (RLAI) versus oral atypical antipsychotic agents (AAP) in persons with bipolar disorder. Acta Neuropsychiatrica 2010;22:68‐80. [DOI] [PubMed] [Google Scholar]
- 65. Citrome L, Cucchiaro J, Sarma K et al. Long‐term safety and tolerability of lurasidone in schizophrenia: a 12‐month, double‐blind, active‐controlled study. Int Clin Psychopharmacol 2012;27:165‐76. [DOI] [PubMed] [Google Scholar]
- 66. Gaebel W, Schreiner A, Bergmans P et al. Relapse prevention in schizophrenia and schizoaffective disorder with risperidone long‐acting injectable vs quetiapine: results of a long‐term, open‐label, randomized clinical trial. Neuropsychopharmacology 2010;35:2367‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Macfadden W, Ma YW, Thomas Haskins J et al. A prospective study comparing the long‐term effectiveness of injectable risperidone long‐acting therapy and oral aripiprazole in patients with schizophrenia. Psychiatry 2010;7:23‐31. [PMC free article] [PubMed] [Google Scholar]
- 68. Naber D, Hansen K, Forray C et al. Qualify: a randomized head‐to‐head study of aripiprazole once‐monthly and paliperidone palmitate in the treatment of schizophrenia. Schizophr Res 2015;168:498‐504. [DOI] [PubMed] [Google Scholar]
- 69. Seeman P. Atypical antipsychotics: mechanism of action. Can J Psychiatry 2002;47:27‐38. [PubMed] [Google Scholar]
- 70. Leucht S, Pitschel‐Walz G, Engel RR et al. Amisulpride, an unusual “atypical” antipsychotic: a meta‐analysis of randomized controlled trials. Am J Psychiatry 2002;159:180‐90. [DOI] [PubMed] [Google Scholar]
- 71. Tenback DE, van Harten PN, Slooff CJ et al. Effects of antipsychotic treatment on tardive dyskinesia: a 6‐month evaluation of patients from the European Schizophrenia Outpatient Health Outcomes (SOHO) Study. J Clin Psychiatry 2005;66:1130‐3. [PubMed] [Google Scholar]
- 72. Woods SW, Morgenstern H, Saksa JR et al. Incidence of tardive dyskinesia with atypical versus conventional antipsychotic medications: a prospective cohort study. J Clin Psychiatry 2010;71:463‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Leucht S, Cipriani A, Spineli L et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple‐treatments meta‐analysis. Lancet 2013;382:951‐62. [DOI] [PubMed] [Google Scholar]
- 74. Emsley RA. Risperidone in the treatment of first‐episode psychotic patients: a double‐blind multicenter study. Risperidone Working Group. Schizophr Bull 1999;25:721‐9. [DOI] [PubMed] [Google Scholar]
- 75. Kenney C, Hunter C, Davidson A et al. Metoclopramide, an increasingly recognized cause of tardive dyskinesia. J Clin Pharmacol 2008;48:379‐84. [DOI] [PubMed] [Google Scholar]
- 76. Matson JL, Mayville EA, Bielecki J et al. Tardive dyskinesia associated with metoclopramide in persons with developmental disabilities. Res Dev Disabil 2002;23:224‐33. [DOI] [PubMed] [Google Scholar]
- 77. Chouza C, Scaramelli A, Caamano JL et al. Parkinsonism, tardive dyskinesia, akathisia, and depression induced by flunarizine. Lancet 1986;1:1303‐4. [DOI] [PubMed] [Google Scholar]
- 78. Vandewalle W, Boon E, Sienaert P. Movement disorders due to modern antidepressants and mood stabilizers. Tijdschr Psychiatr 2015;57:132‐7. [PubMed] [Google Scholar]
- 79. Van Putten T. Why do schizophrenic patients refuse to take their drugs? Arch Gen Psychiatry 1974;31:67‐72. [DOI] [PubMed] [Google Scholar]
- 80. Czobor P, Van Dorn RA, Citrome L et al. Treatment adherence in schizophrenia: a patient‐level meta‐analysis of combined CATIE and EUFEST studies. Eur Neuropsychopharmacol 2015;25:1158‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Garcia‐Cabeza I, Gomez JC, Sacristan JA et al. Subjective response to antipsychotic treatment and compliance in schizophrenia. A naturalistic study comparing olanzapine, risperidone and haloperidol (EFESO Study). BMC Psychiatry 2001;1:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Manu P, Dima L, Shulman M et al. Weight gain and obesity in schizophrenia: epidemiology, pathobiology, and management. Acta Psychiatr Scand 2015;132:97‐108. [DOI] [PubMed] [Google Scholar]
- 83. Zutshi D, Cloud LJ, Factor SA. Tardive syndromes are rarely reversible after discontinuing dopamine receptor blocking agents: experience from a university‐based movement disorder clinic. Tremor Other Hyperkinet Mov 2014;4:266. [DOI] [PMC free article] [PubMed] [Google Scholar]