

Abstract
Protein aggregation is a key molecular feature underlying a wide array of neurodegenerative disorders, including Alzheimer’s and Parkinson’s diseases. To understand protein aggregation in molecular detail, it is crucial to be able to characterize the array of heterogeneous aggregates that are formed during the aggregation process. We present here a high-throughput method to detect single protein aggregates, in solution, from a label-free aggregation reaction, and we demonstrate the approach with the protein associated with Parkinson’s disease, α-synuclein. The method combines single-molecule confocal microscopy with a range of amyloid-binding extrinsic dyes, including thioflavin T and pentameric formylthiophene acetic acid, and we show that we can observe aggregates at low picomolar concentrations. The detection of individual aggregates allows us to quantify their numbers. Furthermore, we show that this approach also allows us to gain structural insights from the emission intensity of the extrinsic dyes that are bound to aggregates. By analyzing the time evolution of the aggregate populations on a single-molecule level, we then estimate the fragmentation rate of aggregates, a key process that underlies the multiplication of pathological aggregates. We additionally demonstrate that the method permits the detection of these aggregates in biological samples. The capability to detect individual protein aggregates in solution opens up a range of new applications, including exploiting the potential of this method for high-throughput screening of human biofluids for disease diagnosis and early detection.
Self-assembly of amyloidogenic proteins underlies a multitude of neurodegenerative disorders, such as Alzheimer’s disease and Parkinson’s disease (PD). Aggregation of the intrinsically disordered intracellular protein α-synuclein (αS) into Lewy bodies,1−3 the pathological hallmark of PD, plays a central role in the pathogenesis of this disease and other synucleinopathies.1,4,5 The soluble aggregates, known as oligomers, of αS have increasingly been demonstrated to be the most cytotoxic species associated with PD6−8 and are therefore of particular interest for comprehensive characterization. Due to the heterogeneity and low abundance of oligomers, single-molecule techniques are well-suited to quantitatively investigate the kinetics of αS aggregation.
By covalently labeling monomeric amyloidogenic proteins prior to aggregation, single-molecule fluorescence techniques have been used to track the aggregation kinetics of these proteins and obtain information on the size and structure of the intermediate oligomeric species arising. Two-color coincidence detection (TCCD)9 and single-molecule fluorescence resonance energy transfer (smFRET)10 have provided robust data sets used to produce in vitro experimentally derived kinetic models for the aggregation of amyloidogenic proteins such as αS6,11,12 and tau.13 However, the intrinsic labeling of proteins has its drawbacks. At worst, the protein’s propensity to aggregate may be modified and the kinetics altered.14 Other potential issues include off-target labeling, labeling inefficiencies, and the inability to remove all free dye.15 In addition, modification of the overall charge of the molecule, or addition of hydrophobic fluorophores, may lead to increased binding of the sample to surfaces, altering the effective concentration of the species over time. Moreover, it is more difficult to image oligomers in their native environment if intrinsic labeling is required.
A single-molecule total internal reflection fluorescence microscopy (TIRFM) method has recently been developed to detect surface-immobilized individual aggregates of αS, amyloid β, and tau without covalently attached (intrinsic) labels. This was used to investigate the number of αS aggregates in cerebrospinal fluid (CSF) samples from PD patients in comparison with controls.16 This technique does not require a dye to be conjugated to the protein of interest but instead relies upon the utilization of the benzothiazole salt dye thioflavin T (ThT), which binds to the β-sheet structure of protein aggregates.17,18 Upon binding, the quantum yield of the dye dramatically increases by multiple orders of magnitude (e.g., from 10–4 to 0.83 with insulin fibrils),19 allowing it to be added at concentrations above the dissociation constant to provide high signal-to-background fluorescence. The major drawback of TIRFM for the study of protein aggregation is the differential adsorption of oligomers of different sizes or structures to the glass surface.20 Therefore, artifacts from surface effects that alter the apparent aggregation kinetics cannot be ruled out. The low data throughput of TIRFM is another drawback, which can be vastly improved upon by using techniques compatible with sample flow.
Here the evolution of aggregates, formed from unlabeled protein in solution, is tracked by probing aliquots taken at different time points via single-molecule confocal microscopy with four different extrinsic dyes. This novel confocal technique has the ability to reliably track the number of aggregates present, without the need for surface immobilization, and does not require intrinsic labels. By aggregating proteins label-free and only adding extrinsic dyes to aliquots taken at set time points, we avoid perturbing the aggregation process. Using microfluidics, we flow samples through the probe volume of a single-molecule confocal microscope to measure individual aggregates at a high detection rate, as previously reported for intrinsically labeled samples.11 We monitor the number of dye-active aggregates over time and the intensity of each species. In our system, the latter parameter can be related to the β-sheet content and other structural elements of these species to which the particular dye is sensitive, and we show that these data are consistent with corresponding TIRFM measurements.
While ThT is the dye most commonly used to track protein aggregation under ensemble conditions, there are a growing number of amyloid-specific dyes being proposed as alternatives.21 We tested four extrinsic dyes: ThT, a dimer derivative of ThT, diThT-PEG2 (diThT),22 pentameric formylthiophene acetic acid (pFTAA),23−25 and PicoGreen.26 Their structures are illustrated in Figure 1. There is limited evidence to suggest that these extrinsic dyes, including ThT, may increase the formation of fibrils in the aggregation of certain amyloidogenic proteins.27 This underlines the advantages of label-free aggregation with our technique where the dyes are added to extracted aliquots at different time points of the aggregation. We investigate the compatibility of our technique with these various dyes and determine which possess the greatest advantages to probe protein aggregation.
Figure 1.

Chemical structures of four extrinsic dyes used in this work: ThT, diThT, pFTAA, and PicoGreen.
Experimental Section
Preparation of Dye Solutions
Thioflavin T
Stock solution was prepared by dissolving ThT (Sigma–Aldrich) in Tris buffer [25 mM 2-amino-2-(hydroxymethyl)propane-1,3-diol and 100 mM NaCl, pH 7.4] to give a solution of ∼100 μM. The exact concentration of this stock solution was determined by measuring absorbance at 412 nm (NanoDrop 2000c UV–vis spectrophotometer, Thermo Scientific), using an extinction coefficient of 36 000 M–1·cm–1.
Thioflavin T Dimer
Stock solution was prepared by diluting diThT-poly(ethylene glycol) (PEG) chloride salt (Peakdale Molecular, custom synthesis) in dimethyl sulfoxide and further diluting in Tris buffer (25 mM Tris and 100 mM NaCl, pH 7.4). A fresh solution was prepared for each aggregation experiment (filtered through 0.02 μm filter, Whatman), with the exact concentration of this stock solution being determined by measuring absorbance at 410 nm, using an extinction coefficient of 45 800 M–1·cm–1.
Pentameric Formylthiophene Acetic Acid
Solutions of either 1 mM or 30 nM were prepared by diluting a concentrated stock solution in Tris buffer (25 mM Tris and 100 mM NaCl, pH 7.4) (filtered through 0.02 μm filter, Whatman).
PicoGreen
Since the concentration of the provided PicoGreen sample (Quant-IT PicoGreen, Thermo Fisher Scientific) was withheld for commercial reasons, it was decided to refer to the concentration of PicoGreen as a dilution factor from the original neat solution. PicoGreen solutions were prepared by diluting the neat sample by a factor of 300 in Tris buffer (25 mM Tris and 100 mM NaCl, pH 7.4) (filtered through 0.02 μm filter, Whatman).
α-Synuclein
Expression and Purification
Full-length wild-type αS was expressed and purified based on the protocol by Hoyer et al.,28 and provided as aliquots (200–300 μM, Tris, pH 7.6) stored at −80 °C.
Aggregation
Monomeric wild-type αS was prepared in Tris buffer (25 mM Tris and 100 mM NaCl, pH 7.4) with 0.02% (w/v) NaN3 to give a total protein concentration of 70 μM in a sample volume of 300 μL. The aggregation mixtures were incubated at 37 °C either with constant shaking at 200 rpm (New Brunswick Scientific Innova 43), or with constant stirring at 1100 rpm (Micro 8 × 1.5 mm stirrer bar, VWR) in a water bath (Star Lab magnetic stirrer with heating).
Single-Molecule Confocal Measurements
Label-free protein monomer was incubated under aggregating conditions, as described earlier. Aliquots were taken at regular time points and immediately diluted to nanomolar concentrations (20 nM) in buffer with the chosen extrinsic dye. Diluted protein samples were withdrawn through a single-channel microfluidic device (width = 100 μm, height = 25 μm, length = 1 cm), at a flow velocity of 0.56 cm·s–1 to a syringe (1 mL, HSW, Normject) via polyethylene tubing (0.38 mm i.d., Intramedic). Flow control was achieved with syringe pumps (Harvard Apparatus PhD Ultra), and the device was placed on the microscope stage. Single-molecule confocal experiments with pFTAA or PicoGreen as the extrinsic dye were performed on the instrument described by Li et al.9 by use of a 488 nm laser (1.5 mW). For single-molecule confocal experiments with ThT or diThT as the extrinsic dye, an analogous second confocal instrument was used. In brief, a 445 nm laser beam (1.5 mW, OFL68, OdicForce) was directed to the back aperture of an inverted microscope (Nikon Eclipse TE2000-U). The beam was reflected by a dichroic mirror (Di01-R488/543/594, Laser 2000) and focused to a concentric diffraction-limited spot, 10 μm into the solutions in a microfluidic detection channel through a high-numerical-aperture oil-immersion objective (Apochromat 60×, NA 1.40, Nikon). Fluorescence was collected with the same objective, passing through the same dichroic mirror, and imaged onto a 50 μm pinhole (Melles Griot) to remove out-of-focus light. The emission was filtered (FF01–510/84) and directed to an avalanche photodiode (APD, SPCM-14, PerkinElmer Optoelectronics). A custom-programmed field-programmable gate array (FPGA, Colexica), was used to count the signals from the APD and combine these into time bins, which were selected according to the expected residence time of molecules passing through the confocal probe volume. At each time point, data were collected for 10 min (100 000 time bins, bin width 0.2 ms).
Detection Limit of Dye Measurements
αS was aggregated for 24 h (70 μM, in Tris buffer, with shaking), since under these conditions αS was previously shown to be efficiently fibrillized.6 Serial dilutions of each dye were prepared and added to the diluted αS fibrillar sample, and single-molecule confocal measurements were made under flow.
Cerebrospinal Fluid Sample
CSF samples were collected by lumbar puncture from patients who sought medical advice at memory clinics because of cognitive symptoms. The sample used was classified as a non-Alzheimer’s disease, normal healthy individual according to CSF Alzheimer’s disease biomarker results (T-tau, P-tau181, and Aβ42) that are 90% sensitive and specific for Alzheimer’s disease, as previously described by Klyubin et al.29 The study protocol was approved by the regional ethics committee at the University of Gothenburg.
Preparation of Artificial Cerebrospinal Fluid
Artificial CSF stock solutions were prepared containing 148 mM NaCl, 3 mM KCl, 1.4 mM CaCl2·2H2O, 0.8 mM MgCl2·6H2O, 0.8 mM Na2HPO4·7H2O, and 0.2 mM NaH2PO4·H2O.
Detection in Biological Samples
αS was aggregated for 24 h (70 μM, in Tris buffer, with shaking) to produce a fibrillar sample of αS aggregates. This sample was diluted to a final concentration of 1 nM (monomer concentration), with 30 nM pFTAA, in both artificial CSF buffer and the previously described CSF sample (diluted 5-fold). Measurements were performed in triplicate for each sample.
Total Internal Reflection Fluorescence Microscopy
Preparation of Chambered Coverslides
Borosilicate glass coverslides (24 × 50 mm, thickness no. 1, Brand, Wertheim, Germany) were cleaned in an argon plasma cleaner (PDC-002, Harrick Plasma, Ithaca, NY) for at least 1 h. A 50-well multiwell chamber set (CultureWell chambered coverglass, Thermo Fisher Scientific) was sealed onto the coverslide, and each well was coated with poly(l-lysine) (PLL, molecular mass 150–300 kDa; P4832, Sigma-Aldrich) for 30 min. The wells were washed three times with Tris buffer before 10 μL of sample (50 nM) in Tris buffer was applied.
Single-Molecule Measurements
TIRFM imaging was performed on the instrument described by Drews et al.30 Briefly, a 405 nm laser (LBX-LD, Oxxius) and a 488 nm laser (MLD, Cobolt) were aligned and directed parallel to the optical axis at the edge of a TIRF objective (1.49 NA, APON60XO TIRF, Olympus) mounted on an inverted microscope (Olympus IX-73). Fluorescence was collected by the same objective, separated from the laser beam by a dichroic mirror (Di01-405/488/532/635, Semrock) and passed through an appropriate emission filter (FF01-480/40-25 for experiments with ThT/diThT and FF03-525/50-25 for experiments with pFTAA/PicoGreen; Laser2000). Images were recorded on an electron-multiplying charge-coupled device (EMCCD) camera (Evolve 512 Delta, Photometrics) operating in frame-transfer mode [EM gain of 4.4 e/analog-to-digital unit (ADU) and 260 ADU/photon], with a pixel length of 235 nm. This imaging mode restricts the imaging plane to within ∼100 nm above the coverslide. Samples were placed in individual wells and an automated script was run to collect data with either the 405 nm channel (for ThT and diThT experiments) or the 488 nm channel (for pFTAA and PicoGreen experiments). Within each well, the sample was imaged nine times in a 350 μm spaced 3 × 3 grid, and images were recorded for 100 frames with 50 ms exposure time by use of an automated script programmed in bean-shell script (MicroManager).31
Ensemble Thioflavin T Fluorescence Measurements
Fluorescence measurements were performed on a Varian model Cary Eclipse spectrofluorometer (Palo Alto, CA) in a temperature-controlled cell holder, with a cuvette of path length 3 mm × 10 mm. Aliquots of protein were diluted to a final concentration of 7 or 20 μM ThT. The sample was excited at 446 nm, and emission was recorded from 460 to 600 nm.
Fourier Transform Infrared Spectroscopic Measurements
Protein aliquots (3 μL, 70 μM) were loaded and allowed to dry on the measurement surface of a Bruker Platinum Diamond attenuated total reflectance (ATR) accessory on a Bruker Vertex 70 Fourier transform infrared (FT-IR) spectrophotometer (Bruker Optics Ltd.). The buffer background was measured and subtracted from each protein spectrum before curve fitting of the amide region (1720–1580 cm–1) and baseline subtraction were carried out. Deconvolution of the spectra with Gaussian curves was performed with a Levenberg–Marquardt algorithm by use of the Opus software package (Bruker Optics Ltd.). For comparison, all absorbance spectra were normalized.
Data Analysis
Extrinsic Dye Confocal Data
The experimental output data were collected by use of an FPGA card and analyzed in Igor Pro by use of custom-written code. A threshold was manually selected so as to maximize the number of events while removing the noise. Thresholding was set for each extrinsic dye individually, then consistently applied to all time points measured for that dye. The Igor Pro code selects events above the threshold and calculates the total number of events, the total intensity of all events recorded over the 10 min data acquisition time for each time point, and, from these two parameters, the average intensity of events.
Extrinsic Dye Total Internal Reflection Fluorescence Microscopic Data
The image data collected consisted of 100 frames, which were first averaged (ImageJ, National Institutes of Health) and then analyzed by custom-written code. This script first removed the modulated background and camera noise from the averaged images. Intensity was corrected to a normalized intensity by dividing the intensity signal above the background by the background. Individual particles were then identified by establishing their boundaries and fine-tuning size and intensity thresholds. The length of these particles was calculated by use of an algorithm in ImageJ that thinned the boundary of the particles to a one-dimensional skeleton, calibrating the value for length obtained with the detection limit of the camera (previously determined to be 450 nm). The normalized intensity of a given particle is calculated as the sum of the normalized intensity of all pixels within its boundary. Using this analyzed data, we plot two-dimensional contour plots of the normalized intensity per pixel and the length of particles for each time point. This analysis gives information on how the number of aggregates evolves over time with respect to their size above the diffraction limit and intensity density, which may be interpreted as a measure of the compactness of the β-sheet core.
Results and Discussion
To confirm that the single-molecule confocal technique is able to sensitively and reliably detect aggregates down to picomolar concentrations, we optimized each extrinsic dye concentration using the same fibrillar sample of αS aggregates. A binding curve of each dye to αS aggregates (20 nM αS, equivalent monomer concentration) was produced and normalized with respect to the maximal number of dye-active species detected (events) and fitted to obtain a value for the dissociation constant, Kd, of each dye (Figure 2A) by use of the Hill–Langmuir equation (eq 1):32
| 1 |
where θ is the normalized number of dye–aggregate fluorescent complexes and [D] is the total dye concentration.
Figure 2.

(A) Normalized binding curves for extrinsic dyes ThT, diThT, and pFTAA (x̅ ± σ, n = 3). Error bars represent the standard deviation from three separate experiments. Kd was extracted for each dye: 1.37 ± 0.25 μM for ThT, 48.7 ± 8.4 nM for diThT, and 15.7 ± 3.4 nM for pFTAA. (B) Serial dilutions of the αS sample were made to determine the linearity of events detected with concentration. Curves for ThT, diThT, and pFTAA overlap.
The obtained Kd values of 1.37 ± 0.25 μM for ThT (2.3 μM with insulin),22 48.7 ± 8.4 nM for diThT (34 nM with insulin),22 and 15.7 ± 3.4 nM for pFTAA (142 nM with tau)24 correspond well with literature values. At higher concentrations of each extrinsic dye, the number of events detected begins to saturate and a plateau is reached. The beginning of the plateau represents the lowest concentration of each extrinsic dye at which all binding sites on the aggregates are occupied by that dye. This was determined to be 5 μM for ThT, 500 nM for diThT, 30 nM for pFTAA, and a dilution factor of 300 for PicoGreen. These concentrations of dye give the optimal signal-to-noise ratio and thus were used for all subsequent experiments.
To verify the linearity of detection of αS aggregates with the concentration of protein present, serial dilutions of the αS fibrillar sample, ranging from 0.1 nM to 70 nM, were added to each dye at its designated concentration (Figure 2B).
Finally, to determine the limit of detection (LoD), the lowest concentration at which the confocal technique can reliably detect aggregates using each extrinsic dye, the limit of blank (LoB) was determined. Via an approach analogous to that of Horrocks et al.,16 the LoB is defined as the highest apparent number of events expected when samples containing no analyte are measured, given by eq 2:33
| 2 |
where x̅blank is the mean number of events measured for blank samples and σblank is the standard deviation about this mean. If a Gaussian distribution of signal is assumed, by taking the sum of the mean and the standard deviation multiplied by a factor of 1.645, the LoB represents 95% of observed values for blank samples.
The LoD is then calculated from measurements of samples containing a very low concentration of analyte and the value for the LoB, given by eq 3:
| 3 |
where σlow concn sample is the standard deviation about the mean number of events from samples containing very low concentrations of protein. The LoD is calculated by taking the sum of the LoB and the standard deviation multiplied by a factor of 1.645, since at this concentration 95% of measured values will exceed the LoB.
Using these definitions, values for the LoB and LoD were calculated for the detection of αS aggregates with each extrinsic dye (Table 1). The lowest concentration of aggregates detectable was determined with the measured size of the confocal probe volume (1.80 and 1.94 fL for 488 nm and 445 nm laser, respectively; calculation in Supporting Information). The signal-to-noise ratio was calculated by taking the averaged signal across different time points, subtracting the averaged background, and then dividing by the standard deviation of the background.
Table 1. Calculated Limit of Blank and Limit of Detection Values for Detection of α-Synuclein Aggregates with Each Extrinsic Dyea.
| ThT | diThT | pFTAA | Pico Green | |
|---|---|---|---|---|
| LoB, events·bin–1 | 2.88 × 10–3 | 1.05 × 10–3 | 1.27 × 10–3 | 4.46 × 10–2 |
| LoD, events·bin–1 | 8.28 × 10–3 | 1.71 × 10–3 | 3.35 × 10–3 | 6.11 × 10–2 |
| LoD aggregate concn, pM | 10 | 2 | 4 | 60 |
| signal-to-noise ratio | 34.5 | 57.5 | 34.9 | 1.0 |
The lowest concentration of aggregate that could be accurately detected with each dye was also calculated, as well as the signal-to-noise ratio.
The values for the lowest concentration of αS aggregates that can be detected with each extrinsic dye (ThT ≈ 10 pM, diThT ≈ 2 pM, pFTAA ≈ 4 pM, PicoGreen ≈ 60 pM) show that PicoGreen is markedly less sensitive than the other three dyes.
Measuring α-Synuclein Aggregation with Four Extrinsic Dyes
The label-free aggregation method, probed by extrinsic dyes added only to extracted aliquots, allows us to measure not only the number of aggregates but also the intensity of these species over time. The deconvolution of number and intensity data, difficult to achieve with ensemble techniques, provides information about the mechanism of aggregation.
In order to test the ability of our confocal method to track in vitro formation of αS aggregates as described earlier, under shaking conditions, aliquots were taken at set time points and interrogated under flow. Single-molecule measurements monitoring αS aggregation were performed, using one dye at a time to monitor the aggregates produced. The number of aggregates was measured as well as the intensity of each, from which the average intensity at a given time point was calculated.
It was observed that all four dyes tracked the evolution of aggregates as their number and average intensity increased over the course of the aggregation (Figure 3A,B). The average intensity of aggregates tracked by each dye was normalized with respect to its maximum value. This normalization of the average intensity simply scales the intensity values for each dye independently (Figure 3C), such that we may compare the evolution in brightness of aggregates detected by each. A difference in the temporal evolution of the brightness of species would indicate that certain dyes have a higher sensitivity to different types of aggregates.
Figure 3.

(A) Number of species and (B) average intensity, tracked by each of the four extrinsic dyes over the course of aggregation of αS (70 μM Tris, shaking conditions; x̅ ± σ, n = 3). (C) By normalizing the average intensity of species data with respect to the maximum for each dye, we observe a shorter lag time for aggregations tracked by pFTAA and PicoGreen.
The data show that ThT and diThT are similar in both the number of αS aggregate events they track and the evolution of average intensity of monitored species over time. This can be expected since these two dyes are so closely related structurally and thus are likely detecting the same aggregate species. pFTAA also detects approximately the same number of aggregates as ThT and diThT. However, from the normalized average intensity data, we observe that the rise in the intensity of species increases markedly earlier for pFTAA. This indicates that pFTAA may be binding most strongly to earlier or smaller species, to which ThT and diThT do not bind. PicoGreen shares this pronounced earlier increase in the intensity of species; however, the signal-to-noise ratio for this dye is much poorer than for the other three (Table 1). There is no clear trend in either the number of events observed or the magnitude of their average intensity between the beginning and end of the aggregation time course for this extrinsic dye. This suggests that PicoGreen is not a suitable choice for such experiments, where greater sensitivity of detection for aggregates is pivotal.
The intensity values acquired by the confocal technique are a gross measure of the relative size of dye-active species, as larger species would be expected to incorporate more extrinsic dyes overall. However, for confocal measurements, there is a convolution of aggregate size and density of β-sheet content and other structural elements to which the particular dye is sensitive.
Total Internal Reflection Fluorescence Microscopic Measurements
In parallel with the confocal measurements, extracted aliquots of the αS aggregation mixture at selected time points were additionally measured by TIRFM. The throughput of the confocal technique is higher than for TIRFM; for late-stage time points, the rate of detection by the confocal technique was 80 ± 22 aggregates·s–1, compared to 19 ± 7 aggregates·s–1 by the TIRFM method. The single-molecule TIRFM imaging technique allows direct observation and visualization of individual aggregates present at different time points during aggregation (Figure 4A,C). TIRFM images were analyzed such that the number of species with fluorescence above a set intensity threshold was counted, their normalized intensity per pixel was calculated, and the length of each species was measured. This is illustrated with two-dimensional contour plots (Figure 4B,D).
Figure 4.

Two-dimensional contour plots for aggregation of αS monitored by ThT (B) and pFTAA (D) as a function of normalized intensity per pixel and length (n = 3), with corresponding representative TIRFM images (A and C, respectively). The contrast was optimized and fixed for each dye; the scale bar is 10 μm for all images and 1 μm for all zoomed insets. Equivalent data for diThT and PicoGreen are shown in Figure S1.
The structure of aggregates monitored by TIRFM can be characterized by deconvoluting the two properties of β-sheet density and size of aggregates (above the diffraction limit of ∼250 nm). The analysis performed provides information on the distribution of aggregate sizes at any one time and the brightness of these species. The latter parameter is a relative measure of how many dye molecules are bound and so provides an indication of the density of β-sheet content of species of size greater than the diffraction limit.
Two-dimensional contour plots for the aggregation of αS monitored by ThT showed a slight increase in aggregate number from 0 to 2 h. There was then an increase in the average length of species and a striking change in normalized intensity per pixel at 8 h. This implies that the structure of species was evolving rapidly in this time frame. An increase in the length of species below the diffraction limit may account for this increased brightness per pixel, since the length of the β-sheet structure to which the dyes bind would be extended in this instance. However, this augmentation would also be explained by an increasingly dense β-sheet structure. This latter interpretation of our observation fits well with the conversion step reported for labeled αS protein aggregation, where diffuse, transient oligomers transform into aggregate species with a more compact β-sheet core.6,11,12 The intensity did not advance beyond this point, and instead the only parameter that continued to increase was the average length of species detected. This is likely to correspond to the growth of αS aggregates via elongation from this point onward.
The evolution of aggregates tracked by TIRFM and the behavior of the extrinsic dyes was broadly equivalent to that observed with the confocal method, reinforcing the validity of both techniques to probe aggregates. Further evidence for the validity of our single-molecule techniques was provided by ensemble ThT fluorescence measurements (Figure S2) and FT-IR measurements (Figure S3).
Measuring the Number of Aggregates Gives Deeper Insights Than Ensemble Kinetics
Unlike ensemble kinetic experiments, which report on the total amount of aggregated monomer by measuring, for example, the total ThT fluorescence, our assay also allows one to monitor the number of aggregated species as a function of time. Thus, this type of assay is able to monitor effects that are not accessible through ensemble kinetics, such as the fragmentation of fibrils once the aggregation reaction has reached completion and the total amount of aggregated material remains unchanged. In order to demonstrate this application, the agitation mode with which the aggregation mixtures were incubated was changed from constant shaking at 200 rpm in an incubator to continual stirring at 1100 rpm in a water bath. Both of these modes of agitation induce a shear force on sample particles, but that of the stirring mode is expected to be of greater magnitude, hence enabling observation of the fragmentation within a shorter time scale. The shear force applied to the αS sample can lead to the fragmentation of existing fibrils. This process speeds up the aggregation reaction by increasing the number of growth-competent aggregates, and additionally we expect to be able to directly observe the increase in the number of fibrils even after all monomer has been consumed.34,35
The number of dye-active species was monitored (Figure 5A) by ThT and pFTAA (selected since the two dyes had shown different behaviors) over the course of aggregation, and the average intensity was calculated and normalized for each dye independently with respect to the maximum value for that dye (Figure 5B). The graphs for αS aggregation monitored by both dyes show that initially the average intensity of species rapidly increased to a peak at 2–4 h before steeply declining until 8 h, when it began to decrease much more gradually. The number of events increased from 2 to 4 h, increased rapidly between 4 and 8 h, and then continued to increase more gradually from 8 h. The lag time tracked by both dyes was approximately 2 h. Thus, agitation with stirring leads to a much faster rate of aggregation than agitation by shaking, as was expected due to the increased shear force from stirring resulting in increased fragmentation and accelerated aggregation.
Figure 5.

(A) Number of dye-active species, tracked over the course of aggregation of αS with agitation by stirring in Tris buffer. (Inset) Number concentration data above 8 h, fitted by use of eq 4 for ThT (dark blue dashed line) and pFTAA (gray dashed line). (B) Average intensity calculated and normalized, independently, to the maximum value for both ThT and pFTAA (x̅ ± σ, n = 3).
Furthermore, for these data, there is some correlation between the decline of average intensity of aggregates and the rapid rise in number of species detected. Most likely this reflects an initial increase in size of strongly dye-binding early-stage aggregates, which are then converted to mature, less strongly dye-binding aggregates, leading to a rapid decrease in intensity per aggregate. These aggregates subsequently fragment, decreasing the average size and leading to a slow decrease in average intensity.
To estimate the fragmentation induced by stirring, we investigated the increase in fibril number after completion of the aggregation reaction (in this case at 8 h), that is, after soluble monomer has reached its equilibrium concentration. In this limit, the rate of nucleation processes is negligible due to the low free monomer concentration, and therefore fragmentation is the main process responsible for increases in aggregate number. The number concentration of aggregates, P(t), is then initially given by eq 4:
| 4 |
where M is the mass concentration of aggregates, P0 is the number concentration of aggregates at completion of the aggregation reaction, and k– is the fragmentation rate constant.
By fitting eq 4 to the number concentration data above 8 h in Figure 5A, we obtain the fragmentation rate induced by stirring as (3.4 ± 1.2) × 10–8 s–1 for ThT and (2.1 ± 1.7) × 10–8 s–1 for pFTAA. This rate of fragmentation is comparable to that of prion protein (PrP) under shaking conditions (1 × 10–8 s–1) but significantly higher than for αS under shaking conditions (2 × 10–10 s–1).36 This finding highlights how the measurement of the number concentration of aggregates, as achieved by our technique, offers an orthogonal approach to conventional ensemble measurements, thus yielding information on processes that are inaccessible in such ensemble studies.
Measurements in a Biological Sample
To verify that the confocal technique can accurately detect unlabeled αS aggregates in a complex biological mixture, we performed experiments in human cerebrospinal fluid (CSF), which contains multiple proteins and other macromolecules. To do this, we diluted a fibrillar sample of αS aggregates (70 μM, 24 h with shaking) 7000-fold to a final concentration of 1 nM (equivalent monomer concentration) with pFTAA (30 nM) spiked in a sample (diluted 5-fold) of CSF from a normal healthy individual. Under these conditions, the oligomer concentration is approximately 1% of the total monomer concentration, that is, 10 pM,6 close to the reported physiological levels of oligomers of 1–10 pM.37,38 We measured the number of dye-active events compared to that of the unspiked CSF sample (diluted 5-fold) with pFTAA (Figure 6). Additionally, using pFTAA, we compared the events detected in an artificial CSF buffer with and without addition of the diluted αS fibrillar sample. The composition of artificial CSF corresponds to the typical electrolyte concentrations and physiological compatibility of endogenous CSF.39 Our results show that the technique can clearly detect picomolar levels of αS aggregates above background for both the CSF sample and artificial CSF buffer. This experiment shows the potential of the confocal technique to make measurements in complex biofluids such as cerebrospinal fluid.
Figure 6.
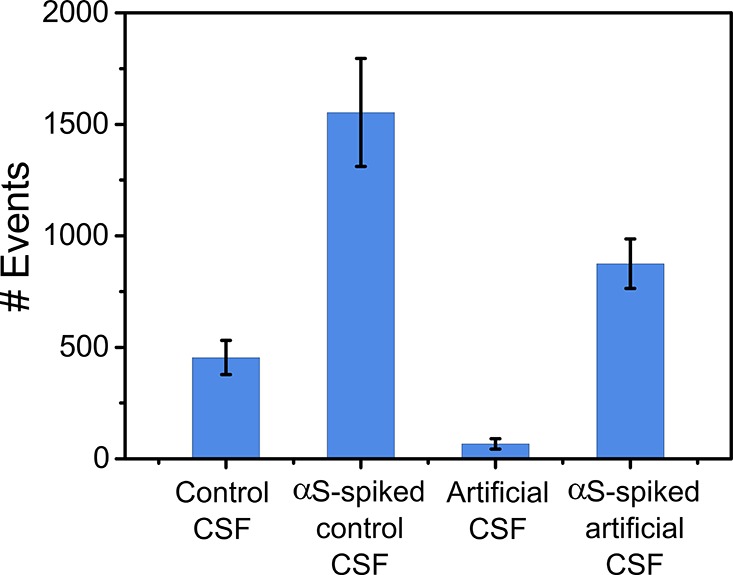
Detection of aggregates above background in biological samples by use of pFTAA (x̅ ± σ, n = 3 measurements).
Conclusions
In conclusion, the single-molecule confocal technique developed here not only overcomes ensemble-averaging limitations but also permits measurements of truly label-free aggregation. We confirm the effectiveness of the technique by comparing with a single-molecule TIRFM method, and we also investigate its ability to detect processes inaccessible by ensemble techniques, such as direct measurement of fragmentation of aggregates.
The use of extrinsic dyes removes the necessity for covalently bound fluorescent labels, which disrupt the kinetics of the aggregation under investigation. By avoiding the need for these intrinsic labels, we open the way for subsequent studies of protein aggregation, in vivo or in human biofluid samples, to accurately detect and quantify aggregates at very low concentration in complex heterogeneous systems.
Acknowledgments
We thank Swapan Preet for expression and purification of α synuclein protein, Francesco Aprile for help with FT-IR measurements, and Franklin Aigbirhio for providing the pentameric formyl thiophene acetic acid dye. C.G.T. was supported by the Frances and Augustus Newman Foundation, the Royal Society and European Research Council (669237). G.M. was supported by the European Research Council and Sidney Sussex College, Cambridge. M.H.H. was a recipient of a Herchel Smith Fellowship and was supported by Christs College, Cambridge. H.Z. was a Wallenberg Academy Fellow and was additionally funded by the Swedish Research Council (2013-2546) and the European Research Council (681712). T.P.J.K. was supported by the Biotechnology and Biological Sciences Research Council, the Frances and Augustus Newman Foundation and the European Research Council (337969) and the Cambridge Centre for Misfolding Diseases. D.K. is a Royal Society Glaxo Wellcome Professor of Molecular Medicine and was funded by the European Research Council (669237).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.8b02226.
Additional text and equations describing confocal probe volume; three figures showing 2D contour plots for aggregation of αS with shaking, monitored by diThT and PicoGreen, and ensemble ThT fluorescence trace and FT-IR spectra of αS aggregation with shaking and with stirring (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Spillantini M. G.; Crowther A. C.; Jakes R.; Hasegawa M.; Goedert M. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 6469–6473. 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi K.; Tanji K.; Mori F.; Takahashi H. Neuropathology 2007, 27, 494–506. 10.1111/j.1440-1789.2007.00803.x. [DOI] [PubMed] [Google Scholar]
- Xia Q.; Liao L.; Cheng D.; Duong D. M.; Gearing M.; Lah J. J.; Levey A. I.; Peng J. Front. Biosci., Landmark Ed. 2008, 13, 3850–3856. 10.2741/2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greffard S.; Verny M.; Bonnet A.-M.; Seilhean D.; Hauw J.-J.; Duyckaerts C. Neurobiol. Aging 2010, 31, 99–103. 10.1016/j.neurobiolaging.2008.03.015. [DOI] [PubMed] [Google Scholar]
- Maries E.; Dass B.; Collier T. J.; Kordower J. H.; Steece-Collier K. Nat. Rev. Neurosci. 2003, 4, 727–738. 10.1038/nrn1199. [DOI] [PubMed] [Google Scholar]
- Cremades N.; Cohen S. I. a.; Deas E.; Abramov A. Y.; Chen A. Y.; Orte A.; Sandal M.; Clarke R. W.; Dunne P.; Aprile F. a.; Bertoncini C. W.; Wood N. W.; Knowles T. P. J.; Dobson C. M.; Klenerman D. Cell 2012, 149, 1048–1059. 10.1016/j.cell.2012.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein W. L.; Stine W. B.; Teplow D. B. Neurobiol. Aging 2004, 25, 569–580. 10.1016/j.neurobiolaging.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Walsh D. M.; Klyubin I.; Fadeeva J. V.; Cullen W. K.; Anwyl R.; Wolfe M. S.; Rowan M. J.; Selkoe D. J. Nature 2002, 416, 535–539. 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Li H.; Ying L.; Green J. J.; Balasubramanian S.; Klenerman D. Anal. Chem. 2003, 75, 1664–1670. 10.1021/ac026367z. [DOI] [PubMed] [Google Scholar]
- Orte A.; Birkett N. R.; Clarke R. W.; Devlin G. L.; Dobson C. M.; Klenerman D. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 14424–14429. 10.1073/pnas.0803086105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horrocks M. H.; Tosatto L.; Dear A. J.; Garcia G. A.; Iljina M.; Cremades N.; Dalla Serra M.; Knowles T. P. J.; Dobson C. M.; Klenerman D. Anal. Chem. 2015, 87, 8818–8826. 10.1021/acs.analchem.5b01811. [DOI] [PubMed] [Google Scholar]
- Iljina M.; Garcia G. A.; Horrocks M. H.; Tosatto L.; Choi M. L.; Ganzinger K. A.; Abramov A. Y.; Gandhi S.; Wood N. W.; Cremades N.; Dobson C. M.; Knowles T. P. J.; Klenerman D. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E1206–E1215. 10.1073/pnas.1524128113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shammas S. L.; Garcia G. A.; Kumar S.; Kjaergaard M.; Horrocks M. H.; Shivji N.; Mandelkow E.; Knowles T. P. J.; Mandelkow E.; Klenerman D. Nat. Commun. 2015, 6, 7025 10.1038/ncomms8025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg M.; Stibius K. B.; Ndoni S.; Larsen N. B.; Kingshott P.; Hou X. L. Anal. Biochem. 2007, 361, 120–125. 10.1016/j.ab.2006.11.016. [DOI] [PubMed] [Google Scholar]
- Saxton M. J.; Jacobson K. Annu. Rev. Biophys. Biomol. Struct. 1997, 26, 373–399. 10.1146/annurev.biophys.26.1.373. [DOI] [PubMed] [Google Scholar]
- Horrocks M. H.; Lee S. F.; Gandhi S.; Magdalinou N. K.; Chen S. W.; Devine M. J.; Tosatto L.; Kjaergaard M.; Beckwith J. S.; Zetterberg H.; Iljina M.; Cremades N.; Dobson C. M.; Wood N. W.; Klenerman D. ACS Chem. Neurosci. 2016, 7, 399–406. 10.1021/acschemneuro.5b00324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeVine H. I. I. I. Protein Sci. 1993, 2, 404–410. 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stsiapura V. I.; Maskevich A. A.; Kuzmitsky V. A.; Uversky V. N.; Kuznetsova I. M.; Turoverov K. K. J. Phys. Chem. B 2008, 112, 15893–15902. 10.1021/jp805822c. [DOI] [PubMed] [Google Scholar]
- Kuznetsova I. M.; Sulatskaya A. I.; Uversky V. N.; Turoverov K. K. PLoS ONE 2012, 7, e30724 10.1371/journal.pone.0030724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban T.; Hoshino M.; Takahashi S.; Hamada D.; Hasegawa K.; Naiki H.; Goto Y. J. Mol. Biol. 2004, 344, 757–767. 10.1016/j.jmb.2004.09.078. [DOI] [PubMed] [Google Scholar]
- Groenning M. Journal of Chemical Biology 2010, 3, 1–18. 10.1007/s12154-009-0027-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L.; Vastl J.; Gao J. Mol. BioSyst. 2010, 6, 1791–1795. 10.1039/c005255h. [DOI] [PubMed] [Google Scholar]
- Åslund A.; Sigurdson C. J.; Klingstedt T.; Grathwohl S.; Dickstein D. L.; Glimsdal E.; Prokop S.; Lindgren M.; Holtzman D. M.; Hof P. R.; Heppner F. L.; Gandy S.; Jucker M.; Aguzzi A.; Hammarström P.; Nilsson K. P. R.; Bolmont T.; Konradsson P. ACS Chem. Biol. 2009, 4, 673–684. 10.1021/cb900112v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brelstaff J.; Spillantini M. G.; Tolkovsky A. M. Neural Regener. Res. 2015, 10, 1746–1747. 10.4103/1673-5374.165298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingstedt T.; Shirani H.; Åslund A.; Cairns N. J.; Sigurdson C. J.; Goedert M.; Nilsson P. Chem. - Eur. J. 2013, 19 (31), 10179–10192. 10.1002/chem.201301463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora A. K.; Singh P. K.; Patro B. S.; Nath S.; et al. Chem. Commun. 2016, 52, 12163–12166. 10.1039/C6CC05600H. [DOI] [PubMed] [Google Scholar]
- D'Amico M.; Di Carlo M. G.; Groenning M.; Militello V.; Vetri V.; Leone M. J. Phys. Chem. Lett. 2012, 3, 1596–1601. 10.1021/jz300412v. [DOI] [PubMed] [Google Scholar]
- Hoyer W.; Antony T.; Cherny D.; Heim G.; Jovin T. M.; Subramaniam V. J. Mol. Biol. 2002, 322, 383–393. 10.1016/S0022-2836(02)00775-1. [DOI] [PubMed] [Google Scholar]
- Klyubin I.; Betts V.; Welzel A. T.; Blennow K.; Zetterberg H.; Wallin A.; Lemere C. A.; Cullen W. K.; Peng Y.; Wisniewski T.; Selkoe D. J.; Anwyl R.; Walsh D. M.; Rowan M. J. J. Neurosci. 2008, 28, 4231–4237. 10.1523/JNEUROSCI.5161-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drews A.; Flint J.; Shivji N.; Jönsson P.; Wirthensohn D.; De Genst E.; Vincke C.; Muyldermans S.; Dobson C.; Klenerman D. Sci. Rep. 2016, 6, 31910 10.1038/srep31910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein A. D.; Tsuchida M. A.; Amodaj N.; Pinkard H.; Vale R. D.; Stuurman N. Journal of Biological Methods 2014, 1, 10–10. 10.14440/jbm.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goutelle S.; Maurin M.; Rougier F.; Barbaut X.; Bourguignon L.; Ducher M.; Maire P. Fundam. Clin. Pharmacol. 2008, 22, 633–648. 10.1111/j.1472-8206.2008.00633.x. [DOI] [PubMed] [Google Scholar]
- Armbruster D. A.; Pry T. Clin. Biochem. Rev. 2008, 29 (Suppl 1), S49–S52. (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2556583/). [PMC free article] [PubMed] [Google Scholar]
- Cohen S.; Rajah L.; Yoon C.; Buell A.; White D.; Sperling R.; Vendruscolo M.; Terentjev E.; Dobson C.; Weitz D.; Knowles T. Phys. Rev. Lett. 2014, 112, 098101 10.1103/PhysRevLett.112.098101. [DOI] [PubMed] [Google Scholar]
- Knowles T. P. J.; White D. A.; Abate A. R.; Agresti J. J.; Cohen S. I. A.; Sperling R. A.; De Genst E. J.; Dobson C. M.; Weitz D. A. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 14746–14751. 10.1073/pnas.1105555108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang J. C.; Hong L.; Thackray A. M.; Meisl G.; Ponjavic A.; Knowles T. P. J.; Bujdoso R.; Klenerman D., Manuscript submitted for review.
- Hansson O.; Hall S.; Öhrfelt A.; Zetterberg H.; Blennow K.; Minthon L.; Nägga K.; Londos E.; Varghese S.; Majbour N. K.; Al-Hayani A.; El-Agnaf O. M. A. Alzheimer's Res. Ther. 2014, 6, 25–25. 10.1186/alzrt255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuda T.; Qureshi M. M.; Ardah M. T.; Varghese S.; Shehab S. A. S.; Kasai T.; Ishigami N.; Tamaoka A.; Nakagawa M.; El-Agnaf O. M. A. Neurology 2010, 75, 1766–1772. 10.1212/WNL.0b013e3181fd613b. [DOI] [PubMed] [Google Scholar]
- Davson H.Physiology of the Cerebrospinal Fluid; J & A Churchill Ltd.: London, 1967. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.