

Abstract
Human embryonic stem cells (hESCs) can be captured in a primed state resembling the postimplantation epiblast or in a naïve state resembling the preimplantation epiblast. Naïve conditions allow the study of preimplantation development ex vivo but reportedly lead to chromosomal abnormalities, compromising their utility in research and potential therapeutic applications. Although MEK inhibition is essential for the naïve state, here we show that reduced MEK inhibition facilitates the establishment and maintenance of naïve hESCs that retain naïve-specific features, including global DNA hypomethylation, HERVK expression and X chromosome reactivation. We further show that hESCs cultured under these modified conditions proliferate more rapidly, accrue fewer chromosomal abnormalities and display changes in the phosphorylation levels of MAPK components, regulators of DNA damage/repair, and cell cycle. We thus provide a simple modification to current methods to enable robust growth and reduced genomic instability in naïve hESCs.
INTRODUCTION
Human embryonic stem cells (hESCs) self-renew indefinitely while retaining the capacity for multilineage differentiation, providing a valuable tool for research and potential therapeutic applications. Conventional hESC culture conditions include Activin A and basic FGF (abbreviated as A/F) and capture pluripotent cells in a “primed” pluripotent state that resembles the postimplantation epiblast1, 2. Several laboratories have recently developed protocols to capture pluripotent cells in a more primitive or “naïve” state that resembles the preimplantation epiblast3–5. Naïve stem cells offer a useful system to study preimplantation development6, 7 and are more efficient at producing certain specialized cell types, such as primordial germ cells8.
Culture conditions to convert primed hESCs to a naïve state typically rely on a combination of growth factors and small molecules that suppress specific protein kinases involved in differentiation, cell adhesion, and survival3–5. Two culture methods appear to be particularly effective9: The “t2iLGö” protocol involves transient overexpression of the transcription factors KLF2 and NANOG in the presence of the MEK inhibitor (MEKi) PD0325901 and titrated amounts of GSK3β inhibitor (CHIR99021), supplemented with the PKC inhibitor Gö6983 and human LIF (hLIF)4, 10. The “5i/LAF” protocol requires treatment of primed hESCs with inhibitors targeting the GSK3β, ROCK, BRAF, MEK, and SRC kinases in addition to hLIF and A/F5, 7. Inhibitors of the mitogen-activated protein kinase (MAPK/ERK) pathway are common to all currently available protocols.
Suppression of the MAPK pathway via the MEK1/2 inhibitor PD0325901 (PD03) has previously been shown to erode genomic imprints, lead to chromosomal abnormalities, and compromise the developmental potential of mouse ESCs11, 12. However, titration of PD03 from 1 μM to 0.3–0.4 μM or replacement with a SRC inhibitor is reportedly sufficient to improve the epigenetic and genomic stability of mouse ESCs as well as their in vitro and in vivo differentiation potential11–13. Considering the impact of MAPK inhibition on mouse ESCs, we examined the consequences of titrating PD03 or replacing PD03 with alternative MEKi’s on the maintenance of naïve hESCs cultured in 5i/LAF or t2iLGöY.
RESULTS
Reduced MEK inhibition maintains naïve hESCs
We tested whether reduced MEK inhibition maintains naïve colony morphology within the 5i/LAF culture system by titrating PD03 in the presence of constant amounts of BRAF, SRC, GSK3β and ROCK inhibitors using WIBR3 hESCs carrying a naïve-specific ΔPE OCT4-GFP reporter5 (Fig. 1a). Specifically, we used 0.3 μM, 0.5 μM, 0.6 μM and 0.8 μM PD03 as these concentrations are lower than the originally used 1 μM5, 7. Complete omission of PD03 (4i/LAF condition) resulted in downregulation of GFP expression and a concomitant increase in differentiated colonies after ~8 days, consistent with previous observations5 (Fig. 1a and Supplementary Fig. 1a). By contrast, hESCs cultured in 4i/LAF and supplemented with reduced amounts of PD03 showed robust GFP expression and undifferentiated colony morphology (Fig. 1a and Supplementary Fig. 1a). Of note, we were only able to maintain undifferentiated colonies upon continuous passaging of WIBR3 hESCs in 0.5 μM, 0.6 μM or 0.8 μM PD03 whereas hESCs in 0.3 μM PD03 lost their typical dome-shaped morphology (Supplementary Fig. 1b). These results show that reduction of MEKi from 1 μM to 0.5 μM preserves undifferentiated colony morphology and OCT4-GFP expression of hESCs cultured in 5i/LAF. We will refer to this modified culture condition as “modified 5i/LAF” (m5i/LAF) in all subsequent experiments.
Figure 1. Attenuated MEK1/2 inhibition maintains naïve pluripotency in hESCs.
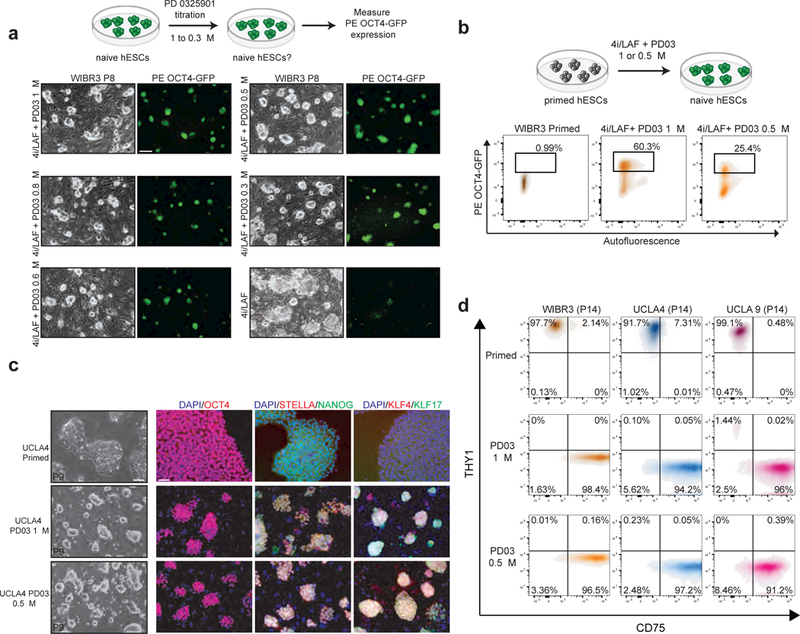
(a) PD0325901 (PD03) titration strategy (upper panel). Representative phase and fluorescence images of WIBR3 ΔPE OCT4GFP hESCs at P8 grown in the indicated media (lower panel). Scale bar 250 μm. (b) Flow cytometric analysis of the proportion of ΔPE OCT4GFP+ cells after reversion of WIBR3 primed hESCs to a naïve state. (c) Representative bright field and immunofluorescence images for P9 UCLA4 hESCs cultured in the indicated media (scale bar: 100 μm, left panel; 50 μm, right panels). (d) Flow cytometric analysis of CD75 and THY-1 protein expression levels in hESC lines cultured as indicated.
We next exposed primed WIBR3 ΔPE OCT4-GFP hESCs to m5i/LAF to determine whether this culture condition also facilitates conversion to a naïve state. Treatment of primed hESCs with either 5i/LAF or m5i/LAF for 10 days led to the activation of the ΔPE OCT4-GFP reporter in 60.3% and 25.4% of cells, respectively (Fig. 1b). Despite the lower efficiency of conversion, we could establish stable, naïve hESC lines from the m5i/LAF culture condition by serially passaging bulk cultures, which expressed the ΔPE OCT4-GFP reporter homogeneously (Supplementary Fig. 1c). We exposed 5 additional primed hESC lines, UCLA1, 3, 4, 5, 9 and 1714 (Supplementary Fig. 1d), to either 5i/LAF or m5i/LAF before scoring for naïve colony morphology and marker expression. We were again able to derive naïve cell lines from UCLA4 and UCLA5 hESCs by serial bulk passaging in m5i/LAF. However, the establishment of naïve hESC lines from UCLA1, 9 and 17 in m5i/LAF was less efficient and could only be achieved after picking and expanding individual colonies from heterogeneous cultures (Supplementary Fig. 1d, e). Of note, UCLA3 hESCs were resistant to naïve conversion in m5i/LAF medium. For all other examined cell lines, the generation of stable, naïve hESC cultures exhibiting homogeneous colony morphology took approximately 5–6 passages in either 5i/LAF or m5i/LAF media, which was confirmed by uniform expression of the pluripotency markers NANOG and OCT4 as well as the naïve markers KLF4, KLF17 and DPPA3 (STELLA) in two representative hESC lines (Fig. 1c and Supplementary Fig. 1f). We found that naïve hESC lines downregulated the primed-specific surface marker THY-115 and induced the naïve-specific surface marker CD7515 to a similar degree using either 5i/LAF or m5i/LAF conditions (Fig. 1d). Finally, we confirmed the ability of naïve hESCs derived in m5i/LAF conditions to access all 3 germ layers by inducing well-differentiated teratomas (Supplementary Fig. 1g). We conclude that m5i/LAF treatment facilitates not only the maintenance of naïve hESCs but also the conversion of primed hESCs to a naïve state.
To assess whether titrated PD03 also maintains naïve hESCs cultured in t2iLGöY, we attempted to directly transfer naïve hESCs from the m5i/LAF to the t2iLGöY medium4, 16. Starting with WIBR3 hESCs stably grown in 5i/LAF (for >8 passages) we switched medium to t2iLGöY and maintained naïve hESCs for at least 4 passages without extinguishing the ΔPE OCT4-GFP reporter, lowering expression of naïve markers (KLF2, REX1, DPPA5, KLF4 and TFCP2L1) or losing self-renewal potential (Supplementary Fig. 2a, b). However, reduction of PD03 from 1 μM to 0.5 μM in t2iLGöY media was insufficient to maintain pluripotency of transferred hESCs (Supplementary Fig. 2c). Consistent with this, H9 hESCs carrying a naïve-specific reporter (EOS-C(3+)-GFP/puroR)4 could not be expanded after naïve conversion in t2iLGöY medium supplemented with 0.5 μM PD03 even when NANOG and KLF2 were exogenously expressed (Supplementary Fig. 2d). We also note that only those primed UCLA lines that efficiently converted to a naïve state in m5i/LAF (Supplementary Fig. 1d and data not shown) could be adapted to t2iLGöY medium, pointing to cell line-dependent factors that influence the response to MEK inhibition. We conclude that t2iLGöY cultured hESCs are more sensitive to PD03 concentration than 5i/LAF cultured hESCs as they cannot be maintained long-term in titrated MEKi despite expression of exogenous transcription factors.
Alternative MEK inhibitors maintain hESCs in a naïve state
We next investigated whether alternative MEKi’s are effective in sustaining naïve pluripotency in hESCs. Specifically, we tested a panel of 9 MEK inhibitors targeting either MEK1 alone (TAK-733 and Cobimetinib), MEK1 and MEK2 together (Selumetinib, Trametinib, Pimasertib, Refametinib, Binimetinib, AZD8330) or MEK1 along with BRAF and CRAF (RO5126766). We replaced PD03 within the 5i/LAF condition using two different concentrations for these inhibitors (1 μM and 0.5 μM) and then monitored expression of the ΔPE OCT4-GFP transgene (Fig. 2a). All 9 inhibitors maintained GFP expression at levels observed in PD03 treated cells (Fig. 2b). However, we detected a loss of dome-shaped morphology in cells cultured with Trametinib, AZD8330 or Selumentinib; we excluded these small molecules from subsequent experiments. To assess whether these inhibitors also facilitate conversion, we exposed the primed hESC line UCLA4 to 5i/LAF, m5i/LAF or 4i/LAF supplemented with 1 or 0.5 μM of TAK-733, Refametinib, Cobimetinib, Pimasertib or RO526766 (Supplementary Fig. 2e). We obtained homogenous cell cultures with naïve-like morphology and these could be expanded for multiple passages using all examined MEKi’s at a concentration of 1 μM, but only TAK-733, Refametinib or Cobimetinib maintained naïve colony morphology at 0.5 μM (Supplementary Fig. 2f). Importantly, UCLA4 hESCs cultured under these conditions expressed the naïve-associated transcription factors KLF4, KLF17, NANOG and STELLA (Fig. 2c). We quantified conversion efficiency by monitoring reactivation of ΔPE OCT4-GFP by flow cytometry. Replacement of PD03 with TAK-733, Refametinib or Cobimetinib did not affect the percentage of GFP+ cells obtained by day 8 of conversion (Fig. 2d). Moreover, treatment of hESCs with TAK-733 and Cobimetinib at low concentration (0.5 μM) activated the ΔPE OCT4-GFP reporter to a similar degree as treatment with high concentrations (1 μM) of PD03 (~40%) (Fig. 2d). Finally, we determined the potential of alternative MEKi’s to replace PD03 during the maintenance of naïve hESCs in t2iLGöY. TAK-733, Refametinib and Cobimetinib sustained cell proliferation, clonal growth and pluripotency gene expression to a similar extent as PD03 in the context of t2iLGöY culture (Supplementary Fig. 3a-d). Overall, these results suggest that (i) PD03 can be substituted by other MEKi’s using two different naïve culture systems, (ii) targeting MEK1 alone is sufficient to sustain naïve pluripotency, and (iii) low concentrations of TAK-733 and Cobimetinib endow primed cells with a naïve phenotype.
Figure 2. MEK1 inhibition is sufficient to sustain naïve pluripotency in 5i/LAF.
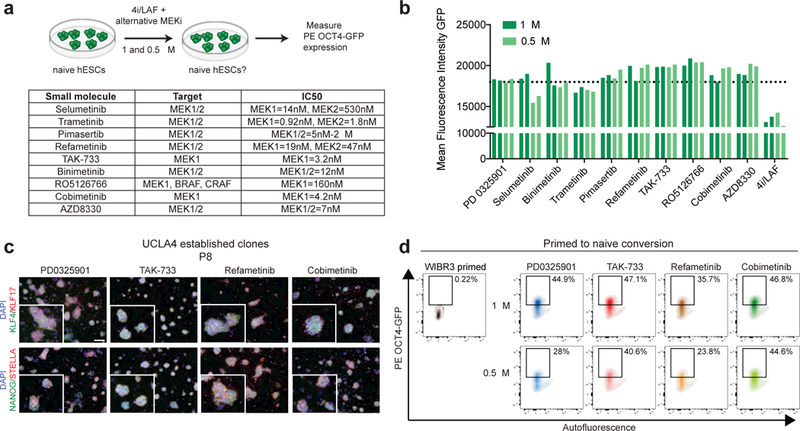
(a) Alternative MEK inhibitors used in this study. (b) Flow cytometric analysis of the proportion of WIBR3 ΔPE OCT4GFP+ hESCs cultured as indicated. (c) Representative immunofluorescence images of UCLA4 hESCs grown in the indicated media (P8) (scale bar: 100 μm). (d) Flow cytometric analysis of the proportion of ΔPE OCT4GFP+ cells after primed-to-naive conversion of WIBR3 hESCs in 5i/LAF supplemented with the indicated MEKi.
A naïve-like transcriptome and proteome in modified 5i/LAF
We performed RNA-seq of hESC lines derived and maintained in primed conditions as well as 6 naïve conditions (i) 5i/LAF, (ii) m5i/LAF, (iii) 4i/LAF plus 0.5 μM TAK-733, (iv) 4i/LAF plus 0.5 μM Cobimetinib, (v) 4i/LAF plus 0.5 μM Refametinib, and (vi) hESCs transitioned from 5i/LAF to t2iLGöY for 3 passages. PCA analysis of these samples showed that the major transcriptional changes along principal component 1 (86% total variability) separated all primed cell lines from all naïve cell lines, regardless of culture condition (Fig. 3a and Supplementary Fig. 4a). Principal component 2 appears to detect differences in the genetic background of cell lines regardless of MEKi or cell state (i.e. primed vs. naïve) (Fig. 3a). This is consistent with the previous finding that genetic background drives transcriptional variation among different human pluripotent stem cells17, 18.
Figure 3. Transcriptome and proteome analysis of hESCs cultured in modified conditions.
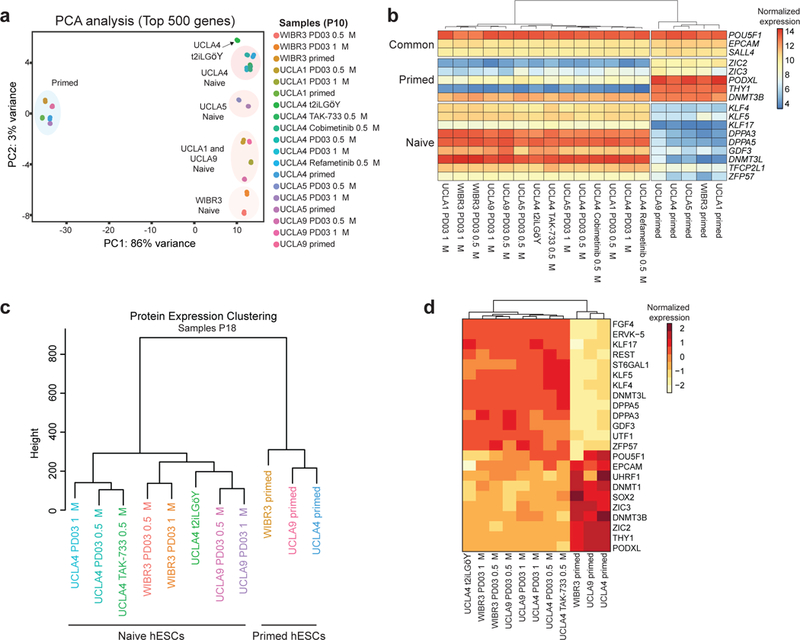
(a) PCA analysis of RNA-seq data for the indicated hESCs samples (P10) using the top 500 most variable genes. (b) Heatmap for selected primed and naïve pluripotency genes. (c) Unsupervised hierarchical clustering of proteomic samples (P18). (d) Heatmap for selected primed and naïve pluripotency proteins.
Genes associated with naïve pluripotency (KLF4/5/17, TFCP2L1, DPPA5) were highly expressed in all naïve cell lines, while genes associated with primed pluripotency (ZIC3, THY1, PODXL) were expressed only in primed hESCs (Fig. 3b and Supplementary Fig. 4b). Induction of naive specific genes was independently confirmed by qRT-PCR in UCLA17 hESCs cultured in either 5i/LAF or m5i/LAF conditions (Supplementary Fig. 4c). Of note, UCLA4 naïve cells cultured in 4i/LAF and supplemented with TAK-733 expressed slightly higher levels of naïve markers compared to UCLA4 cells cultured in 5i/LAF (Supplementary Fig. 4d). Reactome pathway enrichment analysis revealed an overrepresentation of MAPK targets among the upregulated genes in the m5i/LAF compared to the 5i/LAF condition (Supplementary Fig. 4e). Together, these results demonstrate that hESCs cultured in m5i/LAF or with alternative MEKi’s preserve and in some cases boost transcription patterns characteristic of naïve pluripotent cells.
We next performed large-scale quantitative proteomic analysis of UCLA4, UCLA9 and WIBR3 hESC lines derived and maintained in either primed conditions, 5i/LAF or m5i/LAF. In addition, we analyzed the proteome of UCLA4 hESCs cultured in t2iLGöY or in 4i/LAF supplemented with 0.5 μM TAK-733. We quantified >8,649 proteins that were highly expressed in primed and naïve cells (Supplementary Fig. 5a and Supplementary Table 1). We further observed a good correlation (Pearson Correlation=~0.56) between RNA and protein expression in our samples, in line with previous reports using other cell lines19 (Supplementary Fig. 5b). Unsupervised hierarchical clustering of proteome data accurately separated naïve from primed samples regardless of culture condition (Fig. 3c). In further agreement with the RNA-seq data, naïve hESCs upregulated naïve-specific proteins including FGF4 and KLF17, and downregulated proteins associated with the primed state, such as ZIC3 and THY1 (Fig. 3d and Supplementary Fig. 5c). Higher expression of some naïve markers (e.g. DNMT3L, DPPA5) was again observed in hESCs cultured in the presence of TAK-733 (Fig. 3d). Interestingly, we found that ~40% of genes that were upregulated at the protein level in either naïve or primed conditions did not change at the RNA level when comparing proteomics with RNA-seq data, suggesting that a large fraction of genes are regulated posttranscriptionally (Supplementary Fig. 5d). These proteins included chromatin regulators, transcription factors and RNA binding proteins, including known pluripotency regulators such as SOX2 and MBNL1 (Supplementary Fig. 5d, e).
Collectively, these data show that our modification of the naïve culture condition have minimal effects on the proteome of naïve hESCs.
Modified 5i/LAF maintains HERVK expression, genomic hypomethylation and X chromosome inactivation state
Our preparation of RNA-seq libraries from ribodepleted RNA allowed us to measure expression of transposable elements (TEs). TEs have been proposed to be sensitive discriminators between primed and naïve hESCs20. TE transcriptional patterns clearly separated primed samples from all the naïve samples (Supplementary Fig. 6a). Inspection of TE subclasses in our cell lines further revealed that members of the HERVK family were exclusively induced in the naïve state, consistent with a recent report7 and supports the interpretation that our conditions endow hESCs with a bona fide naïve state (Fig. 4a). hESCs cultured in medium containing 0.5 μM TAK-733 showed the highest levels of HERVK expression among all tested naïve conditions, including 5i/LAF (Fig. 4a). This finding correlates with higher levels of naïve transcription factors in TAK-733 cultured hESCs and suggests that suppression of MEK1 with this small molecule is most effective at inducing ‘naïve-like’ molecular characteristics.
Figure 4. hESCs cultured in m5i/LAF are hypomethylated and have two active X chromosomes.
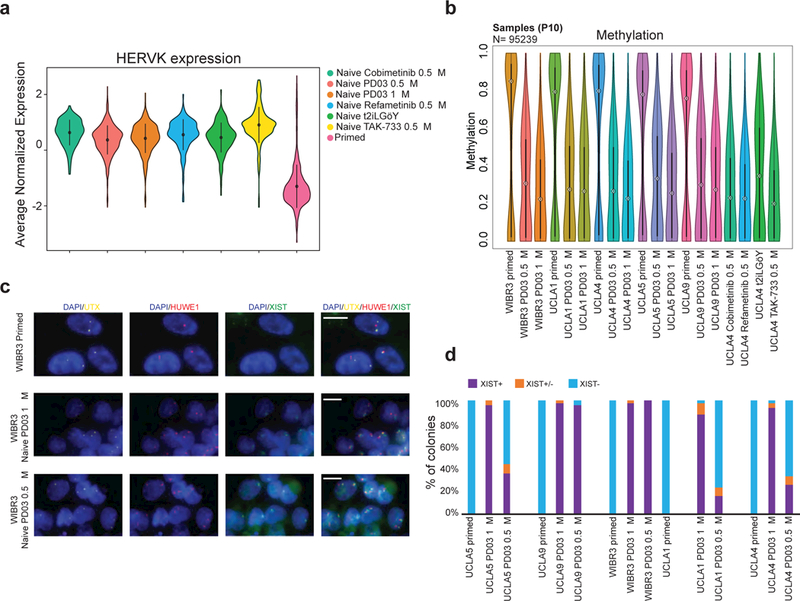
(a) Violin plot representation of HERVK expression (n=64) in hESCs expanded in the indicated culture conditions (P10). For primed, naïve PD03 1μM and naïve PD03 0.5 μM n=5 independent hESC lines (UCLA1, 4, 5, 9 and WIBR3), for naïve Cobimetanib 0.5 μM, Refametinib, TAK-733 and t2iLGöY n=1 (UCLA4). (b) Global methylation analysis of primed and naive hESCs by RRBS analysis using violin plot representation. (c) Representative RNA FISH images for primed and naïve hESCs at P16, detecting the indicated transcripts. (scale bar: 10 μm). (d) Quantification of XIST RNA FISH patterns.
Global DNA hypomethylation is a key epigenetic hallmark of naïve pluripotent cells 21, 22. We used reduced representation bisulfite sequencing (RRBS) to assess the impact of naïve culture conditions on global DNA methylation relative to primed hESCs. All examined naïve samples exhibited global hypomethylation compared to primed counterparts (Fig. 4b and Supplementary Fig. 6b). In agreement, PCA analysis separated primed from naïve samples based on 95,239 1kb tiles (Supplementary Fig. 6c) or promoter regions (Supplementary Fig. 6d). We also observed that imprint control regions (ICRs) become demethylated in naive conditions (Supplementary Fig. 6e), confirming recent reports15, 23. We found that naïve cells cultured in m5i/LAF had slightly higher global methylation levels compared 5i/LAF, consistent with our previous finding that PD03 drives genomic hypomethylation in naïve mouse ESCs11. Closer inspection of these regions revealed 984 sites that were specifically hypermethylated in m5i/LAF relative to 5i/LAF (Supplementary Fig. 6f). Most of these regions were associated with repetitive DNA elements of the SINE and LINE families (Supplementary Fig. 6g). UCLA4 hESCs exposed to 4i/LAF plus 0.5 μM TAK-733 showed the lowest overall methylation levels of any tested condition including 5i/LAF (Fig. 4b and Supplementary Fig. 6b), which concurs with the relative upregulation of naïve-specific markers in TAK-733 treated hESCs (Fig. 3d, 4a and Supplementary Fig. 4d).
X-chromosome inactivation (XCI) is another tightly regulated epigenetic process that distinguishes primed from naïve female hESCs6, 24. We used RNA FISH for X-linked transcripts to assess how the m5i/LAF condition influences XCI status. While all tested female primed hESCs lacked XIST expression due to XCI erosion, we confirmed the presence of two X chromosomes using RNA FISH for the X-linked gene UTX, which generally escapes XCI (Fig. 4c and Supplementary Fig. 7a, b). We detected monoallelic transcription of HUWE1, an X-linked gene commonly subject to XCI, in 4 of 5 primed hESC lines (UCLA1, 4, 5 and WIBR3) (Fig. 4c and Supplementary Fig. 7a-c). However, UCLA9 cells showed HUWE1 signal from both X chromosomes, as previously observed for this line (Supplementary Fig. 7c and ref. 14). Importantly, adaptation of all primed hESC lines to either 5i/LAF or m5i/LAF conditions resulted in biallelic transcription of HUWE1, indicative of X chromosome reactivation in the naïve state (Fig. 4c and Supplementary Fig. 7a-c). We next sought to quantify XIST activation in primed, 5i/LAF and m5i/LAF cultured hESCs. Colonies in which XIST signal was present or absent in over 80% of cells were considered XIST+ and XIST- respectively. Colonies with heterogeneous patterns of XIST expression were designated XIST+/−. The vast majority (~95%) of naïve colonies we derived and expanded in regular 5i/LAF media expressed XIST from one of the active X chromosomes (XIST+), consistent with a previous report6. Naïve cells cultured in m5i/LAF showed a lower fraction (20–40%) of XIST+ colonies (Fig. 4d). Collectively, these results indicate that the m5i/LAF culture condition captures a bona fide naïve state that extends from the gene expression and proteomic level to the level of TE transcription and epigenetic modifications.
Modified 5i/LAF condition preserves hESC genomic stability
We examined functional consequences of continual treatment of naïve hESCs with titrated PD03 by measuring growth potential, apoptosis and genomic stability. hESC lines cultured in m5i/LAF proliferated more rapidly than hESCs cultured in 5i/LAF (n=3 representative hESC lines), indicating that strong MAPK inhibition limits the growth potential of naïve hESCs (Fig. 5a). These differences in growth behavior were more pronounced in some hESC lines (e.g., UCLA5; ~4-fold) than in others (e.g., UCLA4; ~2-fold), pointing once more to cell line-intrinsic responses to MAPK modulation. We found that m5i/LAF cultured WIBR3 hESCs gave rise to twice as many colonies as 5i/LAF cultured hESCs using a clonal colony formation assay, suggesting increased viability of dissociated hESCs that were maintained in low MEKi (Supplementary Fig. 8a). Consistent with these observations, we detected a two-fold decrease in the fraction of Annexin V+ cells using m5i/LAF cultured WIBR3 hESCs (Supplementary Fig. 8b). NhESCs cells cultured in t2iLGöY and subsequently plated in reduced concentrations of PD03 (0.6 μM) for 7 days also exhibited a higher clonogenic potential (Supplementary Fig. 8c). Together, these results show that reduced concentrations of MEKi using two naïve culture conditions leads to increased proliferation and decreased apoptosis.
Figure 5. Naïve hESCs cultured in m5i/LAF retain a more stable karyotype.
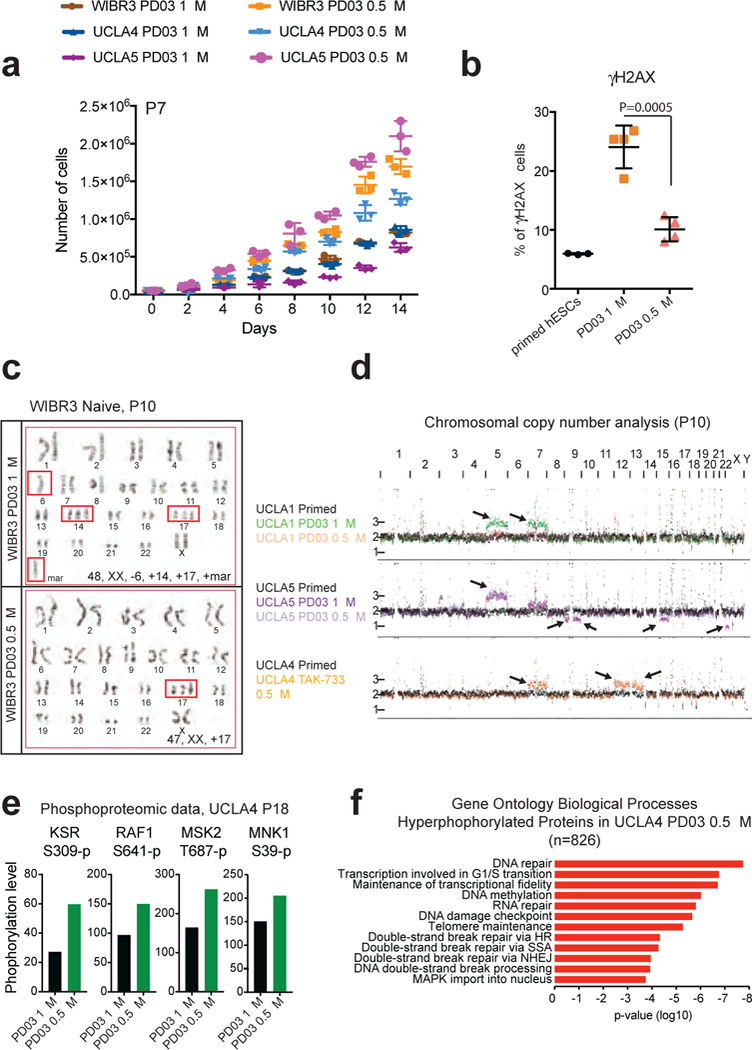
(a) Cell numbers during the expansion of naive hESCs in the indicated conditions. Error bars indicate mean ± s.d. (n=3 biologically independent samples). (b) Flow cytometric analysis for γH2AX+ cells using primed UCLA1 hESCs. Error bars indicate mean ± s.d. (n=3 biologically independent samples), naïve UCLA1 hESCs cultured in 5i/LAF (n=4 independent clones) or m5i/LAF (n=4 independent clones). Statistical significance was determined using a two-tailed unpaired Student’s t-test (P=0.0005). (c) Representative karyotypes at p10 of hESCs converted and cultured as indicated. Red boxes highlight chromosomal abnormalities. (d) Chromosomal copy number analysis by whole-genome sequencing. Black arrows indicate abnormalities. (e) Phosphorylation levels at the indicated residues assessed by phosphoproteomics in UCLA4 cells cultured as shown. (f) Gene ontology analysis of hyperphosphorylated proteins (>1.5-fold, n=826) in UCLA4 hESCs cultured as shown.
Recent studies have shown that suppression of MAPK/ERK signaling compromises genomic stability in mouse ESCs25 and this is accompanied by an increased number of γH2AX foci, which correlates with DNA double strand breaks26. We used flow cytometry to examine the frequency of γH2AX foci in primed hESCs and naïve hESCs cultured in either 5i/LAF or m5i/LAF. Human ESCs expanded in 5i/LAF showed a 4-fold increase in γH2AX foci (~20%) than hESCs maintained in primed conditions (~5%) (Fig. 5b). Decreasing the amount of PD03 from 1 μM to 0.5 μM reduced the percentage of γH2AX foci (~10%; P-value=0.0005), supporting the interpretation that strong MAPK inhibition via PD03 triggers a DNA damage response.
Based on our observations, we hypothesized that naïve hESCs in m5i/LAF may preserve a relatively normal karyotype due to reduced selective pressure. Indeed, we found that naïve WIBR3 hESCs cultured in m5i/LAF for 10 passages retained a largely normal karyotype that was comparable to the parental hESCs cultured in primed medium (Fig. 5c and Supplementary Fig. 8d, e). By contrast, hESCs cultured in conventional 5i/LAF media for the same period of time acquired multiple additional chromosomal abnormalities and none of the karyotyped cells (n=20) were normal (Supplementary Fig. 8f).
To test the generality of our findings, we performed shallow whole-genome sequencing of UCLA1, 4, 5 and 17 hESCs in primed media, 5i/LAF, m5i/LAF and 4i/LAF media supplemented with TAK-733 following continual propagation of these cell lines for 10 passages. We found that UCLA5 hESCs cultured in 5i/LAF conditions accrued multiple chromosomal aberrations (trisomy 5, partial amplification of chromosome 7, loss of one X chromosome and partial loss of chromosomes 8, 9, 15) relative to the m5i/LAF conditions (trisomy 5 and partial amplification of chromosome 7) (Fig. 5d). Similarly, UCLA1 and UCLA17 hESCs cultured in 5i/LAF showed trisomy 5 and 7, while they remained karyotypically normal in m5i/LAF (Fig. 5d and Supplementary Fig. 8g). However, naïve hESCs cultured in 4i/LAF and TAK-733, which show higher HERVK transcription, increased RNA and protein expression of naïve markers as well as reduced global methylation become highly abnormal after 10 passages (trisomy 7, 12 ,13) (Fig. 5d). These results imply that reduced MAPK inhibition relieves selection pressure for acquiring chromosomal abnormalities that promote the growth and/or survival of naïve hESCs.
Molecular effects of reduced MEK inhibition
In an effort to understand the mechanisms by which increased MEK activity affects naïve hESC properties, we generated phosphoproteomic data for UCLA4 hESCs cultured in 5i/LAF, m5iLAF and t2iLGöY (Supplementary Table 2). We confirmed that reducing PD03 to 0.5 μM results in increased ERK phosphorylation in UCLA4 hESCs, which was independently verified by Western blot analysis (Supplementary Fig. 9a, b and Supplementary Fig. 10). We also observed increased phosphorylation of other components of the MAPK pathway (Fig. 5e).
We next focused on proteins that gained phosphorylation in m5i/LAF relative to 5i/LAF but were not directly related to MAPK signaling. Intriguingly, most of these proteins were associated with functional categories related to “DNA repair”, “DNA methylation”, “DNA damage checkpoints” and “G1/S transition of cell cycle”, implying that MAPK inhibition by PD03 affects molecular regulators of genomic stability and DNA repair (Fig. 5f). We confirmed overrepresentation of these functional categories using an independent hESC line (UCLA9) (Supplementary Fig. 9c, d). We finally selected differentially phosphorylated proteins between hESCs cultured in 5i/LAF and t2iLGöY and performed Gene Ontology enrichment analysis. Surprisingly, we also observed a clear enrichment of categories related to the MAPK pathway. Considering that equal concentrations of PD03 are present in 5i/LAF and t2iLGöY, this observation points to MEKi-independent modulation of the MAPK pathway in one of the two naïve culture conditions (Supplementary Fig. 9e).
DISCUSSION
Through systematic characterization of primed and naïve hESCs generated using different culture conditions, we uncovered an unexpected sensitivity of naïve pluripotent stem cells to MAPK signal inhibition, which affects their proliferation rate, survival and genome integrity. We propose that minimal MAPK activity is beneficial to preserve both robust growth potential and genomic stability in naïve human pluripotent stem cells maintained in 5i/LAF (see model Supplementary Fig. 9f). Our data also suggest that different hESC lines respond differently to MEK inhibition. For instance, we found that PD03 can be reduced to 0.5 μM in WIBR3 hESCs while PD03 can be further decreased to 0.25 μM in UCLA4 hESCs (data not shown). We suggest that MEK inhibitors should ideally be titrated in a cell line dependent fashion. The observation that attenuated MEK inhibition impacts phosphorylation levels of proteins regulating DNA damage and repair and leads to a reduced number of γH2AX foci provides a possible molecular explanation for the observed phenotypes.
Our observation that naïve hESCs cultured in 5i/LAF and t2iLGöY exhibit differences in MAPK activity despite equivalent concentrations of MEKi suggests that the BRAF and SRC inhibitors within 5i/LAF media further dampen the MAPK pathway compared to t2iLGöY media. The notion that MAPK signaling is less repressed in t2iLGöY cultured hESCs than in 5i/LAF cultured hESCs, which parallels our observations, is consistent with the following. First, t2iLGöY cultured cells, like m5i/LAF cultured cells, divide more rapidly and are karyotypically more stable than 5i/LAF cultured hESCs10, 27. Second, PD03 cannot be reduced to 0.5 μM in hESCs continually cultured in t2iLGöY without inducing differentiation (Supplementary Fig. 2c, d). Third, UCLA lines that are refractory to naïve conversion using bulk passaging in 0.5 μM PD03 could not be transferred from 5i/LAF to t2iLGöY, suggesting a requirement for stronger suppression of the MAPK pathway in certain hESC lines. Fourth, the majority of cells cultured in t2iLGöY media have two active X chromosomes but lack XIST transcription6, 10, similar to cells expanded in m5i/LAF medium. Finally, hESCs maintained in this alternative condition exhibit higher global methylation levels than naïve cells expanded in 5i/LAF medium but similar levels to m5i/LAF cultured hESCs (Fig. 4b, Supplementary Fig. 6b and ref. 27).
The transcriptional, proteomic, phosphoproteomic and genomic methylation data we have generated here will provide a useful resource for future studies aimed at dissecting the mechanisms underlying naïve and primed pluripotency. Our results may also have a bearing on potential therapeutic applications of naïve pluripotent stem cells since chromosomal stability is of utmost importance when considering cell and gene therapy applications.
ONLINE METHODS
Mice
All procedures involving mice adhered to the guidelines of the approved Massachusetts General Hospital Institutional Animal Care and Use Committee (IACUC) protocol no. 2006N000104. NOD/SCID mice were acquired from Jackson laboratory.
Cell culture
Conventional human primed ESCs (WIBR3 cell line, ΔPE-OCT4GFP5, UCLA1, UCLA3, UCLA4, UCLA5, UCLA9 and UCLA17 cell lines) were maintained on Matrigel (Corning) coated dishes in E8-medium (Stem Cell Technologies) and passaged using Versene solution (Life Technologies). For maintenance, cells were passaged every 4–5 days. Primed H1 and H9 hESC lines were culture as previously reported4. To achieve reversion to a naïve state, hESCs that had been passaged 6 days earlier were washed once with 1X PBS (Life Technologies) and treated for 3 minutes with TrypLE express enzyme (1X, Life Technologies). Cell were dissociated into a single-cell suspension and plated at a density of 30.000 cells per 9.5cm2 on irradiated CF-1 MEFs (MTI Globalstem) in E8 medium supplemented with 10 μM Y-27632 (Axon Medchem). Two days later, medium was changed to 5i/LAF and then changed daily. 5i/LAF medium contained a 50:50 mixture of DMEM/F-12 (Life Technologies) and Neurobasal medium (Life Technologies) containing 1× N2 supplement (Life Technologies), 1× B27 supplement (Life Technologies), 10 ng/mL bFGF (Peprotech), 1% nonessential amino acids (Life Technologies), 1mM GlutaMAX (Life Technologies), penicillin-streptomycin (Life Technologies), 0.1 mM beta-mercaptoethanol (Life Technologies), 50 μg/mL BSA (Life Technologies), 0.5 μM IM-12 (Axon Medchem), 0.5 μM SB590885 (Axon Medchem), 1 μM WH-4–023 (Axon Medchem), 10 μM Y-27632 (Axon Medchem), 20 ng/mL Activin A (Peprotech), 20 ng/mL rhLIF (Peprotech), 0.5% KSR (Life Technologies) and 0.5 or 1 μM PD0325901 (Axon Medchem) as indicated in the main text. After roughly 8–10 days, cells were dissociated using Accutase (Life Technologies) and centrifuged in fibroblast medium [DMEM (Life Technologies) supplemented with 10% FBS (Hyclone), 1 mM GlutaMAX (Life Technologies), 1% nonessential amino acids (Life Technologies), penicillin-streptomycin (Life Technologies), 0.1 mM beta-mercaptoethanol (Life Technologies)] and replated after passing through a 40 μm cell strainer in 5i/LAF medium on irradiated CF-1 MEFs.
Established naïve hESC lines were cultured on irradiated CF-1 MEFs (2.5×106 cells per 9.5 cm2) in 4i/LAF medium with the indicated concentration of PD0325901 and passaged every 6–7 days. Cells were feeded daily with fresh medium.
Human naïve hESCs were also cultured in t2iLGöY conditions for the experiments indicated in the text. t2iLGöY medium contained a 50:50 mixture of DMEM/F-12 (Life Technologies) and Neurobasal medium (Life Technologies) containing 1× N2 supplement (Life Technologies), 1× B27 supplement (Life Technologies), 10 ng/mL bFGF (Peprotech), 1% nonessential amino acids (Life Technologies), 1 mM GlutaMAX (Life Technologies), penicillin-streptomycin (Life Technologies), 0.1 mM beta-mercaptoethanol (Life Technologies), 50μg/mL BSA (Life Technologies), 1 μM or 0.5 μM PD0325901 (Axon Medchem), 1 μM CHIR99021 (Axon Medchem), 2.5 μM Gö6983 (Axon Medchem), 10 μM Y-27632 (Axon Medchem), 20 ng/mL rhLIF (Peprotech) and 50 μg/ml ascorbic acid (Sigma-Aldrich).
H9 hESCs and H9 hESCs carrying the EOS-C(3+)-GFP/puroR piggyBac reporter (EOS) were reset to the naïve state as reported earlier4. Briefly, cells were reset by over-expression of NANOG and KLF2 under t2iL+Dox conditions. After circa 5 passages, medium was switched to t2iLGö and cells were cultured for at least 5 additional passages. For colony formation assay of reset hESCs, 3,500 cells were seeded on irradiated MEFs and cultured under various concentration of PD0325901 (-Dox). Cells were stained at day 7 for alkaline phosphatase expression using Alkaline Phosphatase Kit (SIGMA 86R).
All experiments in this study were performed under low oxygen conditions (5% O2).
Human naïve and primed hESCs were sent for karyotyping to the Cytogenomics Core Laboratory at the Brigham and Women’s Hospital in Boston.
A detailed protocol for the primed to naïve conversion experiments can be found at protocol exchange28.
MEK inhibitors
The following MEKi were used to culture naïve hESCs at the concentrations indicated in the manuscript: PD0325901 (Axon Medchem), Selumetinib (Selleck Chemicals), Binimetinib (Selleck Chemicals), Trametinib (Selleck Chemicals), Pimasertib (Selleck Chemicals), Refametinib (Selleck Chemicals), TAK-733 (Selleck Chemicals), RO5126766 (Selleck Chemicals), Cobimetinib (Selleck Chemicals), AZD8330 (Selleck Chemicals).
RNA FISH and Microscopy
RNA FISH was performed as previously described6. Briefly, cells grown on gelatinized glass coverslips were fixed with 4% formaldehyde, permeabilized with 0.5% Triton X-100 (10 min each), and serially dehydrated with 70%–100% ethanol. Coverslips were hybridized with labeled DNA probes generated from bacterial artificial chromosomes (BACs). The Imager M1 microscope (Zeiss) was used for acquiring and ImageJ software (NIH) for processing z stack images.
Western blot analysis
In preparation for Western blot analysis, cells were lysed in modified RIPA buffer (50 mM Tris-HCL (pH8) (Sigma-Aldrich), 150 mM NaCl (Sigma-Aldrich), 0.1% SDS (Sigma-Aldrich), 0.5% sodium deoxycholate (Sigma-Aldrich), 1% Triton X-100 (Sigma-Aldrich), 1 mM EDTA (Sigma-Aldrich), 1X protease inhibitor cocktail (Roche Diagnostics), 1X PhosSTOP phosphatase inhibitors (Roche Diagnostics), and 0.01 U/μl benzonase (Novagen)). Lysates were sonicated for 5 minutes using a Bioruptor sonication device (Diginode Diagnostics). The following antibodies were used for Western blot: β-ACTIN (Cell Signaling Technology, clone 13E5, cat. #5125); ERK1/2 (Cell Signaling Technology, 137F5, 4695); p-ERK1/2 (Cell Signaling Technology, D13.14.4E, 4370).
AP Staining
Alkaline phosphatase staining was performed with the Vector Red substrate kit (Vector Labs). Colony quantifications were performed using ImageJ.
RNA preparation
RNA isolation was performed using the miRNeasy mini kit (Qiagen). RNA was eluted from the columns using RNase-free water and quantified using a Nanodrop ND-1000. cDNA was produced with the High Capacity RNA-to-cDNA kit (Applied Biosystems). To generate material for sequencing cells were sorted for GFP expression and cell viability (DAPI staining).
qRT-PCR analyses
qRT-PCR reactions were set up in triplicate with the Brilliant III SYBR Master Mix (Agilent Genomics) with primers listed in Supplementary Table 3. Reactions were run on LightCycler 480 (Roche) PCR machine with 40 cycles of 30s at 95 °C, 30s at 60 °C and 30s at 72 °C.
DNA preparation
DNA was extracted using the DNeasy Blood & Tissue Kit (Qiagen) and quantified using the Qubit dsDNA High Sensitivity Kit (Life Technologies).
Reduced representation bisulfite sequencing (RRBS)
RRBS libraries were prepared using a commercial kit (Ovation RRBS Methyl-Seq System, NuGen, San Carlos, CA) following the manufacturer’s protocol except that we pooled 12 individually barcoded reactions after the final repair step and performed the bisulfite conversion and library amplification as a pool. Libraries were sequenced on an Illumina HiSeq2500 high-output flowcell without a PhiX spike-in using Nugen’s custom sequencing primer for read 1 (50 bases) and standard Illumina sequencing primers to read the 8-base sample barcodes. Sequencing reads were trimmed using timgalore (default parameters, http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), as well as a NuGEN-provided script for diversity trimming and filtering. Trimmed reads were aligned to the hg19 human genome using BSmap29 with flags -v 0.05 -s 16 -w 100 -S 1 -p 8 -u. The methylation status of CpGs were called by observing bisulfite conversion in reads at locations of cytosines in the reference sequence. Region methylation averages were called using CpGs that were covered with at least 3 reads in at least 80% of samples. Imprinting control regions were defined as in30.
FACS analyses
GFP, Annexin V (APC, BD Bioscience 550474), γH2AX (Alexa Fluor 647, BD Bioscience 560447), THY-1 (PE anti-human CD90 (Biolegend 328110)) and CD75 (Purified anti-human CD75 (LN-1, 326901 Biolegend) levels were analyzed with an LSR II FACS (BD Biosciences) using Diva v6.1.2 (BD Biosciences) and FlowJo software v10 (TreeStar). Cell permeabilization was performed using the Fix and Perm Cell fixation and cell permeabilization Kit (ThermoFisher Scientific, GAS003) following the manufacturer instructions. Goat anti-Mouse IgM Heavy Chain Secondary Antibody, Alexa Fluor® 647 conjugate (1:500) (A-21238, Thermo Fisher) was used as secondary antibody.
Immunofluorescence
For immunostaining, the cells were fixed with 4% paraformaldehyde, blocked and incubated with primary antibodies overnight at 4°C. They were then stained with secondary antibodies Goat anti-Rabbit IgG (H+L) (1:500) (Alexa Fluor 488, A11008, Thermo Fisher) and Goat anti-Mouse IgG (H+L) (1:500) (Alexa Fluor 546, A11003, Thermo Fisher) at RT for one hour. Nuclear staining was performed with DAPI (1:1000) (BD Bioscience, 564907). The following primary antibodies were used in this study: Anti-KLF17 antibody produced in rabbit (1:100) (HPA024629, SIGMA), GKLF/EKLF/LKLF Antibody (F-8) (1:100) (sc-166238, Santa Cruz), Mouse Anti-Stella Antibody (1:100) (MAB4388, EMD Millipore), Nanog (D73G4) XP® Rabbit mAb #4903 (1:300) (4903S, Cell Signaling), Oct-3/4 Antibody (C-10) (1:300) (sc-5279, Santa Cruz).
Teratoma assay
2 × 106 cells were resuspended in 300 μl of Matrigel (Corning) and injected subcutaneously into the flanks of an immunocompromised NOD/SCID mouse. Teratomas were monitored and removed once tumour size reached 1 cm or tumours became ulcerated. Tumours were then weighed and processed for haemaytoxylin and eosin staining analysis. The tumor sections were blindly scored for the presence of each germ layer. Human naïve cells were re-primed in hES primed media and culture as primed cells before injection.
Chromosomal integrity analysis by shallow-coverage whole-genome resequencing
Chromosomal aneuploidy and partial amplification/deletion was examined by shallow-coverage whole genome resequencing as described in our preceding study with modifications11. Briefly, genomic DNA (1.1 ug in 55 uL) was sonicated using a Covaris S2 sonicator (Covaris, Woburn, MA) to ~350 bp peak fragments and subjected to PCR-free synthesis of Illumina NextSeq deep sequencing libraries using the TruSeq DNA PCR-free LT Sample Preparation Kit - Set A (Illumina FC-121–3001) following the protocol provided by the manufacturer (Illumina FC-121–9006DOC, Part# 15036187 Rev. D). During the library synthesis, the adaptor without index sequences was provided from the NEBNext Multiplex Oliogs for Illumina Dual Index Primer Set 1 (7600S, New England Biolabs, Ipswich, MA), and the loop of the adapter ends were cut with the USER enzyme provided in the kit. The index-free libraries were amplified using adapters including the i7 and i5 dual index sequences for 12 cycles, quantified using KAPA Illumina library quantification qPCR kit (KAPA Biosystems, Wilmington, MA), and equal concentrations (4 nM) of libraries were pooled. Size distributions of gDNA fragments and deep sequencing libraries were examined using Agilent Tapestation with Genomic DNA screening tapes (Agilent Technologies, Santa Clara, CA) and passed our standard quality control criteria. Single-read deep sequencing (76 nt) was performed using Illumina NextSeq 500 sequencer with the high output v2 kit, and the machine-generated base call (bcl) files were converted to raw read sequence files (fastq) using the Illumina bcl2fastq tool (2.17.1.14). Fastq files were subjected to a quality control process using Trim Galore! and fastQC (Babraham Institute, Cambridge, UK), which excluded raw quality reads (Phred score < 20) and deleted the Illumina adaptor sequences and the last one nucleotide at the 3’ end to avoid nucleotide base biases. The trimmed/QCed fastq reads were aligned to the mouse GRCm38/mm10 reference genome sequence using the STAR aligner (ver. 2.4.2.a) with the genomic DNA alignment configuration, generating the bam format aligned reads files. The bam files generated from the four lanes of the sequencer were sorted, merged, and subjected to uniquely mapped reads extraction using the sambamba toolkit31. The deduplication criteria used by sambamba markdup are identical to those used by the Broad Institute toolkit Picard. Numbers of the uniquely mapped reads were 9.7–13.3 million per sample, which corresponded to about 0.25–0.35X average depth of genome coverage. Quality control measurements of the uniquely mapped reads (bam files) using fastQC did not detect any bias with the cell types, either. The aligned unique reads files (.merged.sorted.uniq.bam) were converted into normalized bigWig files, which were visualized by the Integrative Genomics Viewer (ver. 2.3.25). Reads assigned to each chromosome are counted to generate .cnt files. The chromosome count data were subjected to ploidy analysis as described by Treff et al. who reported successful evaluation of chromosomal aneuploidy in early-stage mouse embryos with only 0.04X ~ 0.16X average depth of coverage32.
Library preparation and sequencing
RNA sequencing libraries were prepared using Ribo-Zero rRNA Removal Kit (H/M/R) (Illumina, MRZH11124) and NEBNext Ultra™ Directional RNA Library Prep Kit for Illumina (E7420). The total RNA input amount for the RiboZero kit was 1 μm total. The rRNA input for library construction was 50 ng total. Libraries were amplified for 14 cycles. Post library constructions, the samples were validated using 2200 Tapestation System and High Sensitivity D1000 ScreenTape kit. Libraries were quantified using the Kapa Biosystems Library Quantification kit (KK4828) and the BioRad CFX96 instrument.
Each lane of sequencing was pooled into a 19-plex (19 samples per lane) with unique barcodes. Pooled libraries are also quantified using the Kapa Biosystems Library Quantification kit (KK4828) and the BioRad CFX96 instrument. These pools are then denatured to 16pM with 1% phix and sequenced on the Illumina HiSeq2000 instrument. The sequencing length read was Paired-End 50bp.
Bioinformatic analysis
RNA sequencing library reads were mapped to the human hg19 reference genome using TopHat v2.1.033. The annotation of aligned reads with genomic features was preformed using HTSeq34. Repbase database annotations for transposable element (TE) superfamilies ERV1, ERVK and ERVL were used to intersect the alignment and generate counts using the multiBamCov tool from Bedtools v.2.2735. TEs which overlapped exons were discarded. TEs which did not have at least one sample with 20 reads and genes which did not have at one sample with 5 reads were removed from all downstream analysis. Count normalization was performed with size factors to correct for differences in sequencing depth followed by Variance Stabilizing Transformation (VST) scaling and differential expression analysis performed by DESeq236. A gene (or TE) was considered to be differentially expressed when abs(log2(fold_change)) > 3 and FDR < 0.01. Statistically significant (P-value<0.01) proteins for primed and naïve conditions were determined using a paired T-test and Pearson correlation was used as a metric to compare differential fold changes between genes and proteins. R studio (https://www.rstudio.com/) was used to run custom R scripts to perform principal component analysis (PCA), hierarchical clustering (stats package), variance analysis and to construct heat maps (pheatmap package), scatter plots, violin plots, and dendrograms. Generally, the ggplot2 package was used to visualize data graphs. Gene Ontology analysis were performed using EnrichR.
Proteomics and phosphoproteomics
Cells were syringe-lysed in a buffer containing 2% SDS, 150 mM NaCl, 50 mM Tris pH 8.5, 2 mM sodium vanadate, and protease and phosphatase inhibitors. Protein concentrations in the clarified lysates were estimated using the bicinchoninic acid (BCA) protein assay (Thermo Fischer Scientific). Lysates were reduced with 5 mM dithiothreitol (DTT) for 30 minutes at 60°C. After cooling to room temperature, cysteine residues were alkylated with 14mM iodoacetamide in the dark for 45 minutes at room temperature. Excess iodoacetamide was quenched with 5 mM DTT for 15 minutes in the dark. For each sample, approximately 1 mg of total protein was precipitated using methanol/chloroform and washed 3× with methanol. Protein pellets were dried and solubilized in 4 M urea, 25 mM HEPES pH 8.5 prior to digestion with 15 ug LysC for 1.5 hours at 37°C. Digestion proceeded with another 15 ug LysC overnight at room temperature. Samples were diluted to 1 M urea with 25 mM HEPES pH 8.5 and trypsin added to a final 1/125 enzyme/protein ratio for 12 hours at 37°C. Peptides were desalted with 50 mg Sep-Paks (Waters) and quantified using the Pierce Quantitative Colorimetric Peptide Assay. Fifty micrograms of each sample were reserved for protein-level quantitation prior to vacuum centrifugation.
Phosphopeptide Enrichment
Phosphopeptides were enriched using the High-Select Fe-NTA Phosphopeptide Enrichment Kit (Thermo Scientific). Briefly, 750 ug of peptides were solubilized in 200uL binding/wash buffer and clarified via centrifugation prior to binding equilibrated spin columns for 40 minutes with intermittent gentle mixing. Phosphopeptide-bound columns were washed 3× with 200 uL binding/wash buffer and 1× 200 uL HPLC grade water. Phosphopeptides were eluted 3× with 200 uL elution buffer into trifluoroacetic acid (0.1% final concentration) and immediately dried via vacuum centrifugation. Phosphopeptides were desalted with SOLA HRP 10mg Sep-Pak cartridges (Thermo Fischer Scientific) prior to TMT labelling.
Tandem Mass Tagging (TMT) Labelling
Peptides and phosphopeptides were resuspended in 200mM EPPS pH 8.5 prior to labelling. TMT-11plex reagents (0.8mg) were dissolved in 40uL anhydrous acetonitrile. For protein-level and phosphoproteome-level samples, 5 uL of appropriate TMT reagent was added per sample with anhydrous acetonitrile (final 30%; v/v). The labelling reaction was carried out for 1 hour at room temperature. All labeling reactions were quenched using 0.5% hydroxylamine, combined, acidified with formic acid (final 1.0%), and desalted using 50 mg and 10mg Sep-Paks (Waters) for unenriched and enriched phosphopeptides, respectively. Labeled phosphopeptides were mixed according to protein-level normalization factors rather than phosphopeptide abundance.
Offline basic pH reversed-phase (BPRP) fractionation
TMT-labeled peptides were fractionated using BPRP HPLC using an Agilent 1260 Infinity pump equipped with a degasser and a single wavelength detector (set at 220 nm). Using an Agilent 300 Extend C18 column (3.5 μm particles, 4.6 mm ID and 250 mm in length), peptides were separated with a 50 min linear gradient from 8% to 40% acetonitrile (proteome) or 5% to 35% acetonitrile (phosphoproteome) in 10 mM ammonium bicarbonate, pH 8 at a flow rate of 0.6 mL/min. For each experiment, a total of 96 fractions were collected and consolidated into 24 and vacuum dried. Twelve of the 24 proteome fractions and all 24 phosphoproteome fractions were desalted via StageTip, dried via vacuum centrifugation, and reconstituted in 5% acetonitrile, 5% formic acid for LC-MS/MS processing.
Liquid chromatography and tandem mass spectrometry
All mass spectra were collected using an Orbitrap Fusion Lumos mass spectrometer (Thermo Fischer Scientific) equipped with a Proxeon EASY-nLC 1000 liquid chromatography (LC) system (Thermo Fisher Scientific). Peptides and phosphopeptides were separated on a 75 μm inner diameter microcapillary column packed with ~35 cm of Sepax GPC18 resin (1.8 μm, 150 Å, Thermo Fisher Scientific). Approximately 2ug peptides and 2.5 ug phosphopeptides were separated using a 3 h and 2 h gradient, respectively, of acidic acetonitrile. We used the multinotch MS3-based TMT method37.
The proteome-wide analysis (12 fractions) and phohsphoproteome-wide analysis (24 fractions) scan sequence began with a full MS spectrum (MS1) acquired in the Orbitrap (resolution 120,000; mass range 400−1400 Th), followed by up to 10 MS/MS spectra (MS2). MS2 settings include collision-induced dissociation (CID, CE = 35) with a maximum ion injection time of either 50 ms (proteome) or 200 ms (phosphoproteome) and an isolation window of 0.5 Da. To obtain quantitative information, MS3 precursors were fragmented by high-energy collision-induced dissociation (HCD, CE = 55) and analyzed in the Orbitrap (resolution of 50,000 at 200 Th) with a maximum ion injection time of either 110 ms (proteome) or 300 ms (phosphoproteome) and an isolation window of 1.238.
MS Data Analysis
Mass spectra were processed using a SEQUEST-based software pipeline39. A modified version of ReAdW.exe was used to convert spectra to mzXML. All spectra were searched against a database containing the human proteome downloaded from Uniprot in both forward and reverse orientations and common contaminate protein sequences. Database searches were performed using a peptide mass tolerance of 20 ppm and a fragment ion tolerance of 0.9 Da. These wide-mass-tolerance windows were chosen to maximize sensitivity in conjunction with SEQUEST searches and linear discriminant analysis39, 40. TMT tags on lysine residues and peptide N termini (+229.163 Da) and carbamidomethylation of cysteine residues (+57.021 Da) were set as static modifications, while oxidation of methionine residues (+15.995 Da) was set as a variable modification. Phosphoproteome fractions also included variable modifications of phosphorylation on serine, threonine, and tyrosine residues (+79.966 Da).
Peptide-spectrum matches (PSMs) were filtered by linear discriminant analysis to a false discovery rate (FDR) of 1% at the peptide level based on matches to reversed sequences, as described previously40. Linear discriminant analysis considered the following parameters: XCorr, ΔCn, missed cleavages, adjusted PPM, peptide length, fraction of ions matched, charge state, and precursor mass accuracy. Only peptides containing phosphorylation modifications were considered when setting the FDR for phosphopeptides. Filtered peptides were collapsed further to a final protein-level FDR of 1%. Peptides were quantified from MS3 scans after filtering out those with poor quality (required total TMT reporter signal-to-noise ratio > 200 and isolation specificity 0.5). Localization of phosphorylation sites was determined using AScore, and sites with an AScore > 13 were selected for further analysis. For protein quantitation, the signal-to-noise values for all peptides for a given protein were summed, and each TMT channel was summed across all quantified proteins and normalized to enforce equal protein loading. Prior to downstream analysis, relative protein measurements across the 11 samples were normalized to sum to 100. Hierarchical clustering was performed in R on Euclidean distances using the ward.D2 method.
Statistics and Reproducibility
Exact n values are defined and described in the figure legends. Statistical significance was determined using a two-tailed unpaired Student’s t-test using Prism software (GraphPad) and exact P-values are indicated in the figure legends and in the figures. Violin plots in Figure 4a show the distribution of HERVK repeat elements (n=64) for each replicate in each condition. In each violin, the black dot represents the mean normalized expression value and the black line extending from this point represents one standard deviation from the mean. Violin plots in Figure 4b show the density of methylation values (n=95239 1kb tiles) in each individual cell line. In each violin plot, the white dot represents the median value. The box extends from the twenty-fifth to the seventy-fifth percentile of the data (the interquartile range). Whiskers extend from the box to the most extreme data point, which is no more than 1.5 times the interquartile range. Representative data are shown only if results were similar for two independent experiments. RNA-seq, proteomic, phosphoproteomic, RRBS data were generated for each independent hES cell line.
Reporting Summary
Detailed information about the experimental design, materials and reagents is described in the Life Sciences Reporting Summary.
Code availability
Code employed in this study can be obtained from the corresponding author upon reasonable request.
Data Availability
The original RNA-seq and RRBS data have been deposited under the GEO accession number: GSE111020. Proteomic data from this study are included in Supplementary Tables 1 and 2.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Thorold W. Theunissen and Rudolf Jaenisch for sharing the primed WIBR3 hESCs, the MGH next generation sequencing facility for technical assistance and members of the Hochedlinger lab for discussions. K.H. was supported by funds from the MGH, NIH (R01 HD058013, P01 GM099134) and the Gerald and Darlene Jordan Chair in Regenerative Medicine. B.D.S. was supported by an EMBO long-term Fellowship (#ALTF 1143–2015). S.S. was supported by the UCLA Broad Stem Cell Research Center – Rose Hills Foundation Training Award; A.S. by a UCLA Dissertation Year Fellowship, and K.P. by the UCLA Broad Stem Cell Research Center, the David Geffen School of Medicine, the Jonsson Comprehensive Cancer Center, and NIH P01 GM099134. The research of K.P. was also supported in part by a Faculty Scholar grant from the Howard Hughes Medical Institute. S.P.G. was supported by funds from NIH (GM67945). Y.T. was supported by funds from JSPS KAKENHI (JP16K15489 and JP16H02465).
Footnotes
AUTHOR CONTRIBUTIONS
B.D.S. and K.H. conceived the study and wrote the manuscript. B.D.S., M.U., J. B., A. J. H., A. S., K.P. and Y.T. performed the experiments and analyzed the data. S.S performed the bioinformatics analysis, K.C., H.G. and A.M. performed and analyzed the RRBS experiments, K.S. and T.S. performed and analyzed the whole-genome sequencing experiments, K.J.C, A.R.E. and S.P.G. performed and analyzed the proteomic experiments.
COMPETING INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Weinberger L, Ayyash M, Novershtern N & Hanna JH Dynamic stem cell states: naive to primed pluripotency in rodents and humans. Nature reviews. Molecular cell biology 17, 155–169 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Nichols J & Smith A Pluripotency in the embryo and in culture. Cold Spring Harb Perspect Biol 4, a008128 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gafni O et al. Derivation of novel human ground state naive pluripotent stem cells. Nature 504, 282–286 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Takashima Y et al. Resetting transcription factor control circuitry toward ground-state pluripotency in human. Cell 158, 1254–1269 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Theunissen TW et al. Systematic identification of culture conditions for induction and maintenance of naive human pluripotency. Cell stem cell 15, 471–487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sahakyan A et al. Human Naive Pluripotent Stem Cells Model X Chromosome Dampening and X Inactivation . Cell stem cell 20, 87–101 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Theunissen TW et al. Molecular Criteria for Defining the Naive Human Pluripotent State. Cell stem cell 19, 502–515 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Irie N et al. SOX17 is a critical specifier of human primordial germ cell fate. Cell 160, 253–268 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang K, Maruyama T & Fan G The naive state of human pluripotent stem cells: a synthesis of stem cell and preimplantation embryo transcriptome analyses. Cell stem cell 15, 410–415 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo G et al. Epigenetic resetting of human pluripotency. Development 144, 2748–2763 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi J et al. Prolonged Mek1/2 suppression impairs the developmental potential of embryonic stem cells. Nature 548, 219–223 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yagi M et al. Derivation of ground-state female ES cells maintaining gamete-derived DNA methylation. Nature 548, 224–227 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Hayashi K, Ohta H, Kurimoto K, Aramaki S & Saitou M Reconstitution of the mouse germ cell specification pathway in culture by pluripotent stem cells. Cell 146, 519–532 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Patel S et al. Human Embryonic Stem Cells Do Not Change Their X Inactivation Status during Differentiation. Cell Rep 18, 54–67 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Collier AJ et al. Comprehensive Cell Surface Protein Profiling Identifies Specific Markers of Human Naive and Primed Pluripotent States . Cell stem cell 20, 874–890 e877 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo G et al. Naive Pluripotent Stem Cells Derived Directly from Isolated Cells of the Human Inner Cell Mass . Stem cell reports 6, 437–446 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi J et al. A comparison of genetically matched cell lines reveals the equivalence of human iPSCs and ESCs. Nat Biotechnol 33, 1173–1181 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rouhani F et al. Genetic background drives transcriptional variation in human induced pluripotent stem cells. PLoS Genet 10, e1004432 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y, Beyer A & Aebersold R On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 165, 535–550 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Friedli M & Trono D The developmental control of transposable elements and the evolution of higher species. Annual review of cell and developmental biology 31, 429–451 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Lee HJ, Hore TA & Reik W Reprogramming the methylome: erasing memory and creating diversity. Cell stem cell 14, 710–719 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith ZD et al. DNA methylation dynamics of the human preimplantation embryo. Nature 511, 611–615 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pastor WA et al. Naive Human Pluripotent Cells Feature a Methylation Landscape Devoid of Blastocyst or Germline Memory. Cell stem cell 18, 323–329 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petropoulos S et al. Single-Cell RNA-Seq Reveals Lineage and X Chromosome Dynamics in Human Preimplantation Embryos. Cell 165, 1012–1026 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen H et al. Erk signaling is indispensable for genomic stability and self-renewal of mouse embryonic stem cells. Proceedings of the National Academy of Sciences of the United States of America 112, E5936–5943 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuo LJ & Yang LX Gamma-H2AX - a novel biomarker for DNA double-strand breaks. In Vivo 22, 305–309 (2008). [PubMed] [Google Scholar]
- 27.Liu X et al. Comprehensive characterization of distinct states of human naive pluripotency generated by reprogramming. Nat Methods 14, 1055–1062 (2017). [DOI] [PubMed] [Google Scholar]
METHODS-ONLY REFERENCES
- 28.Di Stefano B & Hochedlinger K Reduced MEK inhibition preserves genomic stability in naïve human ES cells. Protocol Exchange, doi: 10.1038/protex.2018.062 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xi Y & Li W BSMAP: whole genome bisulfite sequence MAPping program . BMC bioinformatics 10, 232 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Woodfine K, Huddleston JE & Murrell A Quantitative analysis of DNA methylation at all human imprinted regions reveals preservation of epigenetic stability in adult somatic tissue. Epigenetics & chromatin 4, 1 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tarasov A, Vilella AJ, Cuppen E, Nijman IJ & Prins P Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Treff NR et al. Next Generation Sequencing-Based Comprehensive Chromosome Screening in Mouse Polar Bodies, Oocytes, and Embryos. Biol Reprod 94, 76 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim D et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anders S, Pyl PT & Huber W HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anders S & Huber W Differential expression analysis for sequence count data. Genome Biol 11, R106 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McAlister GC et al. Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal Chem 84, 7469–7478 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Erickson BK et al. Evaluating multiplexed quantitative phosphopeptide analysis on a hybrid quadrupole mass filter/linear ion trap/orbitrap mass spectrometer. Anal Chem 87, 1241–1249 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huttlin EL et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Elias JE & Gygi SP Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods 4, 207–214 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The original RNA-seq and RRBS data have been deposited under the GEO accession number: GSE111020. Proteomic data from this study are included in Supplementary Tables 1 and 2.