

Abstract
trans-Acyltransferase assembly lines possess enzymatic domains often not observed in their better characterized cisacyltransferase counterparts. Within this repertoire of largely unexplored biosynthetic machinery is a class of enzymes called the pyran synthases that catalyze the formation of five- and sixmembered cyclic ethers from diverse polyketide chains. The 1.55 Å resolution crystal structure of a pyran synthase domain excised from the ninth module of the sorangicin assembly line highlights the similarity of this enzyme to the ubiquitous dehydratase domain and provides insight into the mechanism of ring formation. Functional assays of point mutants reveal the central importance of the active site histidine that is shared with the dehydratases as well as the supporting role of a neighboring semiconserved asparagine.
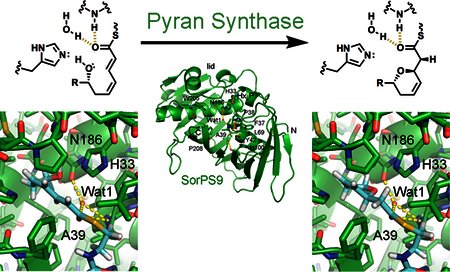
Polyketide natural products offer rich examples of the molecular complexity that can be biosynthetically generated from simple metabolites. This sophistication is realized through the action of megadalton enzymatic assembly lines called modular polyketide synthases (PKSs).1−4 Their multidomain modules perform the two-carbon chain extension and functionalization of a growing acyl chain to help construct molecules with bioactivities that are relevant to the microbes harboring them as well as human health (e.g., erythromycin and mupirocin). PKS modules catalyzing extension minimally include an acyltransferase (AT), an acyl carrier protein (ACP), and a ketosynthase (KS) that select, transfer, and fuse together α-carboxyacyl extender units. They also generally contain a set of processing domains such as ketoreductases (KRs), dehydratases (DHs), and enoylreductases (ERs) that can transform the ketone introduced by the KS into a hydroxyl, a double bond, or a methylene.
trans-AT PKSs are a relatively recently discovered type of PKS that, unlike the cis-AT PKSs, primarily relies on ATs encoded on polypeptides not embedded within the assembly line.2,3 Much remains to be learned about the chemistry performed within these systems. In addition to the processing domains they share with their cis-AT counterparts (KR, DH, and ER), trans-AT modules house largely uncharacterized processing enzymes such as those catalyzing O-methyl transfer, double-bond isomerization, and pyran formation. These additional enzymes allow for greater chemical diversity in the products of trans-AT assembly lines.3
Pyran synthases (PSs) catalyze the formation of many of the five- and six-membered cyclic ethers in the products of trans-AT assembly lines (Figure 1A). They are similar in sequence to DHs but can often be distinguished by an Hx4P motif in place of the DH Hx8P motif. The pyrans in the products of cis-AT assembly lines, such as ambruticin and salinomycin, are not formed by enzymes dedicated to pyran formation. In ambruticin biosynthesis, a DH domain (AmbDH4; the updated module nomenclature is used throughout this report5)performs a sequential dehydration and cyclization to form a pyran ring.6 A structural comparison between AmbDH4 and typical DHs shows only slight differences between their active sites.7 A tailoring enzyme from the salinomycin pathway (SalBIII) homologous to epoxide hydrolase/cyclases is responsible for generating the pyran ring of salinomycin.8 How the pyran in the product of the herboxidiene cis-AT assembly line is installed is unknown.9
Figure 1.

Rings formed by pyran synthases (PSs). (A) The products of trans-AT assembly lines often contain cyclic ethers (indicated by arrows) forged by PS domains. (B) Biosynthetic model for modules 7−9 of the sorangicin assembly line. When the hydroxyl group that forms the ester of the sorangicin macrolactone is installed is unknown and consequently omitted here. A triangle indicates the boundary between the SorA and SorB polypeptides. (C) Examples of PS-catalyzed cyclizations. Either a D-oriented or an L-oriented hydroxyl group can serve as a nucleophile, and either the re or the si face of the trans-α/β-unsaturated thioester can be selectively attacked through conjugate addition. Some DHs are bifunctional, first catalyzing dehydration and then ring formation. See the text for acronyms and abbreviations.
PSs are present in the bryostatin (Bry), diaphorin (Dip), luminaolide (Lum), misakinolide (Mis), oocydin (Ooc), pederin (Ped), phormidolide (Phm), psymberin (Psy), sorangicin (Sor), spliceostatin (Fr9), thailanstatin (Tst), and tolytoxin (Tto) trans-AT assembly lines (all of the currently sequenced PS-containing pathways), with the thailanstatin and sorangicin assembly lines harboring two and three PSs, respectively (Figure 1B,C). Within their biosynthetic pathways, each of these catalyzes the formation of six-membered dihydropyran or tetrahyropyran (oxane) rings through the conjugate addition of a ζ-hydroxyl group to Cβ, except for the oocydin and phormidolide PSs, which produce five-membered tetrahydrofuran (oxolane) rings through an ε-hydroxyl group installed by a trans-acting monooxygenase.10,11 Another unusual nucleophile for the PS reaction is the threonine side chain installed by a nonribosomal peptide synthetase (NRPS) module within the spliceostatin and thailanstatin NRPS/PKS hybrid pathways.12,13 Possibly equivalent to the bifunctional AmbDH4, a DH in the fifth module of the misakinolide trans-AT assembly line (MisDH5) may perform both dehydration and pyran formation. In the currently characterized trans-AT assembly lines, PSs appear in three modular contexts: DH+PS +KR+ACP+KS (BryPS9, DipPS9, Fr9PS7, MisPS16, LumPS16, OocPS8, PedPS9, PhmPS12, PsyPS8, SorPS16, SorPS20, TtoPS16, and TstPS7), DH+PS+ACP+KS (Fr9PS13 and TstPS13), and PS+ACP+KS (SorPS9).
The PS mechanism is currently not well understood. Biosynthetic models for trans-AT pathways predict that the natural substrates of PSs contain a trans-α/β-unsaturated double bond, whose β-carbon is thought to be attacked through an oxa-Michael addition2,3 (Figure 1C). Some PSs activate a D-hydroxyl group as the nucleophile, while others activate an L-hydroxyl group as the nucleophile. Some PSs catalyze addition to the re face of the double bond, while others catalyze addition to the si face. Studies of excised PSs from the misakinolide and pederin assembly lines (MisPS16 and PedPS9, respectively) demonstrated cyclization activity on trans-α/β-unsaturated, ζ-hydroxyacyl N-acetylcysteamine (NAC) thioester substrates (NAC mimics the phosphopantetheine prosthetic group of ACP), with both PSs generating more anti than syn products.14,15 Chiral analysis of the pyrans generated by PedPS9 shows that while the L-ζ-hydroxy substrate is slightly preferred over the D-ζ-hydroxy substrate, the β-carbon atom of each substrate is stereoselectively attacked at its si face. Structural studies are required to learn the PS catalytic mechanism and how stereocontrol is mediated.
Here, we report the 1.55 Å resolution structure of SorPS9 from the Sorangium cellulosum trans-AT assembly line that is responsible for the production of the sorangicin antibiotics (Figure 1B).5,16−18 The SorPS9 active site contains a histidine and an asparagine at positions equivalent to those of the catalytic histidine and active site aspartate of DHs. Functional assays of SorPS9 point mutants with trans-α/β-unsaturated, ζ-hydroxyacyl NAC thioester substrates reveal the critical activity of the histidine and the ancillary role of the neighboring asparagine. At the positive end of a helix dipole, a backbone amide and a water molecule may coordinate the thioester carbonyl of the substrate to facilitate conjugate addition. Restraints provided by the position of the catalytic histidine and the oxyanion hole as well as the stereochemistry of the SorPS9 substrate enabled the construction of a model for ring formation.
RESULTS AND DISCUSSION
Expression and Structure Determination of a PS.
The boundaries for SorPS9 (residues 1360−1641 of SorB) were selected by comparing the sequences of PSs with those of structurally characterized DHs. The DNA encoding this region was amplified from S. cellulosum SoCe12 genomic DNA and cloned into the pET28b expression vector for protein purification from Escherichia coli BL21(DE3) and crystal screening. After optimization, crystals diffracted to a resolution of 1.55 Å. The SorPS9 structure was determined by molecular replacement using a DH from the curacin cis-AT assembly line as a search model [CurDH9, Protein Data Bank (PDB) entry 3KG9] (Table S1).19
Architecture of SorPS9 in Comparison with That of DHs and Related Enzymes.
SorPS9 crystallized as a monomer like half of the structurally characterized trans-AT enzymes with a double hotdog fold [DH, enoyl-isomerase (EI), branching domain (B)] but unlike all of the solved DHs from cis-AT assembly lines, which crystallized as dimers (Figure 2).19−24 The dimeric interaction of trans-AT DHs is slightly different from that of cis-AT DHs and apparently weaker (e.g., PDB entries 5IL6, 5J6O, and 5HQW) (Figure S1). The overall structure of SorPS9 resembles that of canonical DHs, with Cα root-mean-square deviation values of 1.4 and 1.6 Å when compared with two recently solved trans-AT DHs (PDB entries 5HST and 5IL6, respectively) (Figure S2).20,22,23 Despite its origins from an unusual PS+ACP+KS module rather than the more conventional DH+PS+KR+ACP+KS module, SorPS9 appears to be representative of all sequenced PSs based on sequence alignments (Figures 1B, 3, and S3).12,14,15,25
Figure 2.

SorPS9 compared with related enzymes. (A) Stereodiagrams reveal the structural attributes of PSs and DHs (PDB entries 6B2V, 5O15, and 5NJI). The structure of PpsDH4 was determined bound to a trans-α/β-unsaturated thioester that is similar to PS substrates and may reveal how substrates bind to PSs. AmbDH4 is a bifunctional DH that catalyzes both dehydration and ring formation. The lids of both SorPS9 and AmbDH4 are formed by loops rather than the small β-sheet observed in canonical DHs. An Hx4P motif in PSs commonly substitutes for the Hx8P motif that helps form this β-sheet in canonical DHs. Residues discussed in the text are labeled. PpsDH4 is shown with its catalytic histidine (the structure of the phenylalanine mutant was actually determined). (B) A stereodiagram compares the active sites of a trans-AT PS (SorPS9) and DH (MlnDH3, from the third module of the macrolactin synthase, PDB entry 5HST). The PS active site is relatively open. Hydrogen bonds and Wat1 from the MlnDH3 structure are shown. While the DH aspartate is oriented by a neighboring glutamine, the PS asparagine is not oriented by an equivalent residue and is rotated away.
Figure 3.

Sequence alignment of double hotdog fold enzymes from trans-AT assembly lines. The sequences of three PSs, EIs, and DHs are compared, with the secondary structure of SorPS9 depicted on the top and that of MlnDH3 on the bottom.25 The asterisk indicates the universally conserved catalytic histidine. It usually resides in an Hx4P motif in PSs, an Hx8L motif in EIs, and an Hx8P motif in DHs. A circle indicates the position of the DH aspartate. This residue is usually N or H in PSs, N in EIs, and D in DHs. An HPLL motif is often present at the N-termini of DHs and PSs. Abbreviations: SorPS9, sorangicin, S. cellulosum, ADN68477; MisPS16, misakinolide, Candidatus Entotheonella serta, AKQ22696; BryPS9, bryostatin, Candidatus Endobugula sertula, ABM63528; RhiEI13, rhizoxin, Paraburkholderia rhizoxinica, WP_013435478; NspEI8, nosperin, Nostoc sp. N6 (symbiont of Peltigera membranacea), ADA69239; PksEI17, bacillaene, Bacillus subtilis, WP_029317820; ChiDH7, chivosazol, S. cellulosum, AAY89050; RhiDH5, rhizoxin, P. rhizoxinica, WP_013435482; MlnDH3, macrolactin, Bacillus amyloliquefaciens, WP_060675406.
Much can be learned by comparing the active site of SorPS9 with those of DHs (Figure 2). A histidine (H33) is in the same location and orientation as the DH catalytic histidine. A backbone NH and water molecule (Wat1) at the N-terminal end of α1 are positioned equivalent to those of a DH from the phthiocerol cis-AT assembly line (PpsDH4) observed forming hydrogen bonds with the thioester carbonyl of a trans-α/β-unsaturated acyl thioester (PDB entry 5NJI).24 An asparagine (N186) is equivalent to the strictly conserved DH aspartate thought to form a hydrogen bond with the β-hydroxyl group of DH substrates, although it is in a different orientation. The SorPS9 active site is more expansive than the DH active site for several reasons. The Hx4P motif of PSs lacks the β-hairpin of the Hx8P motif that in DHs helps form a lid that covers the active site, a loop substitutes for a β-strand (β8) that in DHs forms a small β-sheet with the β-hairpin and contains bulky aromatic residues that project toward the active site, and the residue analogous to the DH aspartate (N186) rotates away from the active site compared to the aspartate. In contrast to the active site, the tunnel equivalent to that housing the polyketide tail in the PpsDH4 complex structure is relatively constricted in SorPS9, primarily because of a significant bend in α2 and interactions made between it and residues on the following loop, including a tryptophan (W205).24
The presumed PS catalytic histidine is invariant, residing within an Hx4P motif (conserved in all sequenced PSs except the spliceostatin and thailanstatin PSs that possess Hx8P motifs;L can subsitute for the P, as in MisPS16) (Figures 3 and S3). Like the catalytic histidine of DHs, the histidine of SorPS9 stacks with the proline at the end of the motif (Figure 2). Within the related double bond-shifting EIs, the valine or leucine in the same position as this proline does not stack with the EI histidine and may be largely responsible for the different position of the histidine (~1.7 Å from where it resides in DH and PS) (PDB entry 4U3V).21 N186 of SorPS9 is equivalent to the DH aspartate and adjacent to the presumed catalytic histidine. While this residue is an asparagine in half of the sequenced PSs, other residues such as histidine and serine (but apparently not aspartate) can substitute for it.12 Although in the same position on the second long helix, N186 and the DH aspartate are in different rotameric conformations. The carboxylate of the DH aspartate is oriented through a hydrogen bond with a glutamine or histidine four residues downstream, whereas the amide of N186 cannot be oriented by the equivalent residue in SorPS9 because it is a valine (leucine in most PSs). N186 of SorPS9, the equivalent asparagine in EIs, and the equivalent aspartate in DHs all share a hydrogen bond with Wat1.21
The region of SorPS9 adjacent to the active site that is expected to bind the phosphopantetheinyl arm based on the complex structure of PpsDH4 (PDB entry 5NJI) is equivalent to that of DH and EI (Figure 2). The phosphopantetheine NH closest to the thioester is anticipated to form a hydrogen bond with the carbonyl of an invariant proline (P208). The phosphopantetheine carbonyl closest to the thioester is anticipated to form hydrogen bonds with two water molecules: one bound by the carbonyl of the residue before the HxnP proline (F37), the side chain of the residue four residues after the HxnP proline (Y42, highly conserved), and the backbone NH of a structured loop residue (L69) and the other bound by the backbone carbonyl of a histidine (H100).
PS Substrates.
All known PSs are predicted to naturally operate on trans-α/β-unsaturated intermediates (Figure 1C). The configuration of the double bond of the SorPS9 substrate can be inferred from the orientation of the β-hydroxy intermediate generated by the upstream KR because trans-α/β olefins are generated from D-β-hydroxy intermediates and cis-α/β olefins are generated from L-β-hydroxy intermediates.26 As each of these KRs possesses an aspartate diagnostic of a B-type KR that generates a D-β-hydroxy group, the accompanying DH will generate a trans-α/β olefin for the PS (Figure S4).27,28 SorPS9 seems to be an exception to this trend as the KR preceding it is an A-type KR. However, this KR belongs to a type C dehydrating bimodule (KR+ACP+KS0+DH+ACP+DI +KS; the diene isomerase, DI, that isomerizes the cis-α/β, trans-γ/δ diene intermediate contains a cysteine in place of the DH aspartate), which generates a trans-α/β, cis-γ/δ diene.20,29 The presence of the type C dehydrating bimodule immediately upstream of the ninth module of the sorangicin assembly line may help explain the unusual PS+ACP+KS module architecture in which SorPS9 is embedded.
Functional Assays of SorPS9 and Point Mutants.
To assay the conversion of a trans-α/β-unsaturated thioester substrate to a pyran product, SorPS9 was incubated with substrate mimic 1 overnight at 22 °C (Figure 4). Liquid chromatography/mass spectrometry (LC/MS) analysis of the reaction revealed the disappearance of 1 and the appearance of a new peak not observed in negative control reactions. Consistent with pyran formation, the new compound displayed an m/z [M + Na]+ of 268 (the same as 1, expected for an isomeric pyran product) and a maximal absorbance at 230 nm, distinct from from the maximal absorbance of 265 nm of the trans-α/β-unsaturated thioester substrate (Figure S5). These results are consistent with SorPS9 catalyzing pyran ring formation and indicate that the excised domain is an effective biocatalyst. To probe its substrate tolerance, SorPS9 was challenged with the racemic 3a+3b mixture with secondary ζ- hydroxyl groups rather than the primary ζ-hydroxyl group of 1. LC/MS analysis of the reaction showed the disappearance of 3a +3b and the appearance of two new peaks roughly equal in size and with the same m/z as the substrates. The disappearance of 3a+3b reveals that SorPS9 generates pyran products from both D- and L-ζ-hydroxy substrates. When SorPS9 was incubated with potential products of these reactions (2a+2b and 4a+4d), no activity was detected, implying that the equilibrium heavily favors pyran formation (Figure S6).
Figure 4.

Activity assays of SorPS9 and point mutants. (A) LC/MS analysis of incubations with 1 revealed wild-type SorPS9 generated 2a and/or 2b, while the H33A mutant was inactive. The N186H and N186A mutants showed some activity, but no activity was observed from the N186D mutant. Traces show extracted ion current at m/z 268. (B) LC/MS analysis of incubations with 3a+3b revealed wild-type SorPS9 generated syn-pyran products (4a and/or 4d) and anti-pyran products (4b and/or 4c), while the H33A mutant was inactive. Again, the N186H and N186A mutants showed some activity, but no activity was observed from the N186D mutant. Traces show extracted ion current at m/z 282.
To help elucidate the roles of the SorPS9 active site histidine and asparagine (all other residues in the active site are nonpolar), H33A, N186H, N186A, and N186D mutants were generated (Figure S7). Each was incubated with 1 or 3a+3b and analyzed by LC/MS (Figure 4). For each of the substrates, no discernible activity was detected for the H33A mutant, indicating this histidine is indeed catalytic. The N186H mutant retained significant activity, while the N186A and N186D mutants displayed minimal and no activity, respectively.
Mechanism of PS and Related Enzymes.
Understanding how PSs catalyze ring formation will also help elucidate the mechanisms of other double hotdog fold enzymes such as DH, EI, and DI. To model the SorPS9 reaction, L-ζ-hydroxy-trans-α/β-cis-γ/δ-octadienyl-S-NAC (a mimic of its natural substrate; it is unknown when the hydroxyl group that closes the sorangicin macrolactone is inserted3) was positioned in the active site (Figure 5A). Its thioester carbonyl was placed like that of the trans-α/β-dodecenyl-S-CoA substrate bound to PpsDH4, in a putative oxyanion hole formed by the NH of A39 and Wat1 at the positive end of a helix dipole. The L-ζ-hydroxy group was placed as close as possible to Cβ. For the conjugate addition to take place on the re face and be assisted by the catalytic histidine, both the bond between Cα and the thioester carbonyl and the bond between Cβ and Cγ must be in the s-trans geometry. The terminal portion of the phosphopantetheinyl arm was positioned as in the PpsDH4 complex structure. While the tail of the natural polyketide was not modeled, if it binds similarly to the dodecenyl substrate in the PpsDH4 complex structure alongside the α helix, the W205 side chain would need to shift to accommodate it (SorPS20 is the only other known PS with a tryptophan at this position) (Figures 2 and S3).
Figure 5.

Proposed mechanism of ring formation by PSs. (A) Stereodiagrams show relevant portions of the SorPS9 substrate and product modeled in the SorPS9 active site. The thioester carbonyl was positioned next to Wat1 and the NH of A39, as observed in the PpsDH4 complex structure (PDB entry 5NJI), in a putative oxyanion hole.24 This would facilitate a conjugate addition of the hydroxyl group to Cβ. The catalytic histidine H33 is in position to mediate proton transfer to Cα. Whether N186 forms a hydrogen bond with the substrate is not clear; it may interact with only Wat1. As mentioned in Figure 1, the actual SorPS9 substrate may also possess a D-ε-hydroxyl group. (B) For an attack at the re face, like that mediated by SorPS9, the bond between the thioester carbonyl and Cα must be in the s-trans conformation. An attack at the si face may be enabled by positioning a substrate so that this bond is in the s-cis conformation.
With the substrate in place, ring formation can occur. Except for H33, N186, and the putative oxyanion hole made by the NH of A39 and Wat1, the PS active site is hydrophobic, similar to the EI active site in which only a histidine and an oxyanion hole are needed to shift a double bond through the transfer of a proton from Cγ to Cα. 21 Oxyanion formation in the EI reaction suggests oxyanion formation in the PS reaction. An alternative hypothesis is that a four-membered transition state is formed between the OH of the ζ-hydroxyl group and Cα/Cβ, as proposed for eukaryotic hydratase 2 from the crotonase superfamily;30 however, as several other crotonase enzymes, such as Δ3,5,Δ2,4-dienoyl-CoA isomerase, 4-chlorobenzyl-CoA dehalogenase, and methylmalonyl-CoA decarboxylase, apparently conduct catalysis through an oxyanion intermediate, eukaryotic hydratase 2 may also proceed through this route. Without any experimental support for a concerted mechanism in related systems, PS catalysis more likely proceeds through an oxyanion intermediate.31 After the ζ-hydroxyl attacks Cβ to generate the oxyanion, H33 would transfer the proton between the ether oxygen and Cα. As a π bond is converted into a lower-energy σ bond through the reaction, pyran formation is essentially irreversible.
While N186 neighbors the ζ-hydroxyl group of the substrate, whether its side chain forms a hydrogen bond with it is unclear. The main function of this residue (or the equivalent serine, histidine, or threonine of other PSs) may be to form a hydrogen bond with the water molecule, Wat1, that helps build the oxyanion hole. The DH aspartate is thought to form a hydrogen bond with the β-hydroxyl group of its substrate. This residue had been hypothesized to act as a general acid, donating a proton to the hydroxyl group to generate a water leaving group.26 However, it has been argued that the pKa of the aspartate is not likely higher than that of the histidine in the DH active site, and that a one-base mechanism may instead be operative in DHs.32−34 In this mechanism, the DH histidine abstracts the α-proton to generate the oxyanion and then transfers it to the β-hydroxyl group so it can depart as water.
During ring formation, PSs stereoselectively catalyze conjugate addition at Cβ on its re or si face (Figure 5B). While SorPS9 catalyzes addition on the re face, most PSs, including SorPS16 and SorPS20, catalyze addition on the si face. For a hydroxyl group to attack the si face of a substrate whose thioester carbonyl is in the oxyanion hole and for the histidine to assist in the subsequent proton transfer, the bond between the thioester carbonyl and Cα must be in the s-cis geometry. Thus, the nature of a substrate and its preferred conformation in the active site of its PS seem to dictate whether the s-cis or s-trans geometry is bound and consequently whether the si or re face of Cβ will be attacked. Sequence fingerprints that indicate which face is attacked by a PS are not apparent.
The bifunctional DH/PSs AmbDH4 and MisDH5 further inform the requirements for ring formation (Figure 2). The main difference between the structures of AmbDH4 and canonical DHs is how the lid covers the active site (PDB entry 5O15).7 The β-strand that in canonical DHs forms a small β-sheet with the β-hairpin of the Hx8P motif and contains two conserved tyrosines that project toward the active site is replaced by a loop. This additional space in the active site may enable polyketide intermediates to bend back on themselves for the ring-forming reaction. The active site aspartate of AmbDH4 is in the same orientation as the canonical DH aspartate; however, none of the known PSs have an aspartate at this position, and the N186D mutant of SorPS9 is inactive. During the likely evolution of PSs from DHs, the aspartate may have been selected against. It has the potential to impede PS catalysis by promoting the addition of water across the α/β double bond and by forming a hydrogen bond with the nucleophilic hydroxyl group (as the DH aspartate does with the β-hydroxyl group in the DH dehydration reaction) so that the oxygen is held away from Cβ. The fact that the N186 side chain of SorPS9 is relatively farther from the active site suggests that the position of the DH aspartate may sterically inhibit ring formation. Interestingly, MisDH5 possesses an asparagine in place of the DH aspartate, indicating both that the DH aspartate is not essential to dehydration and that the asparagine facilitates pyran formation. The short tails of the intermediates cyclized by AmbDH4 and MisDH5 could also help them access the conformations necessary for catalysis.
Conclusion.
The presented structural and functional studies of SorPS9 shed new light not only on the PS domains from trans-AT assembly lines but also on several related double hotdog fold enzymes from cis- and trans-AT systems. Mutagenic and bioinformatic analysis of SorPS9, guided by the structures of SorPS9 and related enzymes, implicates the histidine in mediating proton transfer and the asparagine in helping create the oxyanion hole through water molecule Wat1. Structural comparison of canonical DHs with SorPS9, PksEI17, and AmbDH4 helps to illustrate how slight alterations of the DH active site lead to diverse chemistry. The expansion of the active site cavity seems to have been particularly important in enabling the ring-forming PS reaction. Thus, modest evolutionary changes to a DH have apparently repurposed this enzymatic scaffold that essentially consists of a histidine and an oxyanion hole to conduct several new types of reactions, including double-bond shifting reactions performed by EIs, the diene isomerization reactions performed by DIs, the bifunctional dehydration/cyclization reactions performed by certain DHs, and the dedicated cyclization reaction performed by PSs.6,20,21,35
As the integration of structural, functional, and bioinformatic research continues to demystify the numerous idiosyncratic features of trans-AT assembly lines, a clearer relationship between their products and the enzymatic components that generate them is emerging. Tapping into the large reservoir of biosynthetic potential of trans-AT assembly lines will enable the employment of enzymes that conduct unusual chemistry as biocatalysts and eventually as components in engineered assembly lines that produce new molecules and medicines.
METHODS
Cloning, Expression, and Purification.
DNA encoding the SorPS9 domain was amplified from S. cellulosum So ce12 genomic DNA with primers DW469 and DW470 and inserted into the pET28b expression vector through Gibson Assembly (New England Biolabs).36 N-Terminally His6-tagged SorPS9 was expressed in E. coli BL21(DE3) in 6 L of LB medium and grown to an OD600 of 0.8 at 37 °C, and then the cells were induced with 500 μM isopropyl β-D-1-thiogalactopyranoside and grown overnight at 15 °C. The protein was purified from the cell lysate using HisPur nickel-NTA resin (Thermo Scientific) followed by gel filtration [GE Superdex 200 Increase 10/300 column equilibrated in 150 mM NaCl and 15 mM HEPES (pH 7.5)]. Active site mutants of SorPS9 were generated using standard site-directed mutagenesis techniques with primers DW504−DW513 (Table S2). Point mutants were purified as described for unmutated SorPS9.
Crystallization and Structure Determination.
SorPS9 was concentrated to 12 mg mL−1, and crystals were obtained in 2−7 days by sitting-drop vapor diffusion at 22 °C with a 1.7 M ammonium sulfate and 0.1 M Bis-Tris (pH 5.5) crystallization condition (1 μL of protein/μL of crystallization condition). Crystals were cryo-protected in the crystallization condition with 15% glycerol prior to being flash-frozen in liquid nitrogen. The structure was determined by molecular replacement using a DH from the curacin cis-AT assembly line as a search model (CurDH9, PDB entry 3KG9) and refined to 1.55 Å resolution with a combination of ARP/wARP, Refmac, and Phenix Refine.37−40
Subtrate/Product Synthesis.
The syntheses of 1, 2a+2b, 3a+3b, and 4a+4d are described in the Supporting Information.
Activity Assays.
All enzyme reaction mixtures were incubated with 15 mM substrate overnight at 22 °C in 150 mM NaCl and 20 mM HEPES (pH 7.5) at an enzyme concentration of 150 μM in 100 μL. After incubation, samples were thrice extracted with 100 μL of ethyl acetate, evaporated, and brought up in 50 μL of methanol. From this methanol solution, 20 μL was diluted into 1.5 mL of LC/MS buffer (5% methanol) for LC/MS analysis (Agilent 1260 HPLC with an Agilent 6120 Quadrupole ESI instrument in positive mode; ZORBAX Eclipse Plus 95 Å C18 column, 2.1 mm × 50 mm, 5 μm; column temperature of 40 °C; flow rate of 0.7 mL/min; gradient from 0 to 12 min of 5 to 100% methanol and from 12 to 15 min of 100% methanol).
Supplementary Material
ACKNOWLEDGMENTS
The authors thank R. Müller for providing S. cellulosum So ce12 genomic DNA. Instrumentation and technical assistance for crystallographic work were provided by A. Monzingo and the Macromolecular Crystallography Facility, with financial support from the College of Natural Sciences, the Offce of the Executive Vice President and Provost, and the Institute for Cellular and Molecular Biology at The University of Texas at Austin. The Advanced Light Source (ALS) is supported by the Director, Offce of Science, Offce of Basic Energy Sciences, of the U.S. Department of Energy (DOE) under Contract DEAC02-05CH11231. The Advanced Photon Source, an Offce of Science User Facility operated for the DOE Offce of Science by Argonne National Laboratory, was supported by the DOE under Contract DE-AC02-06CH11357. The authors are grateful for funding from the EU (ERC Advanced Projects SynPlex to J.P.). The authors also thank the National Institutes of Health (GM106112) and the Welch Foundation (F-1712) for supporting this research (A.T.K.-C.).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.8b00049.
Figures S1–S7, Tables S1 and S2, and the details of chemical synthesis (PDF)
Accession Codes
The coordinates for SorPS9 have been deposited in the Protein Data Bank as entry 6B2V.
The authors declare no competing financial interest.
REFERENCES
- (1).Keatinge-Clay AT (2012) The Structures of Type I Polyketide Synthases. Nat. Prod. Rep 29, 1050–1073. [DOI] [PubMed] [Google Scholar]
- (2).Piel J (2010) Biosynthesis of Polyketides by Trans-at Polyketide Synthases. Nat. Prod. Rep 27, 996–1047. [DOI] [PubMed] [Google Scholar]
- (3).Helfrich EJ, and Piel J (2016) Biosynthesis of Polyketides by Trans-at Polyketide Synthases. Nat. Prod. Rep 33, 231–316. [DOI] [PubMed] [Google Scholar]
- (4).Keatinge-Clay AT (2017) The Uncommon Enzymology of Cis-Acyltransferase Assembly Lines. Chem. Rev 117, 5334–5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Keatinge-Clay AT (2017) Polyketide Synthase Modules Redefined. Angew. Chem., Int. Ed 56, 4658–4660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Berkhan G, and Hahn F (2014) A Dehydratase Domain in Ambruticin Biosynthesis Displays Additional Activity as a Pyran-Forming Cyclase. Angew. Chem., Int. Ed 53, 14240–14244. [DOI] [PubMed] [Google Scholar]
- (7).Sung KH, Berkhan G, Hollmann T, Wagner L, Blankenfeldt W, and Hahn F (2018) Insights into the Dual Activity of a Bifunctional Dehydratase-Cyclase Domain. Angew. Chem., Int. Ed 57, 343. [DOI] [PubMed] [Google Scholar]
- (8).Luhavaya H, Dias MV, Williams SR, Hong H, de Oliveira LG, and Leadlay PF (2015) Enzymology of Pyran Ring a Formation in Salinomycin Biosynthesis. Angew. Chem., Int. Ed 54, 13622–13625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Shao L, Zi J, Zeng J, and Zhan J (2012) Identification of the Herboxidiene Biosynthetic Gene Cluster in Streptomyces Chromofuscus Atcc 49982. Appl. Environ. Microbiol 78, 2034–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Matilla MA, Stockmann H, Leeper FJ, and Salmond GP (2012) Bacterial Biosynthetic Gene Clusters Encoding the Anti-Cancer Haterumalide Class of Molecules: Biogenesis of the Broad Spectrum Antifungal and Anti-Oomycete Compound, Oocydin A. J. Biol. Chem 287, 39125–39138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Bertin MJ, Vulpanovici A, Monroe EA, Korobeynikov A, Sherman DH, Gerwick L, and Gerwick WH (2016) The Phormidolide Biosynthetic Gene Cluster: A Trans-at Pks Pathway Encoding a Toxic Macrocyclic Polyketide. ChemBioChem 17, 164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Eustaquio AS, Janso JE, Ratnayake AS, O’Donnell CJ, and Koehn FE (2014) Spliceostatin Hemiketal Biosynthesis in Burkholderia Spp. Is Catalyzed by an Iron/Alpha-Ketoglutarate-Dependent Dioxygenase. Proc. Natl. Acad. Sci. U. S. A 111, E3376–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Zhang F, He HY, Tang MC, Tang YM, Zhou Q, and Tang GL (2011) Cloning and Elucidation of the Fr901464 Gene Cluster Revealing a Complex Acyltransferase-Less Polyketide Synthase Using Glycerate as Starter Units. J. Am. Chem. Soc 133, 2452–2462. [DOI] [PubMed] [Google Scholar]
- (14).Poplau P, Frank S, Morinaka BI, and Piel J (2013) An Enzymatic Domain for the Formation of Cyclic Ethers in Complex Polyketides. Angew. Chem., Int. Ed 52, 13215–13218. [DOI] [PubMed] [Google Scholar]
- (15).Ueoka R, Uria AR, Reiter S, Mori T, Karbaum P, Peters EE, Helfrich EJ, Morinaka BI, Gugger M, Takeyama H, Matsunaga S, and Piel J (2015) Metabolic and Evolutionary Origin of Actin-Binding Polyketides from Diverse Organisms. Nat. Chem. Biol 11, 705–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zhang L, Hashimoto T, Qin B, Hashimoto J, Kozone I, Kawahara T, Okada M, Awakawa T, Ito T, Asakawa Y, Ueki M, Takahashi S, Osada H, Wakimoto T, Ikeda H, Shin-Ya K, and Abe I (2017) Characterization of Giant Modular Pkss Provides Insight into Genetic Mechanism for Structural Diversification of Aminopolyol Polyketides. Angew. Chem., Int. Ed 56, 1740–1745. [DOI] [PubMed] [Google Scholar]
- (17).Irschik H, Jansen R, Gerth K, Hofle G, and Reichenbach H (1987) The Sorangicins, Novel and Powerful Inhibitors of Eubacterial Rna Polymerase Isolated from Myxobacteria. J. Antibiot 40, 7–13. [DOI] [PubMed] [Google Scholar]
- (18).Irschik H, Kopp M, Weissman KJ, Buntin K, Piel J, and Muller R (2010) Analysis of the Sorangicin Gene Cluster Reinforces the Utility of a Combined Phylogenetic/Retrobiosynthetic Analysis for Deciphering Natural Product Assembly by Trans-at Pks. ChemBio-Chem 11, 1840–1849. [DOI] [PubMed] [Google Scholar]
- (19).Akey DL, Razelun JR, Tehranisa J, Sherman DH, Gerwick WH, and Smith JL (2010) Crystal Structures of Dehydratase Domains from the Curacin Polyketide Biosynthetic Pathway. Structure 18, 94–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wagner DT, Zeng J, Bailey CB, Gay DC, Yuan F, Manion HR, and Keatinge-Clay AT (2017) Structural and Functional Trends in Dehydrating Bimodules from Trans-Acyltransferase Polyketide Synthases. Structure 25, 1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Gay DC, Spear PJ, and Keatinge-Clay AT (2014) A Double-Hotdog with a New Trick: Structure and Mechanism of the Trans-Acyltransferase Polyketide Synthase Enoyl-Isomerase. ACS Chem. Biol 9, 2374–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Bretschneider T, Heim JB, Heine D, Winkler R, Busch B, Kusebauch B, Stehle T, Zocher G, and Hertweck C (2013) Vinylogous Chain Branching Catalysed by a Dedicated Polyketide Synthase Module. Nature 502, 124–128. [DOI] [PubMed] [Google Scholar]
- (23).Keatinge-Clay A (2008) Crystal Structure of the Erythromycin Polyketide Synthase Dehydratase. J. Mol. Biol 384, 941–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Faille A, Gavalda S, Slama N, Lherbet C, Maveyraud L,Guillet V, Laval F, Quemard A, Mourey L, and Pedelacq JD (2017) Insights into Substrate Modification by Dehydratases from Type I Polyketide Synthases. J. Mol. Biol 429, 1554–1569. [DOI] [PubMed] [Google Scholar]
- (25).Robert X, and Gouet P (2014) Deciphering Key Features in Protein Structures with the New Endscript Server. Nucleic Acids Res 42, W320–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Keatinge-Clay AT (2016) Stereocontrol within Polyketide Assembly Lines. Nat. Prod. Rep 33, 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Keatinge-Clay AT (2007) A Tylosin Ketoreductase Reveals How Chirality Is Determined in Polyketides. Chem. Biol 14, 898–908. [DOI] [PubMed] [Google Scholar]
- (28).Piasecki SK, Zheng J, Axelrod AJ, Detelich ME, and Keatinge-Clay AT (2014) Structural and Functional Studies of a Trans-Acyltransferase Polyketide Assembly Line Enzyme That Catalyzes Stereoselective Alpha- and Beta-Ketoreduction. Proteins: Struct., Funct., Genet 82, 2067–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Moebius N, Ross C, Scherlach K, Rohm B, Roth M, and Hertweck C (2012) Biosynthesis of the Respiratory Toxin Bongkrekic Acid in the Pathogenic Bacterium Burkholderia Gladioli. Chem. Biol 19, 1164–1174. [DOI] [PubMed] [Google Scholar]
- (30).Koski MK, Haapalainen AM, Hiltunen JK, and Glumoff T (2004) A Two-Domain Structure of One Subunit Explains Unique Features of Eukaryotic Hydratase 2. J. Biol. Chem 279, 24666–24672. [DOI] [PubMed] [Google Scholar]
- (31).Holden HM, Benning MM, Haller T, and Gerlt JA (2001) The Crotonase Superfamily: Divergently Related Enzymes That Catalyze Different Reactions Involving Acyl Coenzyme a Thioesters. Acc. Chem. Res 34, 145–157. [DOI] [PubMed] [Google Scholar]
- (32).Shah DD, You YO, and Cane DE (2017) Stereospecific Formation of E- and Z-Disubstituted Double Bonds by Dehydratase Domains from Modules 1 and 2 of the Fostriecin Polyketide Synthase. J. Am. Chem. Soc 139, 14322–14330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Schwab JM, Klassen JB, and Habib A (1986) Stereochemical Course of the Hydration Reaction Catalyzed by Beta-Hydroxydecanoylthioester Dehydrase. J. Chem. Soc., Chem. Commun, 357–358. [Google Scholar]
- (34).Schwab JM, and Henderson BS (1990) Enzyme-Catalyzed Allylic Rearrangements. Chem. Rev 90, 1203–1245. [Google Scholar]
- (35).Fiers WD, Dodge GJ, Sherman DH, Smith JL, and Aldrich CC (2016) Vinylogous Dehydration by a Polyketide Dehydratase Domain in Curacin Biosynthesis. J. Am. Chem. Soc 138, 16024–16036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, and Smith HO (2009) Enzymatic Assembly of DNA Molecules up to Several Hundred Kilobases. Nat. Methods 6, 343–345. [DOI] [PubMed] [Google Scholar]
- (37).Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH (2010) Phenix: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr., Sect. D: Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Langer G, Cohen SX, Lamzin VS, and Perrakis A (2008) Automated Macromolecular Model Building for X-Ray Crystallography Using Arp/Warp Version 7. Nat. Protoc 3, 1171–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, and Wilson KS (2011) Overview of the Ccp4 Suite and Current Developments. Acta Crystallogr., Sect. D: Biol. Crystallogr 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Murshudov GN, Vagin AA, and Dodson EJ (1997) Refinement of Macromolecular Structures by the Maximum-Like lihood Method. Acta Crystallogr., Sect. D: Biol. Crystallogr 53, 240– 255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.