

Abstract
The diverse clinical phenotypes of Wolf–Hirschhorn syndrome (WHS) are the result of haploinsufficiency of several genes, one of which, LETM1, encodes a protein of the mitochondrial inner membrane of uncertain function. Here, we show that LETM1 is associated with mitochondrial ribosomes, is required for mitochondrial DNA distribution and expression, and regulates the activity of an ancillary metabolic enzyme, pyruvate dehydrogenase. LETM1 deficiency in WHS alters mitochondrial morphology and DNA organization, as does substituting ketone bodies for glucose in control cells. While this change in nutrient availability leads to the death of fibroblasts with normal amounts of LETM1, WHS‐derived fibroblasts survive on ketone bodies, which can be attributed to their reduced dependence on glucose oxidation. Thus, remodeling of mitochondrial nucleoprotein complexes results from the inability of mitochondria to use specific substrates for energy production and is indicative of mitochondrial dysfunction. However, the dysfunction could be mitigated by a modified diet—for WHS, one high in lipids and low in carbohydrates.
Keywords: LETM1, mitochondrial DNA, mitochondrial morphology, nutrient utilization, Wolf–Hirschhorn syndrome
Subject Categories: Genetics, Gene Therapy & Genetic Disease; Metabolism
Introduction
Mitochondria are major contributors to cellular energy production, converting nutrients to ATP through the oxidative phosphorylation system (OXPHOS). Thirteen essential proteins of OXPHOS are transcribed from mitochondrial DNA (mtDNA) and synthesized by the mitochondrial ribosomes (mitoribosomes), via a highly specialized system that is tightly coupled to the inner mitochondrial membrane (IMM; Liu & Spremulli, 2000). In yeasts, the proposed machinery of co‐translational membrane insertion includes Oxa1, mba1, and mdm38 (Ott & Herrmann, 2010). Mdm38 is an integral inner membrane protein conserved among eukaryotes, and it is suggested to play a role in multiple facets of mitochondrial metabolism, the precise nature of which remains unclear. Under certain conditions, it co‐fractionates with the mitoribosome, on buoyant density gradients (Frazier et al, 2006; Kehrein et al, 2015), and it is proposed to regulate mitochondrial translation (Bauerschmitt et al, 2010). Furthermore, mdm38 regulates mitochondrial morphology and volume by controlling ion homeostasis across the mitochondrial inner membrane (Nowikovsky et al, 2004, 2007; Froschauer et al, 2005).
The human mdm38 homolog, LETM1, is inferred to regulate mitochondrial volume in a similar way to its yeast counterpart, as its ablation results in swollen mitochondria (Dimmer et al, 2002, 2008; Hasegawa & van der Bliek, 2007; Tamai et al, 2008; Jiang et al, 2009; McQuibban et al, 2010), whereas elevated expression produces condensed mitochondria with compact cristae (Hasegawa & van der Bliek, 2007). Whether monoallelic LETM1 deletion results in mitochondrial dysfunction is still uncertain, as some studies described an alteration of the assembly of the respiratory chain supercomplexes (Tamai et al, 2008), while others reported no evident mitochondrial dysfunction or link with the mitochondrial ribosomal machinery (Dimmer et al, 2008).
LETM1 is often deleted in Wolf–Hirschhorn syndrome (WHS; Zollino et al, 2003), a disorder caused by deletions of the short arm of one chromosome 4, encompassing multiple genes. Although LETM1 levels are consistently decreased in fibroblasts and lymphoblastoid cell lines of WHS patients harboring LETM1 deletions (Hasegawa & van der Bliek, 2007; Dimmer et al, 2008; Doonan et al, 2014; Hart et al, 2014), only modest alterations of mitochondrial membrane potential and free radical production have been reported (Doonan et al, 2014; Hart et al, 2014). Moreover, mice with a single copy of Letm1 appear normal, with none of the facial or midline developmental defects typical of WHS LETM1‐deficient patients. However, pyruvate dehydrogenase (PDH) activity was decreased in the brain of fasted Letm1 +/− mice, suggesting a role for the protein in glucose metabolism (Jiang et al, 2013). The uncertainty surrounding LETM1 has left a gap in our understanding of the role of LETM1 protein both in mitochondrial biology and in its clinical relevance to WHS. Therefore, we set out to address several of the outstanding questions: of the role of LETM1 in mitochondrial gene expression in mammals; whether mitochondrial dysfunction in WHS is attributable to LETM1 haploinsufficiency; and whether the effect on glucose metabolism in humans is comparable to the Letm1 +/− mouse (Jiang et al, 2013). Analysis of the form and function of mitochondria in WHS patient‐derived fibroblasts and LETM1 gene‐silenced cells indicates that LETM1 couples pyruvate oxidation to mtDNA metabolism, and that LETM1 deficiency in WHS results in mitochondrial dysfunction that exacerbates the disorder.
Results
LETM1 is required for mitochondrial translation and respiration in mammalian cells
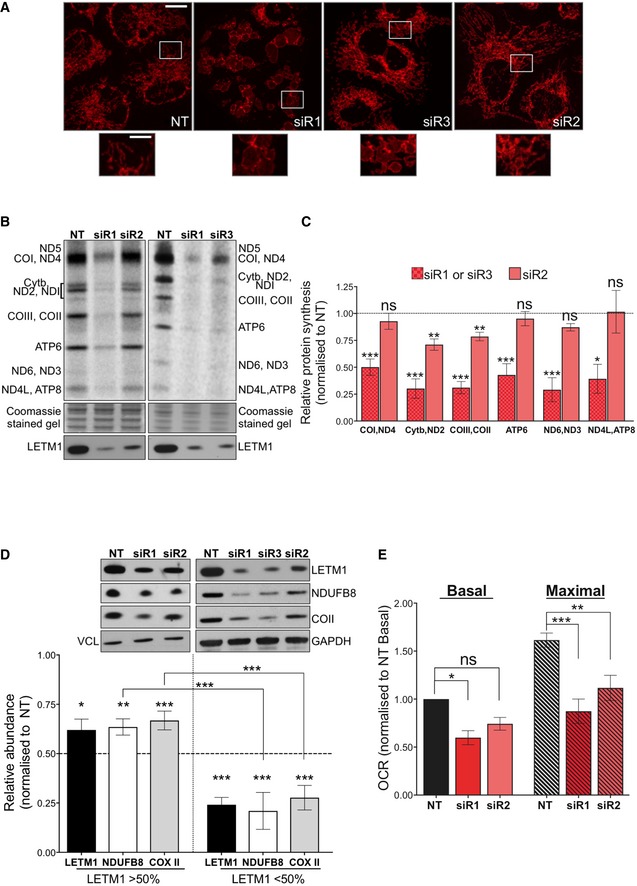
Findings from yeasts suggest that the LETM1 homolog, mdm38, facilitates the translation and assembly of specific mitochondrial proteins into respiratory chain complexes (Bauerschmitt et al, 2010). To address whether LETM1 contributed to mitochondrial protein synthesis (MPS) in mammals, we repressed its expression in HeLa cells via RNA interference. All three siRNAs targeting LETM1 [siR1, siR2, or siR3 (Appendix Fig S1A)] caused mitochondrial swelling (Fig 1A), as previously reported (Dimmer et al, 2002, 2008; Hasegawa & van der Bliek, 2007; Tamai et al, 2008; Jiang et al, 2009; McQuibban et al, 2010), and each siRNA impaired MPS (Fig 1B and C; and Appendix Fig S1B) albeit to different extents. The marked decrease in LETM1 protein induced by siR1 or siR3 produced a more severe impairment of mitochondrial translation than siR2, which was associated with more residual protein (Fig 1B and C). The steady‐state level of selected OXPHOS subunits was also directly proportional to the amount of LETM1 (Fig 1D), and the largest decreases in respiratory capacity were associated with the most pronounced reduction in LETM1 expression (Fig 1E). Thus, LETM1 deficiency impairs mitochondrial translation and restricts respiratory capacity in mammalian cells in a dose‐dependent manner.
Figure 1. LETM1 is required for mitochondrial translation and respiration.
-
AImmunofluorescence analysis of HeLa cells transfected with either a non‐target dsRNA (NT) or one of the three siRNAs targeting LETM1 (siR1, siR2, or siR3) and labeled with anti‐TOM20 antibody. In siR1‐treated cells, the mitochondria formed a “honeycomb” of swollen distinct organelles; siR3 resulted in giant organelles with a central region distinguished by reduced TOM20; siR2 produced relatively little swelling, and the mitochondrial network was generally well preserved. The pronounced swelling induced by siR1 significantly increased circularity (P = 2.32E‐58; ImageJ analysis), siR1 circularity = 0.683 ± 0.012 compared with 0.414 ± 0.008 for the NT, where 1 = a perfect circle. Over 150 mitochondria were quantified in each cell type; data are mean ± SEM. Scale bar represents 12 μm in the main images and 4 μm in offset magnification.
-
B35S labeling of de novo mitochondrial protein synthesis of HeLa cells transfected as in (A). Polypeptide assignments flank the gel images. Coomassie‐stained gels are used as loading controls, and immunoblots indicate the efficiency of LETM1 knockdown.
-
CQuantification of the radiolabeled mitochondrial polypeptides in panel (B) and similar gels, expressed relative to protein synthesis of the NT. Data are expressed as mean ± SEM of n = 6 independent experiments; 1 or 2 rounds of transfection for siR1 and siR3, but exclusively 2 rounds in the case of siR2.
-
DRepresentative immunoblots for the OXPHOS proteins NDUFB8 and COII of HeLa cells transfected with NT or siLETM1 (siR1, siR2, or siR3). Vinculin and GAPDH are shown as loading controls. The mean relative abundances for respiratory subunits COII and NDUFB8 are shown beneath the blots. Data are expressed as mean ± SEM of n = 8 independent experiments.
-
EMitochondrial oxygen consumption rate (OCR) measured using a Seahorse flux analyzer before (basal) and after the addition (maximal) of the uncoupler FCCP, in HeLa cells treated with NT or siLETM1 (siR1 or siR2). Data are expressed as mean ± SEM of n = 5 independent experiments.
LETM1 depletion compromises 55S ribosome maintenance and alters the levels and distribution of mtDNA and mtRNA
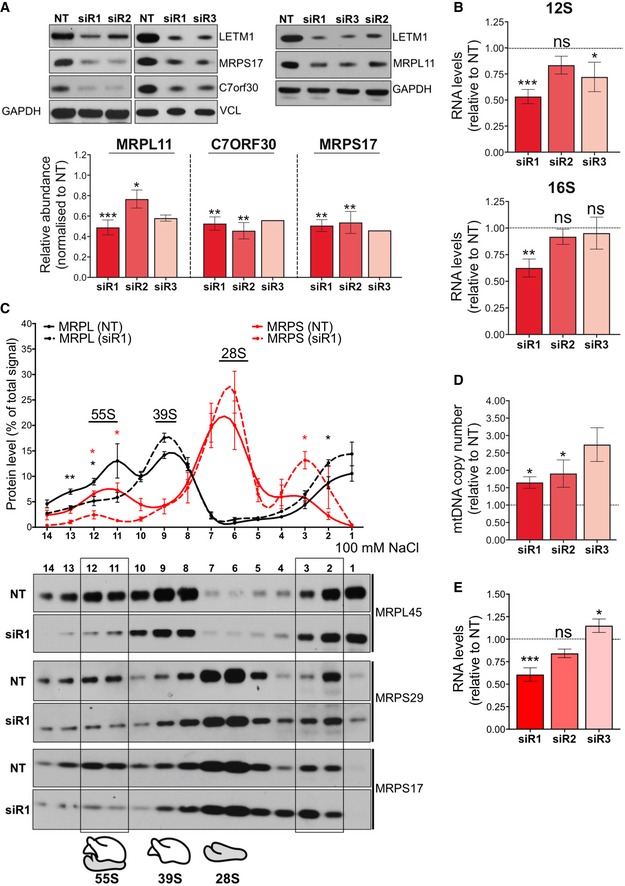
To determine the effect of LETM1 depletion on the mitochondrial ribosome, the abundance of selected mitochondrial ribosomal polypeptides and ribosomal RNAs was determined after LETM1 knockdown. LETM1 silencing decreased the levels of structural components of both mitoribosome subunits, MRPS17 and MRPL11, and the assembly factor C7orf30 (Rorbach et al, 2012; Wanschers et al, 2012; Fig 2A and Appendix Fig S1C), as well as the 12S and 16S ribosomal RNAs (Fig 2B). Furthermore, when lysates were fractionated on sucrose gradients the 55S ribosome was significantly decreased in cells partially depleted of LETM1, with a concomitant increase in the proportion of mitoribosome components at the top of the gradient (Fig 2C). These data suggest that LETM1 is required for 55S ribosome assembly or stability.
Figure 2. LETM1 depletion compromises mitochondrial ribosome maintenance and alters the abundance of mitochondrial DNA and RNAs.
-
ASteady‐state levels of mitochondrial ribosomal structural subunits (MRPL11 and MRPS17) or assembly factor (C7orf30) of HeLa cells transfected with siRNAs for either NT or LETM1 (siR1, siR2, or siR3). Vinculin and GAPDH are shown as loading controls. Data are expressed as mean ± SEM of n = 3 independent experiments for siR1 and siR2, and n = 2 (MRPL11) or 1 (MRPS17 and C7orf30) for siR3.
-
BRelative 12S (upper panel) and 16S rRNA (lower panel) levels in HeLa cells after LETM1 siRNA compared to NT, n = 3 independent experiments.
-
CTotal lysates from HeLa cells treated with NT or a siRNA for LETM1 were separated on 100 mM NaCl, 10–30% isokinetic sucrose gradients, and fractions analyzed by immunoblotting with antibodies to components of the large (39S) and small (28S) subunit of the 55S ribosome. Immunoblots were quantified by ImageJ, and the value for each fraction was expressed as a percentage of the sum of all fractions. Data are expressed as mean ± SEM of n = 3 independent experiments.
-
DRelative mtDNA copy number estimated by qPCR. Data are expressed as mean ± SEM of n = 3 independent experiments except for siR3 (n = 2).
-
EThe combined average mRNA levels of ND1, COII, COIII, Cyt b, and ATP6/8 in HeLa cells treated with either NT or siLETM1 (siR1, siR2, or siR3). Data are mean values ± SEM of n = 3 independent experiments.
Mitochondrial transcription supplies the messenger and ribosomal RNAs for protein synthesis, and ribosomal (small subunit) assembly has been proposed to occur at the mitochondrial nucleoid (He et al, 2012a; Bogenhagen et al, 2014; Dalla Rosa et al, 2014). Moreover, RNA processing and ribosome assembly involve a suite of factors organized in RNA granules in close proximity to the mtDNA, including GRSF1 (Antonicka et al, 2013; Jourdain et al, 2013; Tu & Barrientos, 2015). Hence, mitoribosome dysfunction could be the cause or the consequence of aberrant mtRNA and mtDNA metabolism. Therefore, we next determined the effects of LETM1 depletion on the mitochondrial RNA and DNA levels. LETM1 deficiency consistently increased mtDNA copy number (Fig 2D), whereas it reduced the mitochondrial transcript abundance per mtDNA (Fig 2E and Appendix Fig S1D). The highest mtDNA copy number, 2.6 times that of controls with siR3, was accompanied by transcript levels similar to controls, while the more modest 1.5‐fold increase in mtDNAs induced by siR1 was associated with significantly reduced mRNA levels (Fig 2D and E). Thus, LETM1 deficiency impairs mitochondrial transcription, compensated, fully in the case of siR3, by elevated mtDNA copy number.
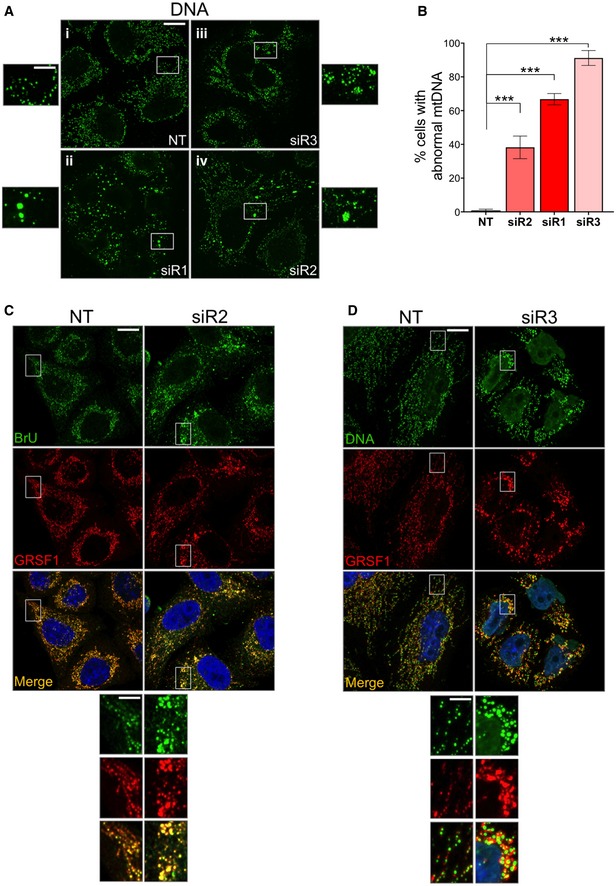
In light of the perturbations to mitochondrial DNA and RNA levels (Fig 2D and E), we next determined the effects of LETM1 silencing on mtDNA and RNA form and distribution. All three siRNAs targeting LETM1 produced mtDNA alterations (Figs 3A and B, and EV1A; and Appendix Fig S2), characterized by an increase in the size of many mtDNA foci (in the case of siR1; Fig 3A‐ii) or by clustering of mtDNA (siR3 and siR2), the extent of which, again, correlated with the level of LETM1 protein (Figs 3A‐iii, 4A‐iv, and EV1A; and Appendix Fig S2). Similarly, the foci formed by newly synthesized RNA and an RNA granule protein, GRSF1, were aberrant in size and distribution in LETM1‐deficient cells (Fig 3C and D).
Figure 3. LETM1 repression perturbs mtDNA and mtRNA organization.
-
ALETM1 expression was suppressed in HeLa cells by transfection with targeted siRNAs (siR1, siR2, or siR3). A non‐target dsRNA (NT) served as control. Cells were fixed and immunolabeled with anti‐DNA antibody (green). A higher magnification of selected mtDNA foci is shown beside each picture.
-
BQuantification of cells in (A) displaying mtDNA abnormalities. At least 50 cells per siRNA were counted from 4 (siR2) and 5 (siR1 or siR3) independent experiments. Data are expressed as mean ± SEM. ***P < 0.001 (one‐way ANOVA).
-
CHeLa cells were labeled with bromouridine (BrU) for 60 mins, 144 h after transfection with NT or siR2, and stained with anti‐BrdU (green) and anti‐GRSF1 (red) antibodies. Cell nuclei were stained with DAPI (blue).
-
DHeLa cells were labeled with anti‐DNA (green) and anti‐GRSF1 (red) antibodies, 144 h after transfection with NT or siR3. Cell nuclei were stained blue with DAPI.
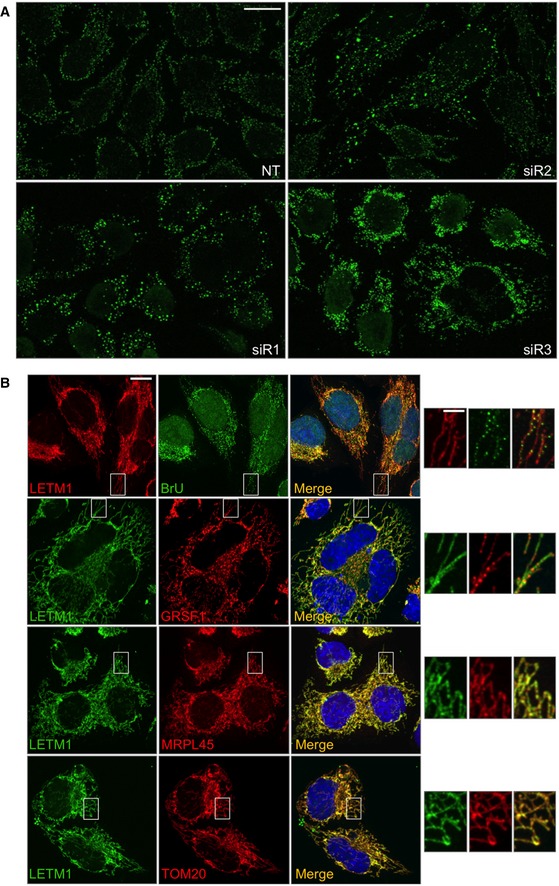
Figure EV1. LETM1 repression perturbs mtDNA organization.
-
AFurther examples of the mtDNA abnormalities are shown in Fig 3A. LETM1 expression was suppressed in HeLa cells by transfection with targeted siRNAs (siR1, siR2, or siR3). A non‐target dsRNA (NT) served as control. Cells were fixed and immunolabeled with anti‐DNA antibody (green). Scale bar: 15 μm.
-
BHeLa cells stained with anti‐LETM1 antibody (red) and anti‐BrdU (green) after labeling with 5 mM BrU for 60 min. In other images, LETM1 was stained green and other proteins stained red using antibodies to the RNA granule protein GRSF1, the 55S ribosome component MRPL45, or the outer mitochondrial membrane protein TOM20. Scale bars are 12 μm in the main images and 3 μm in offset magnification.
Figure 4. LETM1 co‐fractionates with mitochondrial nucleoprotein complexes and is widely distributed in the mitochondrial network, like ribosomes and unlike mtDNA.
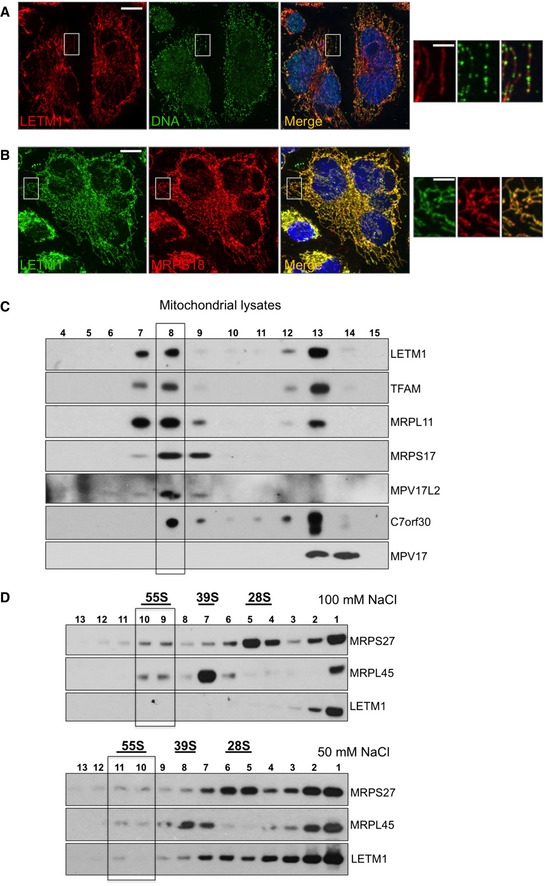
-
A, BHeLa cells were immunostained with anti‐DNA (green) and anti‐LETM1 (red) (A), or anti‐LETM1 (green) and anti‐MRPS18 (red) (B).
-
CMitochondrial lysates from HEK293T cells were fractionated on 20–42.5% iodixanol gradients and the migration patterns of LETM1 and selected nucleoid components determined by immunoblotting.
-
DHeLa mitochondrial lysates were fractionated on 10–30% sucrose gradients, and the distribution of LETM1 and ribosomal proteins was determined by immunoblotting. 55S corresponds to the assembled mitoribosome and 28S and 39S to its small and large subunits, respectively. Mitochondria were lysed in 100 or 50 mM NaCl (top and bottom panels, respectively).
A fraction of LETM1 associates with mitochondrial nucleoprotein complexes
To determine whether the phenotypes associated with LETM1 depletion reflected a direct interaction with the nucleic acids or ribosomes, we first assessed the pattern of LETM1 distribution in mitochondria. Immunofluorescence analysis of endogenous LETM1 indicated that the protein did not form the punctate patterns of either mitochondrial nucleoids (Spelbrink, 2010; Fig 4A) or newly synthesized RNA or GRSF1 (Antonicka et al, 2013; Jourdain et al, 2013; Tu & Barrientos, 2015; Fig EV1B), and was instead distributed throughout the mitochondrial network, comparable to mitochondrial ribosomes, and the outer mitochondrial membrane protein TOM20 (Figs 4B and EV1B). Hence, immunofluorescence lacked the resolution to determine whether LETM1 physically interacts with the mitoribosome; therefore, we next fractionated mitochondrial lysates on two types of density gradient. Mitochondrial ribosomes and nucleoids cluster in one or two fractions on iodixanol gradients, well separated from the majority of mitochondrial proteins (He et al, 2012b), and a portion of LETM1 co‐fractionated with these nucleoprotein complexes (Fig 4C). LETM1 also co‐affinity purified with mitochondrial DNA interacting proteins in a recent study (Matic et al, 2018). For the specific analysis of mitochondrial ribosomes via sucrose gradient sedimentation, different salt concentrations were tested, since it has been proposed that the interaction of the yeast mdm38 with ribosomes is a function of the ionic strength (Kehrein et al, 2015). The 55S mitochondrial ribosome of cultured cells readily dissociates into its constituent 28S and 39S subunits on sucrose gradients; nevertheless, the small amount of intact 55S ribosomes co‐migrated with a fraction of the LETM1 protein when mitochondrial lysates were separated using a 50 mM, but not 100 mM NaCl buffer (Fig 4D). These findings suggest that LETM1 physically interacts with the human mitochondrial ribosome, which can include interaction with mRNA (activators), as has been proposed for the yeast homolog (Bauerschmitt et al, 2010). The effects of LETM1 deficiency on mitochondrial DNA and RNA as well as the 55S ribosome fit with the protein facilitating ribosome assembly, which occurs at the nucleoid (He et al, 2012a; Bogenhagen et al, 2014; Dalla Rosa et al, 2014) and closely associated RNA granules (Antonicka et al, 2013; Jourdain et al, 2013; Tu & Barrientos, 2015). On the other hand, yeast mitochondrial ribosomes and nucleoids are interconnected (Kehrein et al, 2015) and co‐purification analysis is consistent with a similar arrangement in human mitochondria (Rorbach et al, 2008; He et al, 2012b). Thus, LETM1 deficiency could primarily disturb the connections between mitochondrial nucleoids and ribosomes resulting in impaired ribosome maintenance and protein synthesis, when severe. Whatever the details of the interdependence of mtDNA organization and expression, the findings for LETM1 suggest that insufficiency or defects in many components of mitochondrial nucleoids or the mitochondrial translation machinery could produce similar phenotypes; indeed, MPV17L2 depletion causes marked mtDNA aggregation and severe ribosome maintenance defects (Dalla Rosa et al, 2014), and a mutation in the aminoacyl tRNA synthetase, FARS2, causes mtDNA aggregation (Almalki et al, 2014).
Decreased DRP1 activity mitigates the effects of reduced expression of LETM1
We next examined the mitochondrial fission factor DRP1 in the LETM1‐deficient cells, because its repression is known to cause mtDNA clustering (Ban‐Ishihara et al, 2013). Transfection of HeLa cells with siR1 increased the abundance of the active phosphorylated form of DRP1 (DRP1S616; Fig 5A and Appendix Fig S3A), whereas the two LETM1 siRNAs (siR2 and siR3) associated with mtDNA clustering had decreased levels of (active) DRP1 protein (Fig 5A). Therefore, the mtDNA clustering associated with LETM1 silencing might be dependent on DRP1 repression. Moreover, since siR1 produced the most severe impairment of mitochondrial translation, repressing DRP1 when LETM1 is scarce might improve mtDNA expression. Concordant with the latter idea, co‐silencing of DRP1 and LETM1 attenuated the effects of LETM1 silencing alone (siR1), both in respect of MPS and the steady‐state level of selected respiratory chain components (Fig 5B and C; and Appendix Fig S3B). As well as enhancing the expression of OXPHOS complexes, the dual gene silencing altered the mtDNA, producing a phenotype intermediate between that of either DRP1 or LETM1 repression alone (Fig 5D). Hence, silencing of LETM1 and DRP1 has different, possibly opposing, effects on mtDNA organization, which could explain the benefit of co‐repressing DRP1 when there is a shortage of LETM1, as occurred spontaneously with siR3 (and on occasion siR2) targeting LETM1 (Figs 3 and EV1A; and Appendix Fig S2) and in the context of WHS (see below).
Figure 5. Changes to DRP1 in response to LETM1 depletion § .
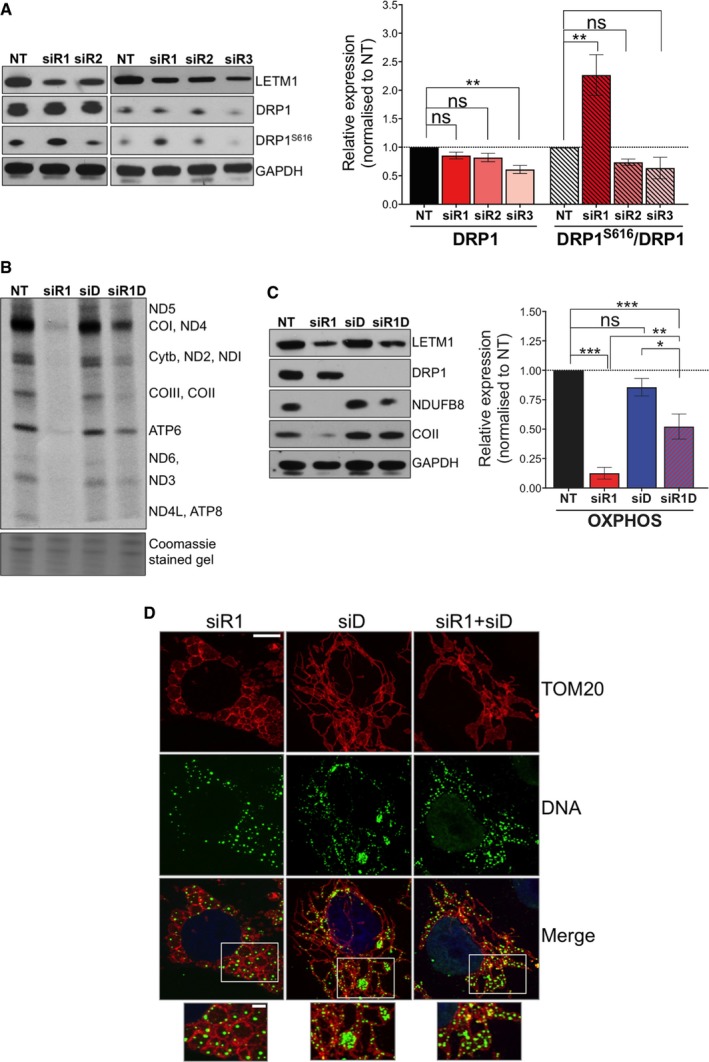
-
ARepresentative immunoblots of LETM1, DRP1 total protein levels, and activating phosphorylation (DRP1S616) in HeLa cells treated with either NT or LETM1 siR1, siR2, or siR3. GADPH is shown as a loading control. Immunoblot signals were quantified using ImageJ; data are displayed as mean ± SEM of n = 4 independent experiments except for siR3 DRP1S616 (n = 2).
-
BOne hour 35S pulse‐labeling of mitochondrial translated proteins in HeLa cells silenced for LETM1 (siR1) or DRP1 (siD), or co‐silenced for LETM1 and DRP1 (siR1D) for 144 h. Coomassie‐stained gels are used as loading controls, and polypeptide assignments are indicated to the right.
-
CImmunoblotting of cell lysates prepared as in (B) with antibodies to respiratory chain subunits NDUFB8 and COII, and GAPDH as loading control. Data are expressed as mean ± SEM of n = 3 independent experiments.
-
DConfocal analysis of HeLa cells silenced for LETM1 (siR1), or for DRP1 (siD), or co‐silenced with both siR1 and siD for 96 h. Mitochondrial network, mtDNA, and cell nuclei were immunostained with anti‐TOM20 (red) anti‐DNA (green), and DAPI (blue), respectively. Scale bars are 6 μm in the main images and 3 μm in offset merged magnifications.
Loss of one allele of LETM1 is associated with mitochondrial swelling and mtDNA clustering
The defined mitochondrial phenotypes caused by LETM1 deficiency in HeLa cells provided points of reference to assess the consequences of LETM1 haploinsufficiency in WHS. Mitochondrial metabolism was investigated in primary fibroblasts of four subjects with a monoallelic deletion on the short arm of chromosome 4 encompassing LETM1 (S1–S4, LETM1 +/−), each of whom presented with typical WHS features and epilepsy (Appendix Table S1 and Appendix Fig S4). Fibroblasts of a fifth subject, S5 (the mother of S3), whose deletion did not include LETM1 (thus LETM1 +/+), were analyzed in parallel. Cells of S1–S4 expressed approximately half as much LETM1 protein as control cells or those of S5 (Figs 6A and EV2A). Hence, LETM1 haploinsufficiency is associated with reduced expression of LETM1 protein in WHS fibroblasts, whereas the adjacent genes deleted in subject S5 had no impact on the expression of LETM1 (Appendix Fig S4). However, with this amount of residual LETM1 protein, mitochondrial abnormalities were expected to be at the milder end of the spectrum observed in HeLa cells silenced for LETM1. Accordingly, there was no appreciable impairment of mitochondrial translation (Appendix Fig S5A), and there was little or no decrease in respiratory chain components in WHS fibroblasts (Appendix Fig S5B); nevertheless, there were mitochondrial perturbations. Confocal microscopy revealed abnormal mitochondrial morphology in all the WHS cell lines lacking one copy of LETM1, and many mitochondria were more elongated than those of control cells and S5 (Appendix Fig S6A) and had numerous distensions (Fig 6B), while the largest mitochondria were often separated from the main network (Fig 6B zoom), suggesting that the swellings caused the mitochondrial network to fragment in places.
Figure 6. LETM1 haploinsufficiency in WHS is associated with abnormal mitochondrial morphology and ultrastructure.
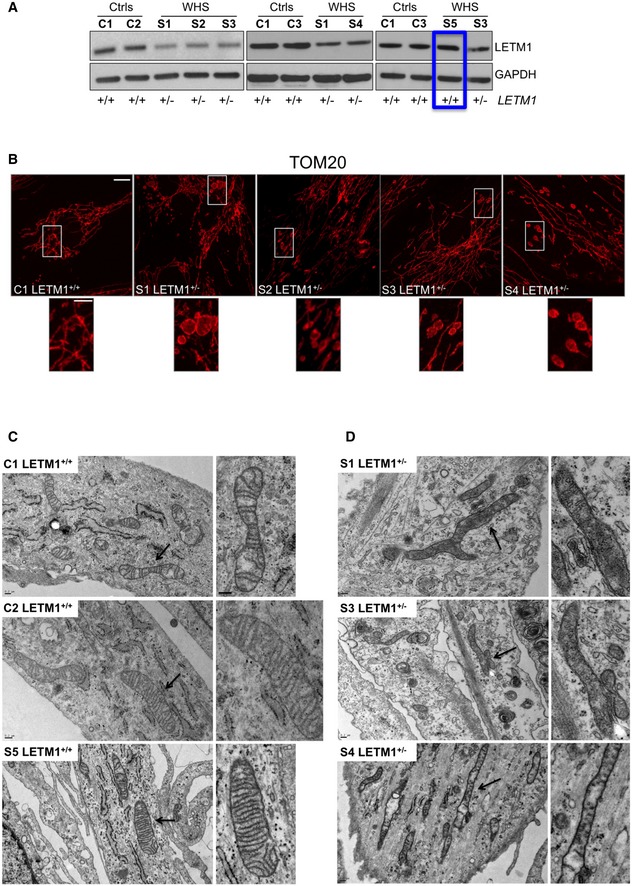
-
ASteady‐state levels of LETM1 protein in fibroblasts from controls (C1–C3), WHS LETM1 +/− (S1, S3, S4), and S5, the sole case of WHS in this study whose deletion does not encompass LETM1 (boxed in blue). GAPDH is shown as a loading control.
-
BImmunofluorescence analysis of control (C1), WHS LETM1 +/− S1‐S4, and fibroblasts with anti‐TOM20 antibody. Scale bars are 8 μm in the main images and 3 μm in offset merged magnifications.
-
CElectron micrographs of controls (C1 and C2) and WHS LETM1 +/+ (S5) cells; side panels show zooms. Scale bar: 0.2 μm. Black arrows indicate the areas shown offset at higher magnification.
-
DElectron micrographs of WHS LETM1+/− S1, S3, and S4 cells. Black arrows indicate elongated and branched mitochondria; side panels show magnifications. Scale bar: 0.2 μm.
Source data are available online for this figure.
Figure EV2. LETM1 haploinsufficiency in WHS is associated with mitochondrial ultrastructure abnormalities.
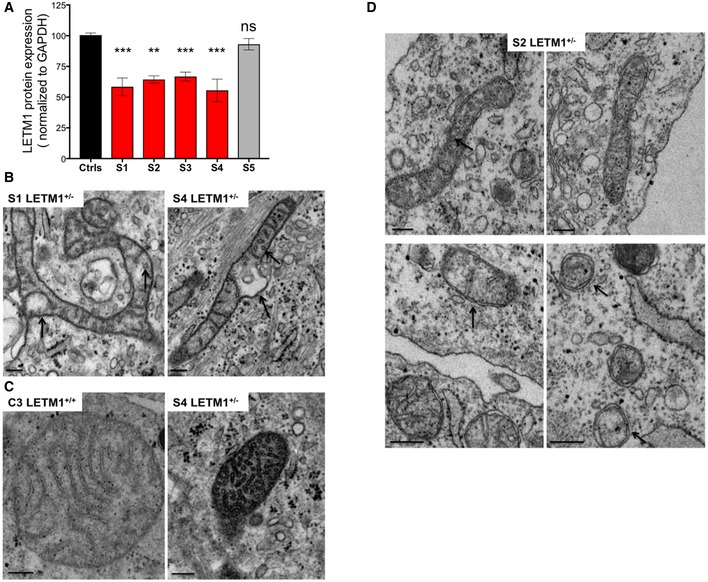
-
AQuantification of LETM1 expression in fibroblasts. Data from control cell lines (ctrls) C1–C3 were pooled. All data are expressed as mean ± SEM (n = 4 or 5). **P < 0.01, ***P < 0.001. ns, not statistically significant (one‐way ANOVA).
-
BMagnification of WHS LETM1 +/− S1 and S4 mitochondria. Black arrows indicate circular cristae and matrix distensions. Scale bar: 0.2 μm.
-
CMagnification of control C3 and WHS LETM1 +/− S4 mitochondria showing dense matrix and circular cristae exclusively in the latter. Scale bar: 0.2 μm.
-
DElectron micrographs of WHS LETM1 +/− (S2) fibroblasts. Black arrows indicate circular cristae in the upper 2 images and mitochondria with sparse cristae in the lower images. Scale bar: 0.2 μm.
The abnormal mitochondrial morphology reported here was marked and not reported in previous studies of WHS fibroblasts (Hasegawa & van der Bliek, 2007; Dimmer et al, 2008; Doonan et al, 2014); therefore, we further analyzed mitochondrial ultrastructure in WHS fibroblasts by transmission electron microscopy. Fibroblasts from three controls (C1–C3) and subject S5 contained mitochondria of normal appearance (Figs 6C and EV2C). In contrast, approximately 70% of mitochondria of the LETM1 +/− WHS patients were branched, thinned, and elongated (Fig 6D); frequently, the cristae were scarce and the remainder were rounded in shape (Figs 6D and EV2B–D), while the mitochondrial matrix in organelles where cristae were present appeared dense and more compact than normal (Fig EV2C). Hence, the loss of one allele of LETM1 in fibroblasts of WHS subjects is associated with mitochondrial morphology abnormalities based on light and electron microscopy.
The distended and swollen mitochondria of the LETM1‐deficient WHS fibroblasts displayed mtDNA clustering (Fig 7A and B; and Appendix Fig S6B) in a significantly greater proportion of cells than controls or subject S5 (Fig 7C), and this was associated with elevated mtDNA copy number in the cell lines tested (S1 and S4; Fig 7D). DRP1 was decreased in all cells of LETM1‐deficient WHS subjects compared to controls, especially the active form of the protein (Fig 7E), which can account for the elongated mitochondria (Appendix Fig S6A). This finding strengthens the earlier conclusion that the repression of DRP1 is a frequent consequence of LETM1 deficiency that causes mtDNA clustering, and the data from HeLa cells suggest that it is a compensatory mechanism that limits the adverse effects of a shortage of LETM1 (Fig 5). Although the repression of DRP1 was associated with elongated mitochondria, there were also many discrete swollen mitochondria in the WHS cells (Fig 6B) suggesting that LETM1 deficiency causes partial fragmentation of the mitochondrial network in a DRP1‐independent manner, as previously proposed (Dimmer et al, 2008). Taken together, these features indicate that WHS fibroblasts lacking one LETM1 allele recapitulate features of synthetic LETM1 deficiency in HeLa cells, including aberrant mitochondrial morphology, mtDNA disorganization, and elevated copy number.
Figure 7. WHS LETM1+/− fibroblasts display increased mtDNA copy number and mtDNA clusters.
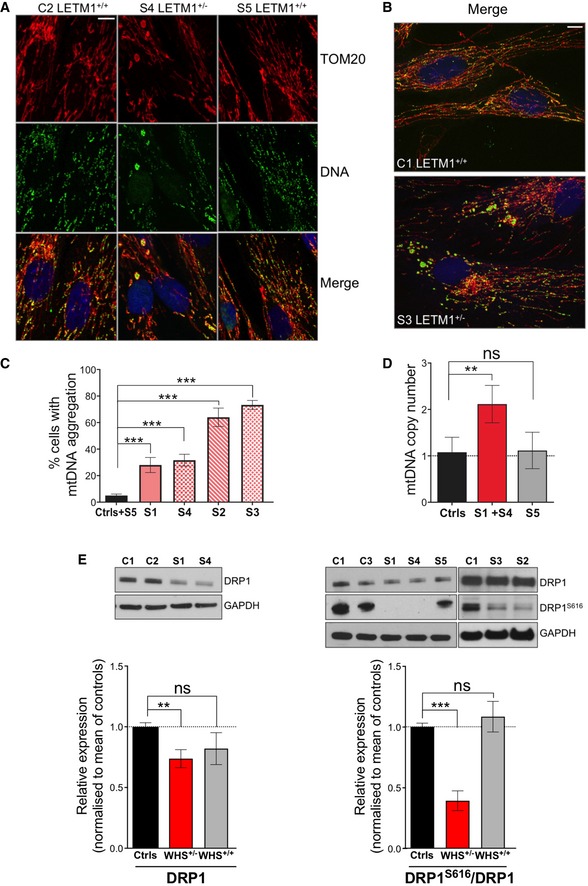
-
AConfocal analysis of control C2, WHS LETM1 +/− S4, and WHS LETM1 +/+ S5 cells, immunolabeled with anti‐TOM20 (red), DNA (green), and DAPI (blue). Scale bar: 8 μm.
-
BConfocal images of control C1 and WHS LETM1 +/− S3 labeled with TOM20, DNA, and DAPI, shown as merge. Scale bar: 8 μm.
-
CQuantification of cells in (A) displaying mtDNA aggregates. At least 50 cells per cell line were counted from three independent experiments. Data are expressed as mean ± SEM.
-
DqPCR quantified mtDNA copy number in WHS and control cells. Values are expressed relative to control cells (C1–C3). Data are expressed as mean ± SEM of n = 3 independent experiments.
-
ERelative abundance of DRP1 and DRP1S616 in whole‐cell extracts of control and WHS fibroblasts, with GAPDH shown as a loading control. Immunoblot signals were quantified using ImageJ, and data are expressed as mean ± SEM of n = 5 independent experiments.
WHS patient fibroblasts remodel their metabolism away from pyruvate oxidation toward ketone body utilization
Mitochondrial morphology and dynamics are altered in response to a number of stimuli, including nutrient deprivation, and it has been proposed that changes in mitochondrial dynamics remodel substrate utilization for bioenergetic purposes (Buck et al, 2016; Wai & Langer, 2016). Hence, the mitochondrial morphology of the WHS fibroblasts lacking one LETM1 allele might be indicative of a nutrient‐restricted state, or increased reliance on substrates other than glucose. Moreover, the brain of Letm1 heterozygous mice, under fasted conditions, displays a reduction in pyruvate dehydrogenase (PDH) activity (Jiang et al, 2013), which is critical for pyruvate utilization by mitochondria. Thus, we judged the regulation of PDH to be a prime potential function of LETM1. Analysis of iodixanol gradients of HEK cells indicated that almost all of the PDH‐E1 subunit co‐fractionated with mitochondrial nucleoprotein complexes (Fig 8A), consistent with its partner subunit (E2) being highly enriched in preparations of amphibian mtDNA (Bogenhagen et al, 2003), and its co‐purification with mtDNA binding proteins and many components of the mitochondrial translation apparatus of HeLa cells (Matic et al, 2018). The inactive phosphorylated form of PDH‐E1 was more abundant in WHS fibroblasts than in controls when cultured in 25 mM glucose and 1 mM pyruvate (Fig 8B and C), which can be attributed to LETM1 deficiency, as phosphorylated PDH‐E1 was also elevated in LETM1 repressed HeLa cells (Appendix Fig S7). Commensurate with the increased phosphorylation of PDH‐E1, WHS fibroblasts had elevated PDH kinase 3 (PDK3; Figs 8B and EV3A), which targets the E1 subunit (Kolobova et al, 2001; Zhang et al, 2014). These results imply that pyruvate oxidation is low in WHS fibroblasts compared to control cells, and thus, WHS mitochondria might be configured to utilize carbon sources other than glucose, such as fatty acids or ketone bodies. In support of this interpretation, fibroblasts cultured in medium containing the ketone body β‐hydroxybutyrate (BHB) in place of glucose, suffered a “metabolic crisis” that led to extensive cell death, from which the fibroblasts of four healthy controls and subject S5 failed to recover, while the cells of all four (LETM1 +/−) WHS subjects continued to divide even after passaging (Figs 8C and EV3B; Appendix Fig S8; Movies EV1 and EV2).
Figure 8. WHS LETM1+/− fibroblasts show reduced PDH phosphorylation and tolerate a strict ketogenic diet.
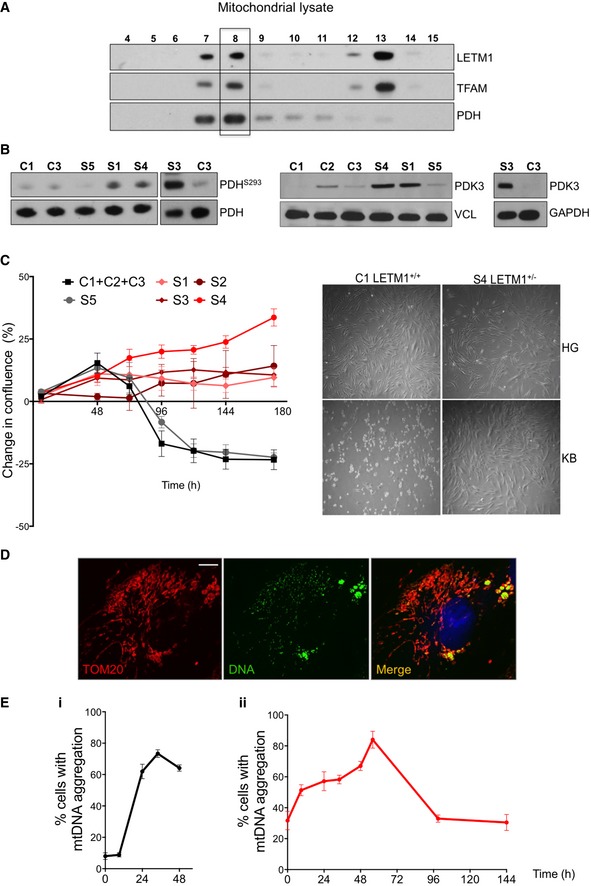
-
ARepresentative immunoblots of mitochondrial lysates of HEK cells separated on a 20–42.5% iodixanol gradients probed for LETM1, TFAM, and PDH.
-
BLeft panel: immunoblot analysis of total PDH protein levels and the inactive phosphorylated (repressed form) PDHS293, in control (C1, C3), WHS LETM1 +/− (S1, S3, S4), and WHS LETM1+/+ (S5) fibroblasts. Center and right panels: Immunoblot analysis of PDK3 in control (C1–C3), WHS LETM1 +/− (S1, S3, and S4), and WHS LETM1 +/+ (S5) fibroblasts.
-
CLeft panel: change in cell confluence of control (C1–C3), WHS LETM1+/− (S1‐S4), and WHS LETM1 +/+ (S5) fibroblasts grown in BHB over the course of 188 h. Data are expressed as mean ± SEM of n = 3 independent experiments. Right panel: images of control and WHS LETM1 +/− S4 cells growing either in the presence of 25 mM glucose (HG) or BHB (KB) for 6 days.
-
DConfocal images of control cells (C1) grown for 24 h in the presence of BHB and immunolabeled with anti‐TOM20 (red) and anti‐DNA (green), and DAPI‐stained (blue). Scale bar 6 μm.
-
EProportion of cells that displayed mtDNA aggregation during the KB treatment (panel i—control cells (C1, C2) and WHS LETM1+/+ (S5) fibroblasts; panel ii—LETM1+/− (S1, S2, S4) fibroblasts). At least 50 cells per cell line were counted at the indicated time points. Data are expressed as mean ± SEM of n = 3 independent experiments.
Source data are available online for this figure.
Figure EV3. PDH is repressed in WHS LETM1 +/− fibroblasts, and unlike controls cells, they are able to grow on medium containing KB in place of glucose and pyruvate.
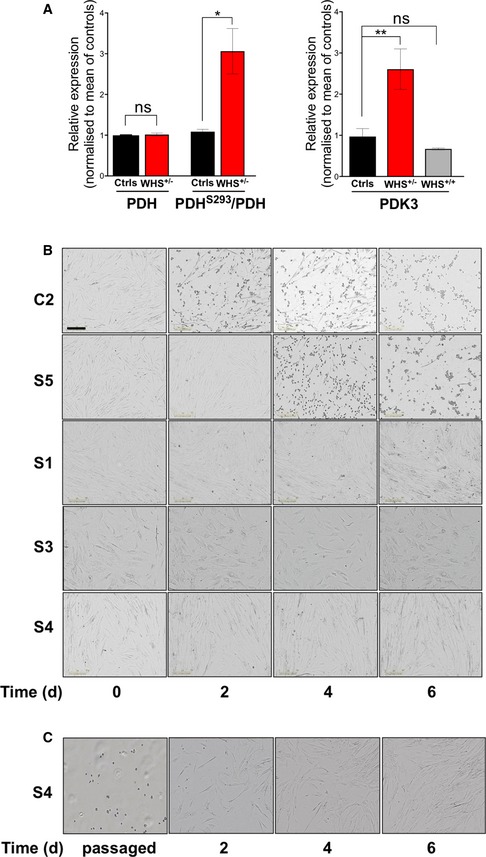
-
AQuantification of total PDH, PDHS293, and PDK3 immunoblots. Data are expressed as mean ± SEM of n = 5 independent experiments. *P < 0.05, **P < 0.01. ns, not statistically significant; t‐test with Welch's correction for PDH and PDHS293; one‐way ANOVA for PDK3.
-
BLight microscope images of control (C2), WHS LETM1 +/+ (S5), and WHS LETM1 +/− (S1, S3, and S4) cells growing in 0.3 mM BHB (KB) for 6 days, after a switch from 25 mM glucose and 1 mM pyruvate at 40% confluence. Scale bar: 300 μm.
-
CS4 cells grown to confluency in KB medium (as per panel A) were passaged and cultured for a further 6 days.
Nutrient availability alters mtDNA organization and expression
The changes in mtDNA organization and nutrient utilization resulting from LETM1 deficiency suggested that the two might be linked. Therefore, the effect on mtDNA distribution of replacing glucose and pyruvate with ketone bodies (KB) was assessed. The number of control fibroblasts with mtDNA aggregates increased sixfold within 24 h of switching to BHB (Fig 8D and E) and remained at this level until the cells died (Fig 8C and E‐i). Replacement of glucose and pyruvate with KB further increased the number of WHS LETM1 +/− fibroblasts displaying mtDNA clustering to a peak of 85% of all the cells after 56 h (Fig 8E‐ii); however, this subsided close to the original level by 99 h and continued to fall thereafter, such that, after 3 weeks of the KB regime, mtDNA organization and distribution appeared similar to control cells (Appendix Fig S9). To further test the link between nutrients and mtDNA aggregation, we cultured control fibroblasts in 5 mM glucose and 0.3 mM BHB with or without 1 mM pyruvate. Neither decreasing the glucose concentration from 25 to 5 mM, nor supplementing the medium with ketone bodies markedly altered mtDNA organization, whereas the withdrawal of pyruvate induced mtDNA clustering in cells to a similar extent as glucose and pyruvate replacement with KB (Fig 9A). Moreover, the mitochondrial translation impairment induced by LETM1 deficiency is influenced by nutrients, as the replacement of glucose with KB (BHB or acetoacetate) increased the amount of newly synthesized mitochondrial protein in HeLa cells depleted of LETM1 compared to the same cells grown in 25 mM glucose and 1 mM pyruvate (Fig 9B). These findings indicate that mtDNA organization, distribution, and expression are influenced directly by nutrient availability.
Figure 9. Mitochondrial DNA re‐organization in response to nutrient changes.
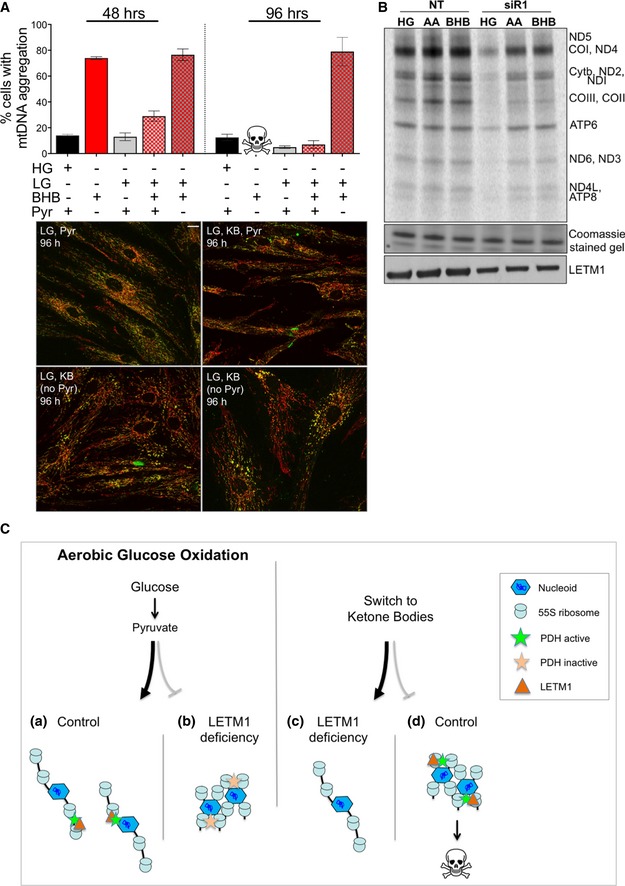
-
AProportion of mtDNA cluster following 48 or 96 h of growth in medium with (+) or without (−) 25 mM glucose (HG), 5 mM glucose (LG), β‐hydroxybutyrate (BHB), or 1 mM pyruvate (pyr). At least 50 cells per cell line were counted per condition from two independent experiments. Data are expressed as mean ± SEM. Representative images appear below the charts. Scale bar: 15 μm.
-
B35S labeling of de novo mitochondrial protein synthesis for 1 h, 72 h after transfection of HeLa cells with either a non‐target dsRNA (NT) or LETM1 siR1; KBs (0.1 mM BHB or acetoacetate (AA)) replaced HG (25 mM glucose, 1 mM pyruvate) 9 h before the labeling reaction. Polypeptide assignments flank the gel image; Coomassie‐stained gels are used as loading controls, and immunoblots indicate the efficiency of LETM1 knockdown.
-
CPerceived or actual nutrient availability alters the organization of mitochondrial nucleoprotein complexes. In yeasts, mitochondrial ribosomes are physically linked to mitochondrial nucleoids (Kehrein et al, 2015), and purification studies of a mitochondrial ribosomal protein (Rorbach et al, 2008) and mitochondrial nucleoid proteins (He et al, 2012a) suggest that the same is true of mammalian mitochondria, as illustrated in (a). The current study indicates that changes in nutrient availability (a switch from glucose to ketone body supplemented culture medium) lead to re‐organization of mitochondrial nucleoprotein complexes (MNPCs; panel A and Fig 8D and E, and illustrated in (b)). We infer that MNPC disorganization in LETM1 deficiency is the result of a failure to utilize pyruvate—a defect in nutrient sensing—that can be attributed to repression of pyruvate dehydrogenase (PDH) via PDK3 (Figs 8B and EV3A). Because the bulk of PDH appears to be physically associated with MNPC (Fig 8A), this could be mediated by the LETM1 bound to mitochondrial ribosomes/MNPC (Fig 4C). The direct and marked impact of nutrients on MNPCs is further evidenced by the adaptation of WHS cells (LETM1 +/−) to KBs (Figs 8E‐ii and EV3; and Appendix Figs S8 and S9), and the demonstration that KBs mitigate the mitochondrial translation impairment caused by LETM1 depletion (Fig 9B). Hence, WHS cells progressively adapt mtDNA organization to the alternative carbon source (c), whereas control cells are not primed for KB utilization, and in their case, MNPC clustering is followed by cell death (d) (Fig 8C and Movie EV1).
Source data are available online for this figure.
Discussion
There has been considerable debate as to the roles of LETM1 in mitochondrial metabolism, the resolution of which was needed to assess its possible contribution to the pathogenesis of WHS. Here, we demonstrate that mitochondrial abnormalities are evident in primary cells of WHS subjects lacking one allele of LETM1 and that reduced LETM1 expression is associated with the remodeling of nutrient metabolism in a disease state. Although WHS deletions involve a number of other genes, the loss of which could contribute to the metabolic and mitochondrial phenotypes, the mitochondrial phenotypes were exclusive to the WHS cells lacking a LETM1 allele and were evident in HeLa cells after LETM1 silencing. Moreover, altered glucose and pyruvate metabolism are features of Letm1 +/− mice, where no other gene is mutated (Jiang et al, 2013). Therefore, we conclude that when LETM1 is scarce, there is a switch to the consumption of ketone bodies, even in the presence of high levels of glucose, and that the decreased reliance of WHS fibroblasts on glucose and pyruvate primes them to survive growth supported on ketone bodies (Figs 8 and EV3B; Appendix Figs S8 and S9; and Movie EV2). The inference that LETM1 promotes the oxidation of glucose over lipid and amino acid catabolism has potentially important clinical implications: WHS patients could benefit from a strict diet comprising high lipid and proteins with low carbohydrate and sugars. A controlled diet of this nature may be particularly important during development (Pliss et al, 2016), including throughout childhood, or to control epilepsy, as has been proposed for PDH deficiency (Sofou et al, 2017).
Mitochondrial DNA organization and nutrient utilization
The re‐organization of the mtDNA and tolerance of a ketone body growth regime in WHS fibroblasts (Figs 8, 9 and EV3; and Appendix Figs S8 and S9), and the association of PDH with mitochondrial nucleoprotein complexes in both human cells (Fig 8A) and Xenopus oocytes (Bogenhagen et al, 2003), suggests that the physical interaction serves a physiological purpose. That is, proteins determining substrate utilization, such as PDH, act directly on mitochondrial nucleoids and ribosomes to couple fuel consumption and energy production. The mechanism of such coupling may well involve ion homeostasis that is known to be important for PDH activity and the TCA cycle in general (Denton et al, 1972; Nichols & Denton, 1995; Llorente‐Folch et al, 2015), and in which LETM1 is heavily implicated (Nowikovsky et al, 2004; Jiang et al, 2009; Hashimi et al, 2013; Nowikovsky & Bernardi, 2014; Tsai et al, 2014). Moreover, the strict link between LETM1 and pyruvate oxidation in mitochondria and increased dependence on alternative fuels in LETM1‐deficient cells may explain why there are contrasting results relating to mitochondrial dysfunction in WHS (Dimmer et al, 2008; Tamai et al, 2008, and this report).
Mitochondrial DNA organization and energy distribution
A growing number of mitochondrial disorders are now understood to feature mtDNA aggregation or clustering (Almalki et al, 2014; Akman et al, 2016; Desai et al, 2017; Nicholls et al, 2018 and this report). Clustering of the different complexes appears to be remarkably well tolerated, in that OXPHOS subunits continue to be produced and assembled correctly in the vicinity of the mtDNA aggregates, as respiratory capacity can remain at normal levels (Appendix Fig S5). Although the failure to distribute mtDNA and its products throughout the cell need not severely compromise respiratory capacity, it can be expected to create problems in tissues such as muscle and brain, which are those most often affected in mitochondrial disorders. Muscle requires a uniform energy supply for maximal efficiency (Glancy et al, 2015), and to function, neurons must deliver mitochondrial cargoes to distant synapses. In the case of mtDNA aggregation and concomitant mitochondrial morphology defects, the mitochondria for transport could have a dearth of mtDNA and OXPHOS complexes, which may explain the severe neurological features in such diseases.
The different outcomes for DRP1 expression in the context of LETM1 deficiency offer further insights into LETM1's role and the architecture of the mtDNA in the organelles. LETM1 deficiency is associated with mitochondrial distensions, but these are mostly within the reticular network when DRP1 activity is co‐repressed (LETM1 siR3 and occasionally siR2), whereas when LETM1 is scarce and DRP1 activity is not repressed, swollen mitochondria are ubiquitous and the network is fragmented (siR1; Fig 1A). Although the latter arrangement maintains a near‐normal mtDNA distribution in the face of LETM1 deficiency (Figs 3A and EV1A—siR1), clustering of the macromolecular complexes might be preferable, to help to counteract the hypotonic environment, and this could explain the frequent spontaneous co‐repression of DRP1 (Figs 5A and 7E), which evidently improves mitochondrial translation and OXPHOS when LETM1 is depleted (Fig 5B and C; and Appendix Fig S3B). Given that the findings suggest that repressing DRP1 attenuates the mitochondrial dysfunction caused by LETM1 deficiency, it will be important to determine whether some cell types of WHS LETM1+/− patients maintain normal levels of DRP1, as, if so, decreasing DRP1 might improve their symptoms or slow disease progression.
Mitochondria are known to adapt physically to nutrient availability (Gomes et al, 2011; Rambold et al, 2011). The current study demonstrates that changes in nutrient availability and utilization remodel the nucleoprotein complexes in mitochondria and thereby indicates how nutrients can modulate gene expression and energy production in the organelle (Fig 9C). An important corollary is that genetic defects in metabolic factors linked to mitochondrial nucleoprotein complexes, or their regulators, can produce a pseudo‐starvation state, owing to an inability to utilize an available nutrient.
Materials and Methods
Human studies
Informed consent was obtained for all the subjects included in the study, and the procedures followed were in accordance with the WMA Declaration of Helsinki and the Department of Health and Human Services Belmont Report.
Cell lines and culture conditions
Primary skin fibroblasts were obtained from healthy controls (n = 4) and from Wolf–Hirschhorn syndrome (WHS) patients (n = 5; Appendix Table S1). Unless stated otherwise, fibroblasts, HeLa, and human embryonic kidney cells (HEK293T, Invitrogen) were grown in Dulbecco's modified Eagle's medium (DMEM, Life Technologies) supplemented with 25 mM glucose, 1 mM pyruvate, 10% fetal bovine serum (FBS, Pan Biotech UK), and 1% penicillin and streptomycin (PS, Life Technologies) at 37°C in a 5% CO2 atmosphere. All cells were tested for mycoplasma infection on regular basis using LookOut Mycoplasma PCR Detection Kit (Sigma).
RNA interference of LETM1 and DRP1 in HeLa cells
For knockdown experiments, HeLa cells were transfected using either siRNAs targeting LETM1 (Appendix Table S2 and Appendix Fig S1A) or no particular gene (NT). All oligos were transfected at a concentration of 10 nM using Lipofectamine RNAiMAX (Life Technologies), according to the manufacturer's specifications. In general, HeLa cells were analyzed after 6 days, following two rounds of siRNA with the second transfection performed 3 days after the first. Where indicated the exceptions were 72 h after one round of transfection. Simultaneous knockdown of LETM1 and DRP1 was performed as described above using the siRNA targeting LETM1 (siR1) and DRP1 (Appendix Table S2).
Cell lysis, SDS–PAGE, Western blotting, and immunodetection
Cells were lysed in PBS, 0.1% n‐dodecyl‐D‐maltoside (DDM, Sigma), 1% SDS, 50 U benzonase (Novagen), 1:50 (v/v) protease inhibitor cocktail (Roche), and 1:100 (v/v) phosphatase inhibitor (Cell Signaling), and protein concentration was measured by Lowry assay (Bio‐Rad). 10–20 μg of proteins was separated by SDS–PAGE on Bis‐Tris NuPAGE gels (Life Technologies) and transferred to polyvinylidene fluoride membrane (Millipore). Membranes were blocked in 5% non‐fat dry milk in PBS with 0.1% (v/v) Tween‐20 (PBST) for 1 h at RT (room temperature) and then incubated overnight with primary antibodies (Appendix Table S3) at 4°C. The day after, membranes were washed three times with PBST and incubated for 1 h at RT with the appropriate secondary antibodies. Membranes were then washed three times with PBST, and signals were developed using ECL (GE Healthcare).
[35S]‐Methionine in cell labeling for mitochondrial protein synthesis
Mitochondrial translation products were labeled using 35S‐methionine as described previously (Dalla Rosa et al, 2014) with some modifications. Fibroblasts or HeLa cells were washed twice with methionine‐/cysteine‐free DMEM (Life Technologies) supplemented with 1 mM l‐GlutaMAX, 96 μg/ml cysteine (Sigma), 1 mM pyruvate, and 5% (v/v) dialyzed FBS, and incubated in the same medium for 10 min at 37°C. 100 μg/ml emetine dihydrochloride (Sigma) was then added to inhibit cytosolic translation, before pulse‐labeling with 100 μCi [35S]‐methionine for 45–60 min. Cell were chased for 10 min at 37°C in regular DMEM with 10% FBS, washed three times with PBS, and collected. Labeled cells were lysed in PBS, 0.1% n‐dodecyl‐D‐maltoside (DDM), 1% SDS, 50 units of benzonase (Novagen), and 1:50 (v/v) Roche protease inhibitor cocktail. Protein concentration was measured by Lowry assay, and 20 μg of protein was separated by 12% SDS–PAGE. Gels were dried and exposed to phosphor plates, and the signal was detected by PhosporImager.
Cellular oxygen consumption
The oxygen consumption rate (OCR) of HeLa cells was assayed with a XF24 Extracellular Flux Analyzer (Agilent). Briefly, 4 × 104 proliferating cells were seeded in microplates (Agilent) in 250 μl of prewarmed growth medium (DMEM, Gibco) and incubated 37°C/5% CO2 for 8 h. Subsequently, the medium was removed and replaced with assay medium (XF Base minimal DMEM (Agilent) complemented with 2 mM glucose, 2 mM GlutaMAX, and 1 mM pyruvate) and cells were incubated for 30 min in a 37°C non‐CO2 incubator. After taking an OCR baseline measurement, 1 μM oligomycin, 1.2 μM carbonyl cyanide‐4‐trifluoromethoxyphenylhydrazone (FCCP), and 1 μM rotenone were added sequentially. For normalization, protein concentration in each well was determined using the Lowry assay (Bio‐Rad).
mtDNA copy number
Quantification of mtDNA was performed as described in Dalla Rosa et al (2016) with some modifications. Briefly, total DNA was isolated from frozen pellets of human fibroblasts or HeLa cells using DNeasy Blood and Tissues Kit (Qiagen), according to the manufacturer’ s protocol. Real‐time quantitative PCR was performed in triplicate on 384‐Well Reaction Plates (Applied Biosystems), in final volumes of 10 μl. Each reaction contained 20 ng DNA template, 1× Power SYBR Green PCR Master Mix (Applied Biosystems), and 0.5 μM of forward and reverse primers. Mitochondrial and nuclear DNAs were amplified using primers specific to regions of human COXII and APP1 genes, respectively. The sequences of the primers used are in Appendix Table S2. Changes in mtDNA amount were calculated using the 2−ΔΔCt method (Schmittgen & Livak, 2008) and represented as fold change relative to the indicated control.
RNA extraction and analysis of mitochondrial transcripts by qPCR
For total RNA extraction, HeLa cells were lysed in TRIzol reagent (Life Technologies) and RNA, DNA, and proteins were separated by the addition of chloroform. After centrifugation (15 min, 12,000 g at 4°C), the upper aqueous phase containing RNA was transferred to a fresh tube and precipitated with equal volume of 70% ethanol. This solution was then loaded onto a PureLink RNA Mini Kit (Invitrogen) column, and RNA extraction was completed as per the manufacturer's instruction. Relative transcript abundance was measured by qPCR on a 7900HT Fast Real‐Time PCR System (Applied Biosystems). Transcript levels were normalized to housekeeping transcript β‐2‐globulin. Primers are detailed in Appendix Table S2.
Mitochondrial isolation, iodixanol, and sucrose gradient fractionation
Mitochondria were isolated as previously described (Dalla Rosa et al, 2014). Briefly, HEK293T cells were disrupted by homogenization in hypotonic buffer (20 mM HEPES pH 8, 5 mM KCl, 1.5 mM MgCl2, and 2 mM DTT) and mixed with a mannitol–sucrose buffer to final concentration of 210 mM mannitol, 70 mM sucrose, 20 mM HEPES pH 8, and 2 mM EDTA (1× MSH), prior to purification of mitochondria by differential centrifugation. For iodixanol gradient fractionation, 1,000 g supernatant from lysed mitochondria was loaded onto 20–42.5% discontinuous iodixanol (OptiPrep, Sigma) gradients (Dalla Rosa et al, 2014) and centrifuged at 100,000 g for 14 h at 4°C. Resulting gradients were fractionated into 0.5 ml fractions collected from the bottom of the tube and subjected to Western blot analysis. For the analysis of the mitochondrial ribosome, mitochondria were isolated as above using EDTA‐free buffers (Dalla Rosa et al, 2014) and subsequently lysed for 20 min on ice. Two different lysis buffers used were as follows: the standard buffer containing 260 mM sucrose, 100 mM NaCl, 20 mM MgCl2, 10 mM Tris–HCl pH 7.5, 1% Triton X‐100, 1× protease inhibitor w/o EDTA (Roche), 0.08 U/μl rRNasin (Promega) in DEPC‐treated water and a second buffer containing a lower concentration of salt (50 mM NaCl). Mitochondrial lysates were cleared by centrifugation (10,000 g for 45 min at 4°C) and loaded on 10–30% linear sucrose gradients made in the appropriate lysis buffer. After centrifugation at 71,000 g for 15 h at 4°C, 15 fractions of 750 μl each were collected from the top of the gradient using an automated gradient harvester (Brandel) and analyzed by Western blotting. In the case of LETM1 silencing, the isokinetic sucrose gradient analysis of mitochondrial ribosomes was performed on total lysates.
Immunofluorescence and imaging
Fibroblasts or HeLa cells grown on coverslips were fixed with 4% paraformaldehyde, permeabilized with 0.3% Triton X‐100, and incubated with indicated primary antibodies (Appendix Table S4) overnight at 4°C. For bromouridine (BrU) labeling, cells grown on coverslips were incubated with 5 mM BrU for 60 min prior to fixation. Anti‐BrdU antibody (Appendix Table S4) was used to detect BrU‐labeled RNA. Following washes, cells were incubated for 1 h at RT with secondary antibody after which coverslips were mounted on glass slides over ProLong® Gold Antifade Reagent, which includes 4′,6‐diamidino‐2‐phenylindole (DAPI). Samples were imaged either on a SP5 TCS Inverted Confocal Microscope (Leica Biosystem) using a 63× oil immersion objective with a numerical aperture of 1.4 (63×/1.4 Oil) or a 100× oil immersion objective with a numerical aperture of 1.46 (100×/1.46 Oil) or on Nikon Ti Inverted Confocal Microscope using 60× immersion objective. Z stack of red, green, and blue images using a step size of either 0.3 or 0.125 μm was acquired sequentially and merged using ImageJ. Confocal images were adjusted for brightness/contrast in ImageJ. Images in each figure were processed equally.
Electron microscopy
WHS and control fibroblast cells were fixed in 2% glutaraldehyde/2% paraformaldehyde for 30 min, and then 1% osmium tetroxide for 1 h, using 0.1 M sodium cacodylate buffer pH 7.2, at RT. The samples were then dehydrated and embedded in Epon resin. Sections were stained with ethanolic uranyl acetate and Reynold's lead citrate, and viewed with a JEOL 100EX transmission electron microscopy (TEM). Images were captured with a Gatan Orius 1000 charge‐coupled devices (CCD).
Ketone body supplementation
Fibroblasts were cultured to reach 40–50% confluency, washed once with PBS (Life Technologies), and incubated with DMEM with glutamine, w/o glucose and w/o pyruvate (Life Technologies) supplemented with 0.3 mM β‐hydroxybutyrate (BHB, Cayman Chemical), 10% dialyzed FBS, and 1% PS. The medium was changed every 48 h. Cell density was determined using an IncuCyte Live Cell Imaging System (Essen Instruments), and phase‐contrast images were acquired at 3‐h intervals over a period of 3 weeks and processed automatically.
Statistics
All statistical calculations were performed using GraphPad Prism 7.0. Analysis of mitochondrial protein synthesis and immunoblots was performed using Fiji ImageJ. The non‐parametric tests used were one‐way ANOVA or t‐test with Welch's correction. The former was used to compare more than two independent groups, while the latter was used to compare two independent groups. All error bars are ± standard error of the mean, which is an estimate of the true variation of the samples extrapolated from the standard deviation of the measured values. P‐values < 0.05 were considered to be statistically significant and labeled as follows: *P < 0.05, **P < 0.01, and ***P < 0.001; ns, not statistically significant. Exact P‐values are shown in Appendix Table S5.
The paper explained.
Problem
The mitochondrial inner membrane protein LETM1 is involved in regulating ion homeostasis and mitochondrial volume. Yet, considerable uncertainty remains around its role in organelle function and cellular metabolism, and it is not known whether mitochondrial dysfunction contributes to the pathology in the many cases of Wolf–Hirschhorn syndrome (WHS) where LETM1 is deficient.
Results
Here, we show that LETM1 is a component of mitochondrial nucleoprotein complexes that supports mitochondrial translation. The loss of one copy of LETM1 produces aberrant mitochondrial morphology and DNA disorganization in WHS LETM1 +/− fibroblasts, phenocopying LETM1 gene silencing. The effect on mtDNA is associated with remodeling of nutrient metabolism toward ketone body utilization.
Impact
The findings establish roles for LETM1 in mitochondrial gene expression and mtDNA organization and show that LETM1 insufficiency causes mitochondrial abnormalities in WHS. Moreover, the results show that the arrangement of mitochondrial nucleoids and ribosomes is tied to nutrient availability and if incorrectly configured, nutrient usage by the mitochondria is no longer matched to availability, which can result in impaired organelle function and disease.
Author contributions
Conceptualization: AS; Methodology: AS and RD; Investigation: RD, ALM, AWEJ, IDR, AM, MM, EMAH, and AS; Writing: AS and IJH; Funding Acquisition: AS; and Resources: HH, GM, SL, PND, and MZ.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie Legends
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Source Data for Figure 9
Acknowledgements
The study was funded by a UK Medical Research Council senior non‐clinical fellowship to AS (MC_PC_13029) and by the European Commission (MEET project grant 317433) to AS and IJH. AM and HH are supported by the UK Medical Research Council. SL, PD, GM, and MZ are supported by the Catholic University of Rome. IJH is supported by the Ikerbasque Science Foundation, The Carlos III Health Program, and Biodonostia Research Institute. We thank all the families with the Wolf–Hirschhorn syndrome and the Italian Association for WHS. We would like to thank Professor Wiesner for TFAM antibody.
EMBO Mol Med (2018) 10: e8550
Footnotes
§Correction added online on 2 August 2018 after first online publication: the labels “siD” and “siR1+siD” in Figure 5D have been corrected.
References
- Akman G, Desai R, Bailey LJ, Yasukawa T, Dalla Rosa I, Durigon R, Holmes JB, Moss CF, Mennuni M, Houlden H et al (2016) Pathological ribonuclease H1 causes R‐loop depletion and aberrant DNA segregation in mitochondria. Proc Natl Acad Sci USA 113: E4276–E4285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almalki A, Alston CL, Parker A, Simonic I, Mehta SG, He L, Reza M, Oliveira JM, Lightowlers RN, McFarland R et al (2014) Mutation of the human mitochondrial phenylalanine‐tRNA synthetase causes infantile‐onset epilepsy and cytochrome c oxidase deficiency. Biochem Biophys Acta 1842: 56–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonicka H, Sasarman F, Nishimura T, Paupe V, Shoubridge EA (2013) The mitochondrial RNA‐binding protein GRSF1 localizes to RNA granules and is required for posttranscriptional mitochondrial gene expression. Cell Metab 17: 386–398 [DOI] [PubMed] [Google Scholar]
- Ban‐Ishihara R, Ishihara T, Sasaki N, Mihara K, Ishihara N (2013) Dynamics of nucleoid structure regulated by mitochondrial fission contributes to cristae reformation and release of cytochrome c. Proc Natl Acad Sci USA 110: 11863–11868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauerschmitt H, Mick DU, Deckers M, Vollmer C, Funes S, Kehrein K, Ott M, Rehling P, Herrmann JM (2010) Ribosome‐binding proteins Mdm38 and Mba1 display overlapping functions for regulation of mitochondrial translation. Mol Biol Cell 21: 1937–1944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogenhagen DF, Wang Y, Shen EL, Kobayashi R (2003) Protein components of mitochondrial DNA nucleoids in higher eukaryotes. Mol Cell Proteomics 2: 1205–1216 [DOI] [PubMed] [Google Scholar]
- Bogenhagen DF, Martin DW, Koller A (2014) Initial steps in RNA processing and ribosome assembly occur at mitochondrial DNA nucleoids. Cell Metab 19: 618–629 [DOI] [PubMed] [Google Scholar]
- Buck MD, O'Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, Qiu J, Kretz O, Braas D, van der Windt GJ et al (2016) Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 166: 63–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalla Rosa I, Durigon R, Pearce SF, Rorbach J, Hirst EM, Vidoni S, Reyes A, Brea‐Calvo G, Minczuk M, Woellhaf MW et al (2014) MPV17L2 is required for ribosome assembly in mitochondria. Nucleic Acids Res 42: 8500–8515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalla Rosa I, Camara Y, Durigon R, Moss CF, Vidoni S, Akman G, Hunt L, Johnson MA, Grocott S, Wang L et al (2016) MPV17 loss causes deoxynucleotide insufficiency and slow DNA replication in mitochondria. PLoS Genet 12: e1005779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton RM, Randle PJ, Martin BR (1972) Stimulation by calcium ions of pyruvate dehydrogenase phosphate phosphatase. Biochem J 128: 161–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai R, Frazier AE, Durigon R, Patel H, Jones AW, Dalla Rosa I, Lake NJ, Compton AG, Mountford HS, Tucker EJ et al (2017) ATAD3 gene cluster deletions cause cerebellar dysfunction associated with altered mitochondrial DNA and cholesterol metabolism. Brain 140: 1595–1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmer KS, Fritz S, Fuchs F, Messerschmitt M, Weinbach N, Neupert W, Westermann B (2002) Genetic basis of mitochondrial function and morphology in Saccharomyces cerevisiae . Mol Biol Cell 13: 847–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmer KS, Navoni F, Casarin A, Trevisson E, Endele S, Winterpacht A, Salviati L, Scorrano L (2008) LETM1, deleted in Wolf‐Hirschhorn syndrome is required for normal mitochondrial morphology and cellular viability. Hum Mol Genet 17: 201–214 [DOI] [PubMed] [Google Scholar]
- Doonan PJ, Chandramoorthy HC, Hoffman NE, Zhang X, Cardenas C, Shanmughapriya S, Rajan S, Vallem S, Chen X, Foskett JK et al (2014) LETM1‐dependent mitochondrial Ca2+ flux modulates cellular bioenergetics and proliferation. FASEB J 28: 4936–4949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier AE, Taylor RD, Mick DU, Warscheid B, Stoepel N, Meyer HE, Ryan MT, Guiard B, Rehling P (2006) Mdm38 interacts with ribosomes and is a component of the mitochondrial protein export machinery. J Cell Biol 172: 553–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froschauer E, Nowikovsky K, Schweyen RJ (2005) Electroneutral K+/H+ exchange in mitochondrial membrane vesicles involves Yol027/Letm1 proteins. Biochem Biophys Acta 1711: 41–48 [DOI] [PubMed] [Google Scholar]
- Glancy B, Hartnell LM, Malide D, Yu ZX, Combs CA, Connelly PS, Subramaniam S, Balaban RS (2015) Mitochondrial reticulum for cellular energy distribution in muscle. Nature 523: 617–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes LC, Di Benedetto G, Scorrano L (2011) During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 13: 589–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart L, Rauch A, Carr AM, Vermeesch JR, O'Driscoll M (2014) LETM1 haploinsufficiency causes mitochondrial defects in cells from humans with Wolf‐Hirschhorn syndrome: implications for dissecting the underlying pathomechanisms in this condition. Dis Model Mech 7: 535–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa A, van der Bliek AM (2007) Inverse correlation between expression of the Wolfs Hirschhorn candidate gene Letm1 and mitochondrial volume in C. elegans and in mammalian cells. Hum Mol Genet 16: 2061–2071 [DOI] [PubMed] [Google Scholar]
- Hashimi H, McDonald L, Stribrna E, Lukes J (2013) Trypanosome Letm1 protein is essential for mitochondrial potassium homeostasis. J Biol Chem 288: 26914–26925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Cooper HM, Reyes A, Di Re M, Kazak L, Wood SR, Mao CC, Fearnley IM, Walker JE, Holt IJ (2012a) Human C4orf14 interacts with the mitochondrial nucleoid and is involved in the biogenesis of the small mitochondrial ribosomal subunit. Nucleic Acids Res 40: 6097–6108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Cooper HM, Reyes A, Di Re M, Sembongi H, Litwin TR, Gao J, Neuman KC, Fearnley IM, Spinazzola A et al (2012b) Mitochondrial nucleoid interacting proteins support mitochondrial protein synthesis. Nucleic Acids Res 40: 6109–6121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, Zhao L, Clapham DE (2009) Genome‐wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 326: 144–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, Zhao L, Clish CB, Clapham DE (2013) Letm1, the mitochondrial Ca2+/H+ antiporter, is essential for normal glucose metabolism and alters brain function in Wolf‐Hirschhorn syndrome. Proc Natl Acad Sci USA 110: E2249–E2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdain AA, Koppen M, Wydro M, Rodley CD, Lightowlers RN, Chrzanowska‐Lightowlers ZM, Martinou JC (2013) GRSF1 regulates RNA processing in mitochondrial RNA granules. Cell Metab 17: 399–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehrein K, Schilling R, Moller‐Hergt BV, Wurm CA, Jakobs S, Lamkemeyer T, Langer T, Ott M (2015) Organization of mitochondrial gene expression in two distinct ribosome‐containing assemblies. Cell Rep 10: 843–853 [DOI] [PubMed] [Google Scholar]
- Kolobova E, Tuganova A, Boulatnikov I, Popov KM (2001) Regulation of pyruvate dehydrogenase activity through phosphorylation at multiple sites. Biochem J 358: 69–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Spremulli L (2000) Interaction of mammalian mitochondrial ribosomes with the inner membrane. J Biol Chem 275: 29400–29406 [DOI] [PubMed] [Google Scholar]
- Llorente‐Folch I, Rueda CB, Pardo B, Szabadkai G, Duchen MR, Satrustegui J (2015) The regulation of neuronal mitochondrial metabolism by calcium. J Physiol 593: 3447–3462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matic S, Jiang M, Nicholls TJ, Uhler JP, Dirksen‐Schwanenland C, Polosa PL, Simard ML, Li X, Atanassov I, Rackham O et al (2018) Mice lacking the mitochondrial exonuclease MGME1 accumulate mtDNA deletions without developing progeria. Nat Commun 9: 1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuibban AG, Joza N, Megighian A, Scorzeto M, Zanini D, Reipert S, Richter C, Schweyen RJ, Nowikovsky K (2010) A Drosophila mutant of LETM1, a candidate gene for seizures in Wolf‐Hirschhorn syndrome. Hum Mol Genet 19: 987–1000 [DOI] [PubMed] [Google Scholar]
- Nicholls TJ, Nadalutti CA, Motori E, Sommerville EW, Gorman GS, Basu S, Hoberg E, Turnbull DM, Chinnery PF, Larsson NG et al (2018) Topoisomerase 3alpha is required for decatenation and segregation of human mtDNA. Mol Cell 69: 9–23 e26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols BJ, Denton RM (1995) Towards the molecular basis for the regulation of mitochondrial dehydrogenases by calcium ions. Mol Cell Biochem 149–150: 203–212 [DOI] [PubMed] [Google Scholar]
- Nowikovsky K, Froschauer EM, Zsurka G, Samaj J, Reipert S, Kolisek M, Wiesenberger G, Schweyen RJ (2004) The LETM1/YOL027 gene family encodes a factor of the mitochondrial K+ homeostasis with a potential role in the Wolf‐Hirschhorn syndrome. J Biol Chem 279: 30307–30315 [DOI] [PubMed] [Google Scholar]
- Nowikovsky K, Reipert S, Devenish RJ, Schweyen RJ (2007) Mdm38 protein depletion causes loss of mitochondrial K+/H+ exchange activity, osmotic swelling and mitophagy. Cell Death Differ 14: 1647–1656 [DOI] [PubMed] [Google Scholar]
- Nowikovsky K, Bernardi P (2014) LETM1 in mitochondrial cation transport. Front Physiol 5: 83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott M, Herrmann JM (2010) Co‐translational membrane insertion of mitochondrially encoded proteins. Biochem Biophys Acta 1803: 767–775 [DOI] [PubMed] [Google Scholar]
- Pliss L, Jatania U, Patel MS (2016) Beneficial effect of feeding a ketogenic diet to mothers on brain development in their progeny with a murine model of pyruvate dehydrogenase complex deficiency. Mol Genet Metab Rep 7: 78–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambold AS, Kostelecky B, Elia N, Lippincott‐Schwartz J (2011) Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA 108: 10190–10195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorbach J, Richter R, Wessels HJ, Wydro M, Pekalski M, Farhoud M, Kuhl I, Gaisne M, Bonnefoy N, Smeitink JA et al (2008) The human mitochondrial ribosome recycling factor is essential for cell viability. Nucleic Acids Res 36: 5787–5799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorbach J, Gammage PA, Minczuk M (2012) C7orf30 is necessary for biogenesis of the large subunit of the mitochondrial ribosome. Nucleic Acids Res 40: 4097–4109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ (2008) Analyzing real‐time PCR data by the comparative C(T) method. Nat Protoc 3: 1101–1108 [DOI] [PubMed] [Google Scholar]
- Sofou K, Dahlin M, Hallbook T, Lindefeldt M, Viggedal G, Darin N (2017) Ketogenic diet in pyruvate dehydrogenase complex deficiency: short‐ and long‐term outcomes. J Inherit Metab Dis 40: 237–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spelbrink JN (2010) Functional organization of mammalian mitochondrial DNA in nucleoids: history, recent developments, and future challenges. IUBMB Life 62: 19–32 [DOI] [PubMed] [Google Scholar]
- Tamai S, Iida H, Yokota S, Sayano T, Kiguchiya S, Ishihara N, Hayashi J, Mihara K, Oka T (2008) Characterization of the mitochondrial protein LETM1, which maintains the mitochondrial tubular shapes and interacts with the AAA‐ATPase BCS1L. J Cell Sci 121: 2588–2600 [DOI] [PubMed] [Google Scholar]
- Tsai MF, Jiang D, Zhao L, Clapham D, Miller C (2014) Functional reconstitution of the mitochondrial Ca2+/H+ antiporter Letm1. J Gen Physiol 143: 67–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu YT, Barrientos A (2015) The human mitochondrial DEAD‐box protein DDX28 resides in RNA granules and functions in mitoribosome assembly. Cell Rep 10: 854–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai T, Langer T (2016) Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab 27: 105–117 [DOI] [PubMed] [Google Scholar]
- Wanschers BF, Szklarczyk R, Pajak A, van den Brand MA, Gloerich J, Rodenburg RJ, Lightowlers RN, Nijtmans LG, Huynen MA (2012) C7orf30 specifically associates with the large subunit of the mitochondrial ribosome and is involved in translation. Nucleic Acids Res 40: 4040–4051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Hulver MW, McMillan RP, Cline MA, Gilbert ER (2014) The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr Metab 11: 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zollino M, Lecce R, Fischetto R, Murdolo M, Faravelli F, Selicorni A, Butte C, Memo L, Capovilla G, Neri G (2003) Mapping the Wolf‐Hirschhorn syndrome phenotype outside the currently accepted WHS critical region and defining a new critical region, WHSCR‐2. Am J Hum Genet 72: 590–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie Legends
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Source Data for Figure 9