

Keywords: nerve regeneration, mitogen-activated protein kinase phosphatase 1, c-Jun N-terminal kinase signaling pathway, Alzheimer's disease, neurons, dementia, apoptosis, RNA interference, lentivirus, inflammation, oxidative stress, neural regeneration
Abstract
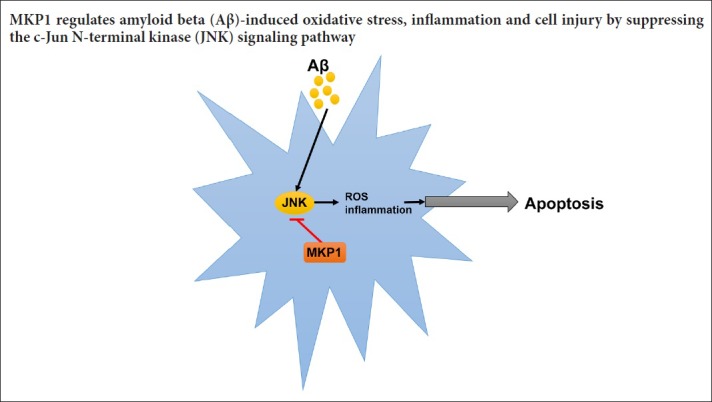
The mitogen-activated protein kinase (MAPK) signaling pathway plays an important role in the regulation of cell growth, proliferation, differentiation, transformation and death. Mitogen-activated protein kinase phosphatase 1 (MKP1) has an inhibitory effect on the p38MAPK and JNK pathways, but it is unknown whether it plays a role in Aβ-induced oxidative stress and neuronal inflammation. In this study, PC12 cells were infected with MKP1 shRNA, MKP1 lentivirus or control lentivirus for 12 hours, and then treated with 0.1, 1, 10 or 100 μM amyloid beta 42 (Aβ42). The cell survival rate was measured using the cell counting kit-8 assay. MKP1, tumor necrosis factor-alpha (TNF-α) and interleukin-1β (IL-1β) mRNA expression levels were analyzed using quantitative real time-polymerase chain reaction. MKP1 and phospho-c-Jun N-terminal kinase (JNK) expression levels were assessed using western blot assay. Reactive oxygen species (ROS) levels were detected using 2′,7′-dichlorofluorescein diacetate. Mitochondrial membrane potential was measured using flow cytometry. Superoxide dismutase activity and malondialdehyde levels were evaluated using the colorimetric method. Lactate dehydrogenase activity was measured using a microplate reader. Caspase-3 expression levels were assessed by enzyme-linked immunosorbent assay. Apoptosis was evaluated using the terminal deoxynucleotidyl transferase dUTP nick end labeling method. MKP1 overexpression inhibited Aβ-induced JNK phosphorylation and the increase in ROS levels. It also suppressed the Aβ-induced increase in TNF-α and IL-1β levels as well as apoptosis in PC12 cells. In contrast, MKP1 knockdown by RNA interference aggravated Aβ-induced oxidative stress, inflammation and cell damage in PC12 cells. Furthermore, the JNK-specific inhibitor SP600125 abolished this effect of MKP1 knockdown on Aβ-induced neurotoxicity. Collectively, these results show that MKP1 mitigates Aβ-induced apoptosis, oxidative stress and neuroinflammation by inhibiting the JNK signaling pathway, thereby playing a neuroprotective role.
Introduction
The economic and social burden of Alzheimer's disease (AD) is an important public health problem, particularly as the prevalence of AD is increasing along with the aging population. The pathological features of AD include neuronal loss, intracellular neurofibrillary tangles and extracellular amyloid beta (Aβ) protein deposition (Goedert, 2015; Amit et al., 2017; Liu et al., 2017). Aβ plays a pivotal role in AD, and aggregated Aβ is observed in brain tissue in the early stage of AD (Puzzo et al., 2015; Golde, 2016; Müller et al., 2017; VanItallie, 2017). Aβ induces neuronal apoptosis, neuroinflammation and oxidative stress by regulating multiple signaling pathways, including mitogen-activated protein kinase (MAPK), protein kinase C and phosphoinositide 3-kinase/Akt (Smith et al., 2006; Petersen et al., 2007; Shafi, 2016; Ridler, 2017). However, the molecular mechanisms by which Aβ regulates these signaling pathways remain largely unknown. Therefore, studies on the complex signaling networks that are altered by Aβ are urgently needed to elucidate the pathogenesis of AD and aid in the development of effective clinical treatments for the disease.
The MAPK signaling pathway plays a critical role in the regulation of cell growth, proliferation, differentiation, transformation and death (Liu et al., 2012; Li et al., 2014; Thouverey and Caverzasio, 2015; Zhou et al., 2015; Yao et al., 2017). In mammalian cells, there are four MAPK signal transduction pathways, including the c-Jun N-terminal kinase (JNK, also known as stress-activated MAPK), p38 kinase (p38 MAPK), extracellular signal-regulated protein kinase (ERK) and large mitogen-activated protein kinase (BMK1/ERK5) pathways (Tiedje et al., 2014; Zhou et al., 2015; Lanna et al., 2017; Latrasse et al., 2017). Numerous studies have shown that the MAPK signaling pathway plays a major role in the pathophysiology of AD (Giraldo et al., 2014; Petrov et al., 2016; Lee and Kim, 2017). McDonald et al. (1998) found that Aβ activates the p38 MAPK pathway in rat glial cells. p38 MAPK activation in the hippocampus and cortical region is associated with neurofibrillary tangles, senile plaques, vacuolar degeneration and dystrophic neurites in post-mortem AD brain tissue (Zhu et al., 2000; Johnson and Bailey, 2003). Moreover, Aβ42 injection into the basal ganglia in rats for 7 days leads to significant activation of p38 MAPK, accompanied by the activation of glial cells and neuroinflammation (Giovannini et al., 2002), whereas inhibition of the p38 MAPK pathway alleviates Aβ-induced neurotoxicity (Xu et al., 2014). Similarly, JNK signaling is activated by Aβ and is involved in Aβ-induced human neuronal damage (Troy et al., 2001; Tare et al., 2011). In addition, it has been reported that JNK activation results in excessive Aβ deposition, which in turn activates a positive feedback loop that accelerates the progression of AD (Zhu et al., 2002). These findings indicate that the p38 MAPK and JNK signaling pathways play important roles in the pathogenesis of AD. However, the molecular mechanisms underlying these processes are largely unknown.
MAPK phosphatase 1 (MKP1) is a member of the MAPK phosphatase family and is the main negative regulator of p38 MAPK, JNK and ERK (Chi and Flavell, 2008; Lawan et al., 2013; Low and Zhang, 2016). MKP1 is involved in oxidative stress, inflammatory reactions and apoptosis, and it plays a key role in the occurrence and progression of many human diseases by regulating the MAPK signaling pathway (Wancket et al., 2012; Ma et al., 2015; Moosavi et al., 2017). However, it is unknown whether MKP1 plays a role in Aβ-induced oxidative stress and inflammation in neurons.
In this study, we investigated the role of MKP1 in Aβ-induced neurotoxicity. To this end, we assessed the production of reactive oxygen species (ROS), expression of tumor necrosis factor-alpha (TNF-α) and interleukin-1 beta (IL-1β), and apoptosis in Aβ-treated PC12 cells. Furthermore, we examined the effect of pharmacological inhibition of JNK and the effects of MPK1 overexpression and siRNA-mediated silencing.
Materials and Methods
Cell culture
PC12 cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum in a 5% CO2 incubator at 37°C. The experiments were performed in accordance with the “Recommendations for the Conduct, Reporting, Editing, and Publication of Scholarly Work in Medical Journals”, published by the International Committee of Medical Journal Editors (http://www.icmje.org/recommendations/).
Aβ42 preparation and cell treatment
Aβ42 peptide (Sigma-Aldrich, St. Louis, MO, USA) was incubated in Tris/HCl (50 mM, pH 7.4) at a concentration of 1 mg/mL at room temperature for a minimum of 2 days for aggregation. For the experiments, Aβ42 solution was diluted and added to culture media at different concentrations (0.1, 1, 10 or 100 μM).
Lentivirus infection
PC12 cells were infected with MKP1 short hairpin RNA (shRNA), MKP1 lentiviral particles or control lentivirus (Hanheng, Shanghai, China) at a multiplicity of infection of 10 in 5 mg/mL Polybrene (Sigma-Aldrich) for 12 hours and treated with 10 µg/mL puromycin (Santa Cruz Biotechnology, Dallas, TX, USA) for 21 days, according to the manufacturer's protocol.
Cell counting kit-8 assay
Cells were plated onto 96-well plates (4000 cells/well) the day before treatment. After treatment with different concentrations (0.1, 1, 10 or 100 μM) of Aβ, 10 μL of cell counting kit-8 reagent (Sigma-Aldrich) was added to each well and cultured for 2 hours. The absorbance at 450 nm was read on a microplate reader (Bio-Rad, Hercules, CA, USA).
Quantitative real time-polymerase chain reaction (qRT-PCR)
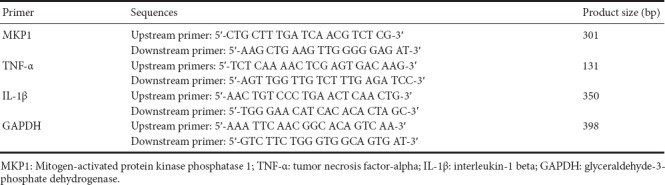
Total RNA was extracted with Trizol (Invitrogen, Carlsbad, CA, USA), and cDNA synthesis was performed using the Superscript III Reverse Transcriptase kit (Invitrogen). The relative levels of target gene mRNA to control GAPDH mRNA were determined by qRT-PCR using TransStartTM SYBR Green qPCR Supermix (Transgene, Beijing, China) on the ABI 7500 PCR instrument (ABI, Foster City, CA, USA). The data were analyzed with the 2-ΔΔCt method. The primer sequences are listed in Table 1.
Table 1.
Primer sequences
Western blot assay
Total cellular protein was extracted with RIPA lysis buffer (Cell Signaling Technology, Boston, MA, USA), and the concentration of total protein was determined by the BCA method (Song et al., 2017). Total protein, 15 μg, was separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and electrotransferred onto polyvinylidene fluoride membranes. Membranes were blocked with 5% bovine serum albumin (Boster, Wuhan, China) at room temperature for 2 hours and immunoblotted overnight at 4°C with primary rabbit anti-rat polyclonal antibodies (1:1000; Abcam) against MKP1, p-JNK (JNK1 + JNK2 + JNK3) and GAPDH, followed by incubation with horseradish peroxidase-conjugated goat anti-rabbit polyclonal secondary antibodies (1:4000; Abcam). After extensive washing, protein bands were visualized by an ECL plus chemiluminescence kit (Beyotime Institute of Biotechnology, Haimen, China). Densitometric analysis was performed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
2′,7′-Dichlorofluorescein diacetate (DCFH-DA) detection of ROS
To measure intracellular ROS levels, 2 × 105 PC12 cells were seeded into 6-well plates. Following treatment, the cells were treated with 20 nM DCFH-DA (Sigma-Aldrich) for 30 minutes. The green fluorescence signal was observed and photographed under a fluorescence microscope (Olympus, Tokyo, Japan).
Detection of mitochondrial membrane potential
Following treatment, the cells were incubated with 10 μg/mL JC-1 (Sigma-Aldrich) at 4°C in the dark for 30 minutes. After washing with PBS, the cells were subjected to flow cytometry with an excitation wavelength of 520 nm and an emission wavelength of 596 nm. The median fluorescence intensity was recorded by the flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Assessment of superoxide dismutase (SOD) activity and malondialdehyde (MDA) levels
Intracellular SOD activity and MDA levels were measured using SOD and MDA assay kits (Jiancheng, Nanjing, China) in accordance with the manufacturer's instructions. In brief, after cell lysis and centrifugation at 12,000 × g for 10 minutes, the supernatant was added to test reagents and incubated at 95°C in a water bath for 40 minutes. After cooling for 10 minutes, the reaction mixture was centrifuged at 4000 × g for 10 minutes, and the OD of the supernatant was measured at a wavelength of 532 nm. The SOD and MDA levels were calculated according to the OD values.
Detection of lactate dehydrogenase (LDH) activity
To assess damage caused by Aβ to cells, LDH activity was measured with the LDH detection kit (Jiancheng) as previously described (Song et al., 2016). Briefly, after Aβ treatment, 20 μL of the cell culture supernatant was mixed thoroughly with 25 μL of detection buffer and 5 μL of coenzyme I, and incubated in a water bath at 37°C for 15 minutes. Thereafter, 25 μL of 2′,4′-dinitrophenylhydrazine was added to the mixture and incubated at 37°C for 15 minutes. Then, 5 minutes after the addition of 250 μL of 0.4 M NaOH, the absorbance was measured at 450 nm with a microplate reader.
Enzyme-linked immunosorbent assay (ELISA)
Cells were added to 500 μL of cold carbonate buffer (100 mM Na2 CO3, 50 mM NaCl with pH 11.5) with protease inhibitors and homogenized by sonication. The cell lysate was centrifuged at 12,000 × g for 45 minutes and the supernatant was used for determination of caspase-3 content with the Caspase-3 ELISA kit (Abcam, Hong Kong, China) using a microplate reader.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining
Cells were fixed with 4% paraformaldehyde at room temperature (25°C) for 20 minutes. After washing three times with PBS, the cells were permeabilized with 1% Triton X-100 and blocked with 3% H2O2 at room temperature for 10 minutes. After three washes with PBS, TdT enzyme reaction solution (Coolrun, Shenzheng, China) containing TRITC-5-dUTP and TdT enzyme was added to the cells and incubated in the dark at 37°C for 60 minutes. Following nuclear staining with 5 μg/mL DAPI, the cells were observed by fluorescence microscopy.
Statistical analysis
SPSS 17.0 for Windows (SPSS, Chicago, IL, USA) was used for data processing. The data are expressed as the mean ± SD. Intergroup comparison was performed using analysis of variance (with α = 0.05).
Results
Aβ42 downregulated MKP1 expression in PC12 cells
To assess the effect of Aβ42 on the viability of PC12 cells, the cells were treated with different concentrations of Aβ42 for 24 hours, and cell viability was assessed. Aβ42 treatment resulted in the loss of cell viability in a dose-dependent manner (Figure 1A). To evaluate the effect of Aβ on MKP1 mRNA and protein expression in PC12 cells, 10 μM Aβ42 was added to the cell culture medium, and MKP1 mRNA and protein expression was assessed by qRT-PCR and western blot assay at different time points. qRT-PCR showed that MKP1 mRNA expression was significantly downregulated 6 hours after Aβ42 addition (P < 0.05), with the lowest expression at 12 hours (P < 0.01; Figure 1B). Western blot assay showed that MKP1 protein expression was significantly downregulated at 12 hours after Aβ42 exposure (P < 0.01), with the lowest expression at 18 hours (P < 0.01) (Figure 1C). These results demonstrate that Aβ42 downregulates MKP1 expression in PC12 cells in a time and concentration-dependent manner.
Figure 1.
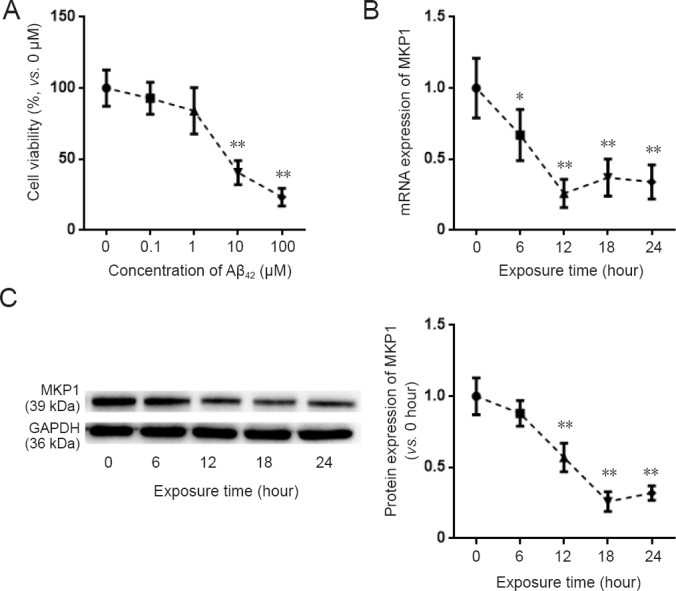
Effect of Aβ42 on MKP1 expression in PC12 cells.
(A) PC12 cells were treated with the indicated concentrations of Aβ42 for 24 hours. Cell viability was assessed with the cell counting kit-8 assay. (B) PC12 cells were treated with 10 μM Aβ42. At the indicated time points, total RNA was extracted for quantitative real time-polymerase chain reaction for MKP1 mRNA expression with GAPDH as the internal control. The result is expressed as a percentage of the value at 0 hours. (C) PC12 cells were treated with 10 μM Aβ42. At the indicated time points, total protein was extracted for immunoblotting of MKP1 protein with GAPDH as the loading control. The relative expression of MKP1 to GAPDH was assessed by densitometric analysis using ImageJ software. MKP1 expression is shown relative to that at time 0. Data are expressed as the mean ± SD (six separate experiments for each time point). Intergroup comparison was performed using analysis of variance. *P < 0.05, **P < 0.01, vs. control group (0 μM Aβ42 or value at 0 hours). Aβ42: Amyloid beta 42; MKP1: mitogen-activated protein kinase phosphatase 1.
MKP1 suppressed Aβ42-induced oxidative stress
To examine the role of MKP1 in Aβ42-induced oxidative stress, we measured intracellular ROS levels in PC12 cells with the DCFH-DA assay. Aβ42 treatment resulted in an increase in ROS generation compared with untreated control cells. In comparison, Aβ42 treatment of PC12 cells with stable MKP1 knockdown with shRNA resulted in substantially higher ROS production compared with control cells. Conversely, cells with stable MKP1 overexpression showed reduced ROS generation compared with control cells (Figure 2A).
Figure 2.

Effect of MKP1 on Aβ42-induced oxidative stress in PC12 cells.
PC12 cells were treated with (Aβ42) or without 10 μM Aβ42 (Control), and PC12 cells stably expressing MKP1 shRNA (Aβ42 + MKP1 KD) or MKP1 lentiviral particles (Aβ42 + MKP1) were treated with 10 μM Aβ42 for 24 hours. (A) ROS levels were detected with DCFH-DA and analyzed by fluorescence microscopy. Scale bars: 50 μm. (B) The mitochondrial membrane potential (ΔΨm) was determined by staining with JC-1 coupled with flow cytometry. The left spectral graph shows the representative median fluorescence intensities for the various groups. (C) SOD activity was assessed with the SOD kit. (D) MDA level was determined with the MDA kit. Data are expressed as the mean ± SD (six separate experiments for each group). Intergroup comparison was performed with analysis of variance. #P < 0.05, ##P < 0.01, vs. Aβ42 group. MKP1: Mitogen-activated protein kinase phosphatase 1; ROS: reactive oxygen species; Aβ42: amyloid beta 42; DCFH-DA: 2′,7′-dichlorofluorescein diacetate; SOD: superoxide dismutase; MDA: malondialdehyde; MFI: mean fluorescence indensity.
Mitochondrial membrane potential (ΔΨm) is a cellular indicator of oxidative stress, and ROS reduce ΔΨm (Vayssier-Taussat et al., 2002). We found that Aβ42 exposure led to a reduction in ΔΨm in PC12 cells, which was significantly enhanced by MKP1 knockdown (P < 0.05), and significantly suppressed by MKP1 overexpression (P < 0.05; Figure 2B). SOD activity and MDA levels in PC12 cells were also measured. Aβ42 treatment reduced SOD activity, and this effect of the peptide was significantly enhanced by MKP1 knockdown (P < 0.01) and significantly suppressed by MKP1 overexpression (P < 0.05; Figure 2C). In addition, Aβ42 significantly increased MDA levels (P < 0.05). However, MKP1 knockdown had no impact on this Aβ42-mediated increase in MDA levels (P > 0.05), whereas MKP1 overexpression significantly diminished the Aβ42-mediated increase in MDA levels (P < 0.01; Figure 2D). These results demonstrate that MKP1 inhibits Aβ42-induced oxidative stress in PC12 cells.
MKP1 prevented Aβ42-induced neuroinflammation
To assess the effect of MKP1 on Aβ-induced neurotoxicity, PC12 cells were treated without (Control) or with 10 μM Aβ42 (Aβ42) for 24 hours. Furthermore, PC12 cells stably expressing MKP1 shRNA (MKP1 KD + Aβ42) or MKP1 (MKP1 + Aβ) were treated with 10 μM Aβ42 for 24 hours. phospho-JNK (p-JNK) levels were then measured by western blot assay, and mRNA levels of the inflammatory cytokines TNF-α and IL-1β were assessed by qRT-PCR. As shown in Figure 3A, Aβ42 increased the p-JNK signal, and this effect was significantly enhanced by MKP1 knockdown (P < 0.05) and significantly diminished by MKP1 overexpression (P < 0.05). Aβ42 increased mRNA levels of TNF-α and IL-1β, and MKP1 knockdown bolstered this effect of the peptide (P < 0.05). In contrast, MKP1 overexpression significantly diminished the Aβ42-mediated increase in expression of these inflammatory cytokines (P < 0.05; Figure 3B).
Figure 3.

Effect of MKP1 on Aβ42-induced neuroinflammation in PC12 cell culture.
(A) PC12 cells were treated without (Control) or with 10 μM Aβ42 (Aβ42), and PC12 cells stably expressing MKP1 shRNA (Aβ42 + MKP1 KD) or MKP1 lentiviral particles (Aβ42 + MKP1) were treated with 10 μM Aβ42 for 24 hours. Total protein was extracted for western blot assay for p-JNK with GAPDH as loading control. The relative expression of p-JNK to GAPDH was assessed by densitometric analysis using ImageJ software. (B) Cells were treated as in A. Total RNA was extracted for quantitative real time-polymerase chain reaction for TNF-α and IL-1β mRNA with GAPDH as internal control. Data are representative images or are expressed as the mean ± SD (six separate experiments for each group of cells). Intergroup comparison was performed with analysis of variance. #P < 0.05, vs. Aβ42 group. MKP1: Mitogen-activated protein kinase phosphatase 1; Aβ42: amyloid beta 42; p-JNK: phospho-c-Jun N-terminal kinase; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; TNF-α: tumor necrosis factor-alpha; IL-1β: interleukin-1 beta.
MKP1 alleviated Aβ42-induced neuronal apoptosis
To evaluate the effect of MKP1 on Aβ42-induced cellular injury, normal PC12 cells and PC12 cells stably expressing MKP1 shRNA or MKP1 were treated with 10 μM Aβ42 for 24 hours, and cell viability was assessed with the cell counting kit-8 assay and by measuring the activity of LDH released into the medium. Aβ42 reduced the viability of PC12 cells, and this effect of the peptide was significantly enhanced by MKP1 knockdown (P < 0.05) and significantly suppressed by MKP1 overexpression (P < 0.01; Figure 4A). Moreover, Aβ42 increased LDH release. MKP1 knockdown enhanced Aβ42-induced LDH release (P < 0.05), whereas MKP1 overexpression diminished Aβ42-induced LDH release (P < 0.01; Figure 4B). In addition, Aβ42 increased caspase-3 levels, which was significantly enhanced by MKP1 knockdown (P < 0.05) and significantly diminished by MKP1 overexpression (P < 0.05; Figure 4C). Furthermore, TUNEL staining demonstrated that Aβ42 treatment led to the apoptosis of PC12 cells. The number of TUNEL-positive cells was significantly increased by MKP1 knockdown (P < 0.05) and significantly decreased by MKP1 overexpression (P < 0.01; Figure 4D).
Figure 4.

Effect of MKP1 on Aβ42-induced neurotoxicity towards PC12 cells.
PC12 cells were treated without (Control) or with 10 μM Aβ42 (Aβ42), and PC12 cells stably expressing MKP1 shRNA (Aβ42 + MKP1 KD) or MKP1 (Aβ42 + MKP1) were treated with 10 μM Aβ42. Then, 24 hours later, cell viability was assessed using the cell counting kit-8 assay (A), the LDH activity assay (B), intracellular caspase-3 content (C), and TUNEL staining (D). Blue fluorescence indicates normal (non-apoptotic) nuclei, and purple fluorescence indicates TUNEL-positive (apoptotic) nuclei. Data are representative images or are expressed as the mean ± SD (six separate experiments for each group). Intergroup comparison was performed with analysis of variance. #P < 0.05, ##P < 0.01, vs. Aβ42 group. MKP1: Mitogen-activated protein kinase phosphatase 1; Aβ42: amyloid beta 42; LDH: lactate dehydrogenase; TUNEL: terminal deoxynucleotidyl transferase dUTP nick end labeling.
Inhibition of JNK abolished the effect of MKP1 knockdown on Aβ42-induced neurotoxicity
The preceding results show that MKP1 attenuates Aβ42-induced oxidative stress, inflammation and cellular injury. To explore the role of the JNK pathway in the neuroprotective action of MKP1, PC12 cells stably expressing MKP1 shRNA were treated with or without the JNK-specific inhibitor SP6001250 (2 μM; Sigma-Aldrich) in the presence of 10 μM Aβ42 for 24 hours, and cell viability, oxidative stress and apoptosis were assessed. SP600125 abrogated the MKP1 knockdown-induced increase in ROS generation and mitochondrial membrane potential. It also increased SOD activity and lowered MDA levels in PC12 cells (Figure 5A). In addition, SP600125 inhibited the MKP1 knockdown-induced upregulation of TNF-α and IL-1β mRNA expression (Figure 5B). Furthermore, SP600125 diminished the MKP1 knockdown-induced loss of cell viability, and alleviated the MKP1 knockdown-mediated increase in LDH release and apoptosis (Figure 5C).
Figure 5.

Inhibition of JNK suppressed the aggravating effect of MKP1 knockdown on Aβ-induced neurotoxicity in PC12 cells.
(A) PC12 cells stably expressing MKP1 shRNA were treated without (Control) or with 2 μM of the JNK specific inhibitor SP6001250 in the presence of 10 μM Aβ42 for 24 hours. DCFH-DA fluorescence, mitochondrial membrane potential (ΔΨm), SOD activity and MDA levels were then analyzed. (B) Cells were treated as in A, and total RNA was extracted for quantitative real time-polymerase chain reaction for TNF-α and IL-1β mRNA with GAPDH as internal control. (C) Cells were treated as in A, and cell viability was assessed using the cell counting kit-8 assay, LDH activity and TUNEL staining. Data are representative images or are expressed as the mean ± SD (six separate experiments for each group). Intergroup comparison was performed using analysis of variance. *P < 0.05, **P < 0.01, vs. Control group. JNK: c-Jun N-terminal kinase; MKP1: mitogen-activated protein kinase phosphatase 1; Aβ: amyloid beta; DCFH-DA: 2′,7′-dichlorofluorescein diacetate; SOD: superoxide dismutase; MDA: malondialdehyde; TNF-α: tumor necrosis factor-alpha; IL-1β: interleukin-1 beta; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; LDH: lactate dehydrogenase; TUNEL: terminal deoxynucleotidyl transferase dUTP nick end labeling.
Discussion
In the early phase of AD, Aβ accumulates in brain tissue and induces activation of MAPK signaling pathways, the generation of ROS and inflammatory reactions, ultimately leading to neuronal apoptosis (Origlia et al., 2009; Ghavami et al., 2014; Jazvinšćak Jembrek et al., 2015; González-Reyes et al., 2016; Jarosz-Griffiths et al., 2016). These phenomena have been verified in AD animal models and in vitro experiments; however, the mechanisms by which Aβ activates the MAPK signaling pathway remained unknown.
In this study, Aβ42 neurotoxicity towards PC12 cells was concentration-dependent, and it decreased both MKP1 mRNA and protein expression. A previous study showed that MKP1 expression is downregulated in the brain tissue of patients with Huntington's disease (Taylor et al., 2013), but it was unknown whether MKP1 expression is affected in AD. Our results revealed that MKP1 expression is downregulated following Aβ exposure, suggesting that MKP1 may be involved in the pathogenesis of AD.
There is substantial evidence that oxidative stress induced by ROS overproduction combined with the low antioxidative capacity of cells plays an important role in AD (Aliev et al., 2014; Luque-Contreras et al., 2014; Ganguly et al., 2017; Rojas-Gutierrez et al., 2017). Excessive oxidative stress leads to lipid peroxidation of the cell and organellar membranes, thereby affecting the function of nerve cells (Shichiri, 2014; Di Domenico et al., 2017). Moreover, oxidative stress results in nitration of proteins and damage to nucleic acids (Stepien et al., 2017; Wu and Tang, 2018). These combined negative effects eventually lead to the functional impairment or loss of neurons. Aβ catalyzes the generation of ROS via the α-helical structure, with evidence demonstrating that the oxidative stress in the brains of AD patients and animal models is mainly produced by Aβ (Prasad and Bondy, 2014). In this study, Aβ induced the generation of ROS in PC12 cells. Intriguingly, ROS generation in PC12 cells was substantially increased after MKP1 knockdown, while it was abolished by MKP1 overexpression. Moreover, ROS reduce ΔΨm, a biological indicator of oxidative stress. We found that MKP1 knockdown enhanced, while MKP1 overexpression inhibited the Aβ42-induced decrease in ΔΨm. In addition, MKP1 knockdown enhanced, while MKP1 overexpression inhibited the Aβ42-induced decrease in SOD activity in PC12 cells. Furthermore, MKP1 overexpression prevented the increase in MDA levels induced by Aβ42. These results indicate that MKP1 plays an important role in Aβ-induced oxidative stress in neurons.
Chronic inflammation in the brain is another important pathological feature of AD (Bagyinszky et al., 2017; Fraga et al., 2017; Shamim and Laskowski, 2017). Epidemiological studies have shown that nonsteroidal anti-inflammatory drugs significantly reduce the incidence of AD, and nonsteroidal anti-inflammatory drugs and corticosteroids reduce Aβ deposition and suppress the activation of glial cells, as well as the release of inflammatory factors and free radicals in the brains of AD animal models (Doost Mohammadpour et al., 2015; Heneka et al., 2015). JNK plays an important role in inflammation (Cano and Mahadevan, 1995) and is involved in the regulation of transcription of inflammatory genes via phosphorylation of the downstream target c-Jun (Coffey, 2014). In this study, we demonstrated that Aβ promotes JNK activation. Furthermore, MKP1 modulated the Aβ-induced activation of the JNK pathway, accompanied with changes in the expression of the inflammatory cytokines TNF-α and IL-1β. JNK is also involved in apoptosis (Chen, 2012), and the inhibition of the JNK pathway alleviates Aβ-induced neuronal apoptosis (Bozyczko-Coyne et al., 2001). In this study, the JNK specific inhibitor SP600125 prevented the MKP1 knockdown-induced increase in ROS generation. It also reduced oxidative stress and the generation of inflammatory factors, and it prevented apoptosis caused by Aβ. These findings suggest that JNK modulates the action of MKP1 in Aβ-induced toxicity.
There are some limitations to this study. The mechanisms by which Aβ downregulates MKP1 need to be further clarified. Furthermore, an in vivo study should be conducted to evaluate the neuroprotective effects of MKP1 in AD.
In summary, Aβ downregulates MKP1 expression. MKP1, in turn, alleviates Aβ-induced oxidative stress, inflammation and cellular injury by suppressing the JNK signaling pathway. Our findings provide insight into the mechanisms by which the MAPK signaling pathway is activated by Aβ. The MKP1/JNK signaling axis may be a promising therapeutic target for the development of novel drugs for AD.
Footnotes
Conflicts of interest: The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Financial support: None.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
(Copyedited by Patel B, Raye W, Yu J, Li CH, Qiu Y, Song LP, Zhao M)
References
- Aliev G, Priyadarshini M, Reddy VP, Grieg NH, Kaminsky Y, Cacabelos R, Ashraf GM, Jabir NR, Kamal MA, Nikolenko VN, Zamyatnin AA, Jr, Benberin VV, Bachurin SO. Oxidative stress mediated mitochondrial and vascular lesions as markers in the pathogenesis of Alzheimer disease. Curr Med Chem. 2014;21:2208–2217. doi: 10.2174/0929867321666131227161303. [DOI] [PubMed] [Google Scholar]
- Amit T, Bar-Am O, Mechlovich D, Kupershmidt L, Youdim MBH, Weinreb O. The novel multitarget iron chelating and propargylamine drug M30 affects APP regulation and processing activities in Alzheimer's disease models. Neuropharmacology. 2017;123:359–367. doi: 10.1016/j.neuropharm.2017.05.026. [DOI] [PubMed] [Google Scholar]
- Bagyinszky E, Giau VV, Shim K, Suk K, An SSA, Kim S. Role of inflammatory molecules in the Alzheimer's disease progression and diagnosis. J Neurol Sci. 2017;376:242–254. doi: 10.1016/j.jns.2017.03.031. [DOI] [PubMed] [Google Scholar]
- Bozyczko-Coyne D, O’Kane TM, Wu ZL, Dobrzanski P, Murthy S, Vaught JL, Scott RW. CEP-1347/KT-7515, an inhibitor of SAPK/JNK pathway activation, promotes survival and blocks multiple events associated with Abeta-induced cortical neuron apoptosis. J Neurochem. 2001;77:849–863. doi: 10.1046/j.1471-4159.2001.00294.x. [DOI] [PubMed] [Google Scholar]
- Cano E, Mahadevan LC. Parallel signal processing among mammalian MAPKs. Trends Biochem Sci. 1995;20:117–122. doi: 10.1016/s0968-0004(00)88978-1. [DOI] [PubMed] [Google Scholar]
- Chen F. JNK-induced apoptosis, compensatory growth, and cancer stem cells. Cancer Res. 2012;72:379–386. doi: 10.1158/0008-5472.CAN-11-1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi H, Flavell RA. Acetylation of MKP-1 and the control of inflammation. Sci Signal. 2008;1:pe44. doi: 10.1126/scisignal.141pe44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey ET. Nuclear and cytosolic JNK signalling in neurons. Nat Rev Neurosci. 2014;15:285–299. doi: 10.1038/nrn3729. [DOI] [PubMed] [Google Scholar]
- Di Domenico F, Tramutola A, Butterfield DA. Role of 4-hydroxy-2-nonenal (HNE) in the pathogenesis of alzheimer disease and other selected age-related neurodegenerative disorders. Free Radic Biol Med. 2017;111:253–261. doi: 10.1016/j.freeradbiomed.2016.10.490. [DOI] [PubMed] [Google Scholar]
- Doost Mohammadpour J, Hosseinmardi N, Janahmadi M, Fathollahi Y, Motamedi F, Rohampour K. Non-selective NSAIDs improve the amyloid-beta-mediated suppression of memory and synaptic plasticity. Pharmacol Biochem Behav. 2015;132:33–41. doi: 10.1016/j.pbb.2015.02.012. [DOI] [PubMed] [Google Scholar]
- Fraga VG, Carvalho MDG, Caramelli P, de Sousa LP, Gomes KB. Resolution of inflammation, n-3 fatty acid supplementation and Alzheimer disease: A narrative review. J Neuroimmunol. 2017;310:111–119. doi: 10.1016/j.jneuroim.2017.07.005. [DOI] [PubMed] [Google Scholar]
- Ganguly G, Chakrabarti S, Chatterjee U, Saso L. Proteinopathy, oxidative stress and mitochondrial dysfunction: cross talk in Alzheimer's disease and Parkinson's disease. Drug Des Devel Ther. 2017;11:797–810. doi: 10.2147/DDDT.S130514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghavami S, Shojaei S, Yeganeh B, Ande SR, Jangamreddy JR, Mehrpour M, Christoffersson J, Chaabane W, Moghadam AR, Kashani HH, Hashemi M, Owji AA, Los MJ. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol. 2014;112:24–49. doi: 10.1016/j.pneurobio.2013.10.004. [DOI] [PubMed] [Google Scholar]
- Giovannini MG, Scali C, Prosperi C, Bellucci A, Vannucchi MG, Rosi S, Pepeu G, Casamenti F. Beta-amyloid-induced inflammation and cholinergic hypofunction in the rat brain in vivo: involvement of the p38MAPK pathway. Neurobiol Dis. 2002;11:257–274. doi: 10.1006/nbdi.2002.0538. [DOI] [PubMed] [Google Scholar]
- Giraldo E, Lloret A, Fuchsberger T, Vina J. Abeta and tau toxicities in Alzheimer's are linked via oxidative stress-induced p38 activation: protective role of vitamin E. Redox Biol. 2014;2:873–877. doi: 10.1016/j.redox.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M. NEURODEGENERATION. Alzheimer's and Parkinson's diseases: The prion concept in relation to assembled Abeta, tau, and alpha-synuclein. Science. 2015;349:1255555. doi: 10.1126/science.1255555. [DOI] [PubMed] [Google Scholar]
- Golde TE. Alzheimer disease: Host immune defence, amyloid-beta peptide and Alzheimer disease. Nat Rev Neurol. 2016;12:433–434. doi: 10.1038/nrneurol.2016.105. [DOI] [PubMed] [Google Scholar]
- González-Reyes RE, Aliev G, Ávila-Rodrigues M, Barreto GE. Alterations in glucose metabolism on cognition: a possible link between diabetes and dementia. Curr Pharm Des. 2016;22:812–818. doi: 10.2174/1381612822666151209152013. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, Herrup K, Frautschy SA, Finsen B, Brown GC, Verkhratsky A, Yamanaka K, Koistinaho J, Latz E, Halle A, Petzold GC, et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol. 2015;14:388–405. doi: 10.1016/S1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarosz-Griffiths HH, Noble E, Rushworth JV, Hooper NM. Amyloid-beta receptors: the good, the bad, and the prion protein. J Biol Chem. 2016;291:3174–3183. doi: 10.1074/jbc.R115.702704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazvinšćak Jembrek M, Hof PR, Šimić G. Ceramides in Alzheimer's disease: key mediators of neuronal apoptosis induced by oxidative stress and Abeta accumulation. Oxid Med Cell Longev. 2015;2015:346783. doi: 10.1155/2015/346783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson GV, Bailey CD. The p38 MAP kinase signaling pathway in Alzheimer's disease. Exp Neurol. 2003;183:263–268. doi: 10.1016/s0014-4886(03)00268-1. [DOI] [PubMed] [Google Scholar]
- Lanna A, Gomes DC, Muller-Durovic B, McDonnell T, Escors D, Gilroy DW, Lee JH, Karin M, Akbar AN. A sestrin-dependent Erk-Jnk-p38 MAPK activation complex inhibits immunity during aging. Nat Immunol. 2017;18:354–363. doi: 10.1038/ni.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latrasse D, Jegu T, Li H, de Zelicourt A, Raynaud C, Legras S, Gust A, Samajova O, Veluchamy A, Rayapuram N, Ramirez-Prado JS, Kulikova O, Colcombet J, Bigeard J, Genot B, Bisseling T, Benhamed M, Hirt H. MAPK-triggered chromatin reprogramming by histone deacetylase in plant innate immunity. Genome Biol. 2017;18:131. doi: 10.1186/s13059-017-1261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawan A, Shi H, Gatzke F, Bennett AM. Diversity and specificity of the mitogen-activated protein kinase phosphatase-1 functions. Cell Mol Life Sci. 2013;70:223–237. doi: 10.1007/s00018-012-1041-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, Kim NJ. Recent advances in the inhibition of p38 MAPK as a potential strategy for the treatment of Alzheimer's disease. Molecules. 2017;22:E1287. doi: 10.3390/molecules22081287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Chen M, Liu H, Yang L, Yang T, He G. The dual role of ERK signaling in the apoptosis of neurons. Front Biosci (Landmark Ed) 2014;19:1411–1417. doi: 10.2741/4291. [DOI] [PubMed] [Google Scholar]
- Liu H, Yang J, Wang L, Xu Y, Zhang S, Lv J, Ran C, Li Y. Targeting beta-amyloid plaques and oligomers: development of near-IR fluorescence imaging probes. Future Med Chem. 2017;9:179–198. doi: 10.4155/fmc-2016-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZD, Wang JC, Yang JD. Current researches of signal pathways involved in bone marrow mesenchymal stem cells differentiation into nerve cells. Zhongguo Zuzhi Gongcheng Yanjiu. 2012;16:1111–1114. [Google Scholar]
- Low HB, Zhang Y. Regulatory roles of MAPK phosphatases in cancer. Immune Netw. 2016;16:85–98. doi: 10.4110/in.2016.16.2.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luque-Contreras D, Carvajal K, Toral-Rios D, Franco-Bocanegra D, Campos-Peña V. Oxidative stress and metabolic syndrome: cause or consequence of Alzheimer's disease? Oxid Med Cell Longev. 2014;2014:497802. doi: 10.1155/2014/497802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller UC, Deller T, Korte M. Not just amyloid: physiological functions of the amyloid precursor protein family. Nat Rev Neurosci. 2017;18:281–298. doi: 10.1038/nrn.2017.29. [DOI] [PubMed] [Google Scholar]
- Ma G, Pan Y, Zhou C, Sun R, Bai J, Liu P, Ren Y, He J. Mitogen-activated protein kinase phosphatase 1 is involved in tamoxifen resistance in MCF7 cells. Oncol Rep. 2015;34:2423–2430. doi: 10.3892/or.2015.4244. [DOI] [PubMed] [Google Scholar]
- McDonald DR, Bamberger ME, Combs CK, Landreth GE. beta-Amyloid fibrils activate parallel mitogen-activated protein kinase pathways in microglia and THP1 monocytes. J Neurosci. 1998;18:4451–4460. doi: 10.1523/JNEUROSCI.18-12-04451.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moosavi SM, Prabhala P, Ammit AJ. Role and regulation of MKP-1 in airway inflammation. Respir Res. 2017;18:154. doi: 10.1186/s12931-017-0637-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Origlia N, Arancio O, Domenici L, Yan SS. MAPK, beta-amyloid and synaptic dysfunction: the role of RAGE. Expert Rev Neurother. 2009;9:1635–1645. doi: 10.1586/ern.09.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen RB, Nunomura A, Lee HG, Casadesus G, Perry G, Smith MA, Zhu X. Signal transduction cascades associated with oxidative stress in Alzheimer's disease. J Alzheimers Dis. 2007;11:143–152. doi: 10.3233/jad-2007-11202. [DOI] [PubMed] [Google Scholar]
- Petrov D, Luque M, Pedros I, Ettcheto M, Abad S, Pallas M, Verdaguer E, Auladell C, Folch J, Camins A. Evaluation of the role of JNK1 in the hippocampus in an experimental model of familial Alzheimer's disease. Mol Neurobiol. 2016;53:6183–6193. doi: 10.1007/s12035-015-9522-6. [DOI] [PubMed] [Google Scholar]
- Prasad KN, Bondy SC. Inhibition of early upstream events in prodromal Alzheimer's disease by use of targeted antioxidants. Current aging science. 2014;7:77–90. doi: 10.2174/1874609807666140804115633. [DOI] [PubMed] [Google Scholar]
- Puzzo D, Gulisano W, Arancio O, Palmeri A. The keystone of Alzheimer pathogenesis might be sought in Abeta physiology. Neuroscience. 2015;307:26–36. doi: 10.1016/j.neuroscience.2015.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridler C. Alzheimer Disease: Misfolded diabetes-mellitus peptide seeds amyloid-beta aggregation. Nat Rev Neurol. 2017;13:128. doi: 10.1038/nrneurol.2017.5. [DOI] [PubMed] [Google Scholar]
- Rojas-Gutierrez E, Muñoz-Arenas G, Treviño S, Espinosa B, Chavez R, Rojas K, Flores G, Díaz A, Guevara J. Alzheimer's disease and metabolic syndrome: A link from oxidative stress and inflammation to neurodegeneration. Synapse. 2017 doi: 10.1002/syn.21990. doi: 10.1002/syn.21990. [DOI] [PubMed] [Google Scholar]
- Shafi O. Inverse relationship between Alzheimer's disease and cancer, and other factors contributing to Alzheimer's disease: a systematic review. BMC Neurol. 2016;16:236. doi: 10.1186/s12883-016-0765-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamim D, Laskowski M. Inhibition of inflammation mediated through the tumor necrosis factor alpha biochemical pathway can lead to favorable outcomes in Alzheimer disease. Journal of central nervous system disease. 2017;9:1179573517722512. doi: 10.1177/1179573517722512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shichiri M. The role of lipid peroxidation in neurological disorders. J Clin Biochem Nutr. 2014;54:151–160. doi: 10.3164/jcbn.14-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WW, Gorospe M, Kusiak JW. Signaling mechanisms underlying Abeta toxicity: potential therapeutic targets for Alzheimer's disease. CNS Neurol Disord Drug Targets. 2006;5:355–361. doi: 10.2174/187152706784111515. [DOI] [PubMed] [Google Scholar]
- Song L, Li D, Gu Y, Li X, Peng L. Let-7a modulates particulate matter (≤ 2.5 μm)-induced oxidative stress and injury in human airway epithelial cells by targeting arginase 2. J Appl Toxicol. 2016;36:1302–1310. doi: 10.1002/jat.3309. [DOI] [PubMed] [Google Scholar]
- Song L, Li D, Li X, Ma L, Bai X, Wen Z, Zhang X, Chen D, Peng L. Exposure to PM2. 5 induces aberrant activation of NF-kappaB in human airway epithelial cells by downregulating miR-331 expression. Environ Toxicol Pharmacol. 2017;50:192–199. doi: 10.1016/j.etap.2017.02.011. [DOI] [PubMed] [Google Scholar]
- Stepien KM, Heaton R, Rankin S, Murphy A, Bentley J, Sexton D, Hargreaves IP. Evidence of oxidative stress and secondary mitochondrial dysfunction in metabolic and non-metabolic disorders. J Clin Med. 2017;6:E71. doi: 10.3390/jcm6070071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tare M, Modi RM, Nainaparampil JJ, Puli OR, Bedi S, Fernandez-Funez P, Kango-Singh M, Singh A. Activation of JNK signaling mediates amyloid-ss-dependent cell death. PLoS One. 2011;6:e24361. doi: 10.1371/journal.pone.0024361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DM, Moser R, Regulier E, Breuillaud L, Dixon M, Beesen AA, Elliston L, Silva Santos Mde F, Kim J, Jones L, Goldstein DR, Ferrante RJ, Luthi-Carter R. MAP kinase phosphatase 1 (MKP-1/DUSP1) is neuroprotective in Huntington's disease via additive effects of JNK and p38 inhibition. J Neurosci. 2013;33:2313–2325. doi: 10.1523/JNEUROSCI.4965-11.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thouverey C, Caverzasio J. Focus on the p38 MAPK signaling pathway in bone development and maintenance. Bonekey Rep. 2015;4:711. doi: 10.1038/bonekey.2015.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiedje C, Holtmann H, Gaestel M. The role of mammalian MAPK signaling in regulation of cytokine mRNA stability and translation. J Interferon Cytokine Res. 2014;34:220–232. doi: 10.1089/jir.2013.0146. [DOI] [PubMed] [Google Scholar]
- Troy CM, Rabacchi SA, Xu Z, Maroney AC, Connors TJ, Shelanski ML, Greene LA. beta-Amyloid-induced neuronal apoptosis requires c-Jun N-terminal kinase activation. J Neurochem. 2001;77:157–164. doi: 10.1046/j.1471-4159.2001.t01-1-00218.x. [DOI] [PubMed] [Google Scholar]
- VanItallie TB. Alzheimer's disease: Innate immunity gone awry? Metabolism. 2017;69S:S41–S49. doi: 10.1016/j.metabol.2017.01.014. [DOI] [PubMed] [Google Scholar]
- Vayssier-Taussat M, Kreps SE, Adrie C, Dall’Ava J, Christiani D, Polla BS. Mitochondrial membrane potential: a novel biomarker of oxidative environmental stress. Environ Health Perspect. 2002;110:301–305. doi: 10.1289/ehp.02110301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wancket LM, Frazier WJ, Liu Y. Mitogen-activated protein kinase phosphatase (MKP)-1 in immunology, physiology, and disease. Life Sci. 2012;90:237–248. doi: 10.1016/j.lfs.2011.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Tang M. Review of the effects of manufactured nanoparticles on mammalian target organs. J Appl Toxicol. 2018;38:25–40. doi: 10.1002/jat.3499. [DOI] [PubMed] [Google Scholar]
- Xu Y, Cao DH, Wu GM, Hou XY. Involvement of P38MAPK activation by NMDA receptors and non-NMDA receptors in amyloid-beta peptide-induced neuronal loss in rat hippocampal CA1 and CA3 subfields. Neurosci Res. 2014;85:51–57. doi: 10.1016/j.neures.2014.05.011. [DOI] [PubMed] [Google Scholar]
- Yao B, Wang S, Xiao P, Wang Q, Hea Y, Zhang Y. MAPK signaling pathways in eye wounds: Multifunction and cooperation. Exp Cell Res. 2017;359:10–16. doi: 10.1016/j.yexcr.2017.06.024. [DOI] [PubMed] [Google Scholar]
- Zhou YY, Li Y, Jiang WQ, Zhou LF. MAPK/JNK signalling: a potential autophagy regulation pathway. Biosci Rep. 2015;35:e00199. doi: 10.1042/BSR20140141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Lee HG, Raina AK, Perry G, Smith MA. The role of mitogen-activated protein kinase pathways in Alzheimer's disease. Neurosignals. 2002;11:270–281. doi: 10.1159/000067426. [DOI] [PubMed] [Google Scholar]
- Zhu X, Rottkamp CA, Boux H, Takeda A, Perry G, Smith MA. Activation of p38 kinase links tau phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer disease. J Neuropathol Exp Neurol. 2000;59:880–888. doi: 10.1093/jnen/59.10.880. [DOI] [PubMed] [Google Scholar]