

Publisher's Note: There is a Blood Commentary on this article in this issue.
Key Points
Injured brain releases hyperadhesive and microvesicle-bound VWF that causes neurological dysfunction and a systemic hypercoagulable state.
ADAMTS-13 protected the blood-brain barrier to prevent TBI-induced neurological dysfunction and systemic coagulopathy.
Abstract
von Willebrand factor (VWF) is an adhesive ligand, and its activity is proteolytically regulated by the metalloprotease ADAMTS-13 (a disintegrin and metalloprotease with thrombospondin type 1 repeat 13). An elevated level of plasma VWF has been widely considered a marker for endothelial cell activation in trauma and inflammation, but its causal role in these pathological conditions remains poorly defined. Using a fluid percussion injury mouse model, we demonstrated that VWF released during acute traumatic brain injury (TBI) was activated and became microvesicle-bound. The VWF-bound microvesicles promoted vascular leakage and systemic coagulation. Recombinant ADAMTS-13 given either before or after TBI reduced the VWF reactivity with minimal influence on VWF secretion. rADAMTS-13 protected the integrity of endothelial cell barriers and prevented TBI-induced coagulopathy by enhancing VWF cleavage without impairing basal hemostasis. Promoting microvesicle clearance by lactadherin had efficacy similar to that of rADAMTS-13. This study uncovers a novel synergistic action between VWF and cellular microvesicles in TBI-induced vascular leakage and coagulopathy and demonstrates protective effects of rADAMTS-13.
Visual Abstract
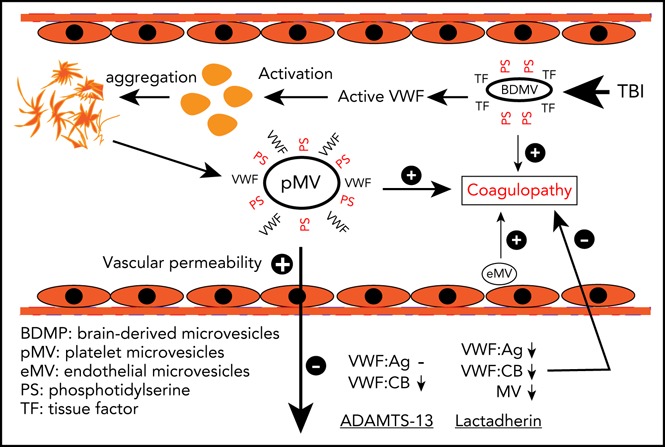
Introduction
von Willebrand factor (VWF) is the largest adhesive ligand in circulation. It is synthesized in megakaryocytes and endothelial cells as a single-chain propolypeptide of 2813 amino acids.1 The pro-VWF monomers dimerize through C-terminal disulfide bonds2 and a variable number of dimers multimerize through N-terminal disulfide bonds.3,4 The newly synthesized VWF multimers are either constitutively released or stored in the Weibel-Palade bodies of endothelial cells and α-granules of platelets.5 VWF multimers in these storage granules are enriched in ultralarge forms that are intrinsically active in activating platelets and promoting thrombosis.6,7 Therefore, the adhesive activity of VWF multimers is determined not only by antigen levels but also by multimeric structures.4,8 ADAMTS-13 (a disintegrin and metalloprotease with thrombospondin type 1 repeat 13) decreases the adhesive activity of VWF multimers by cleaving the Y1605-M1606 peptide bond in the A2 domain to reduce their size,9-11 demonstrating an important balance between the adhesive activity of VWF and the proteolytic activity of ADAMTS-13 in maintaining VWF hemostatic activity.
We hypothesize that VWF promotes TBI-induced and microvesicle (MV)-mediated blood-brain barrier (BBB) disruption and coagulopathy and that ADAMTS-13 protects the endothelial integrity and prevents a TBI-induced consumptive coagulopathy. The development of these hypotheses is based on several earlier observations. First, VWF is a known acute phase reactant that is rapidly released upon injury and is associated with poor outcomes of TBI.12-14 Second, VWF is detected in thrombosis formed within hours after TBI in the lesion boundary zone and remote regions of cortex, and reducing plasma VWF decreases the thrombosis.15 Third, VWF deficiency upregulates the tight junction protein claudin 5 to improve the BBB integrity, but paradoxically sensitized the mice for hypoxia-induced BBB disruption.16 Fourth, an antibody against the VWF A1 domain blocks inflammation-induced vascular leakage.17 Finally, we have recently shown that anionic phospholipid-rich membrane microvesicles (BDMVs) and extracellular mitochondria released from traumatically injured brains disrupt the BBB integrity so that they could rapidly be released into systemic circulation, where they trigger consumptive coagulopathy.18,19 The microvesicle-endothelial cell interaction could be mediated by hyperadhesive VWF.20,21
Here we discuss the results of a study designed to test these hypotheses by investigating the synergistic interaction between VWF and cellular microvesicles in disrupting BBB and propagating TBI-induced coagulopathy. Specifically, we investigated whether a high basal ADAMTS-13 activity could prevent TBI-induced disruption of BBB integrity and coagulopathy by preconditioning mice with the metalloprotease before exposing them to TBI. rADAMTS-13 was also given after TBI to test its therapeutic potential.
Materials and methods
Mouse model of TBI
Adult male C57BL/6J mice (12-16 weeks and 22-29 g; Jackson Laboratory, Bar Harbor, ME) were subjected to fluid percussion injury (FPI; Custom Design & Fabrication, Richmond, VA).18,19,22 FPI was induced at a controlled pressure of 1.9 ± 0.2 atm by rapidly injecting saline from a Plexiglas syringe into an unstrained mouse through a 3 mm hole with the dura matter intact. ADAMTS-13-deficient mice on CASA/Rk background (ADAMTS-13−/−) and their littermates23 were similarly tested to study the effect of VWF proteolysis on FPI-induced cerebral and systemic changes. The ADAMTS-13−/− mice were kindly provided by David Ginsburg of the University of Michigan and were generated by crossing ADAMTS-13−/− mice with CASA/Rk mice. They were genotyped as previously described.23
For the preconditioning regimen, a mouse received a single bolus infusion of 200 μg/kg rADAMTS-13, which was produced in Chinese Hamster Ovary cells transfected with human ADAMTS-13 cDNA (supplemental Methods, available on the Blood Web site) or an equal volume of the vehicle phosphate-buffered saline (PBS) through the tail vein 30 minutes before FPI. This dose was chosen to achieve 5 times the normal plasma concentration of ADAMTS-13.24 A sham mouse underwent craniectomy without FPI. For the therapeutic regimen, a mouse was infused with 200 μg/kg rADAMTS-13 or PBS 30 min after FPI. Blood samples were collected at baseline and at multiple points after FPI to measure VWF antigen and adhesive activity, MVs, blood cell counts, fibrinogen, D-dimer, clotting time, and tail bleeding. The mice were also evaluated for TBI-induced neurological dysfunction using the modified Neurological Severity Scores25 and monitored for 3-day survival.
The study was approved by the Institutional Animal Care and Use Committee (IACUC) of the Bloodworks Research Institute.
Vascular permeability
The FPI-induced BBB and pulmonary vascular permeability were measured by Evans blue dye extravasation.25,26 Briefly, a mouse received 100 μL 2% Evans blue (Sigma Aldrich, St. Louis, MO) through the tail vein 2 hours after FPI or sham surgery. It was sacrificed 2 hours after the dye injection and immediately perfused with PBS to dissect the brain and the lungs. The brain was photographed from topical and coronal views to show dye leakage. The brain and the lungs were then snap-frozen in liquid nitrogen, homogenized in formamide (1:20 wt/vol), and incubated at 60°C overnight. The brain and lung homogenates were centrifuged at 16 000g for 30 minutes. Evans blue dye in the supernatant was detected at optical density (OD) 620 nm, using a SpectraMax M5 plate-reader (Molecular Devices, Sunnyvale CA).
Endothelial permeability was also measured in vitro, using a transwell assay.19,25 Human umbilical cord endothelial cells (ATCC, Manassas, VA) were grown in 24-well plates coated with collagen in endothelial growth medium (Lonza, Allendale, NJ) until confluent. They were then incubated with purified VWF, MV-free plasma, or purified MVs for 3 hours at 37°C, followed by incubation with 100 μg/mL fluorescein isothiocyanate (FITC)-Dextran for 60 minutes at 37°C. Aliquots of the endothelial growth medium-2 from the bottom wells were collected to measure FITC fluorescence in a plate-reader (Molecular Devices).
Microvesicle measurement by flow cytometry
VWF-bound MVs of different cell origins were detected in the peripheral blood samples of FPI and sham mice, using flow cytometry (LSR II, Beckon Dickinson, San Jose, CA). These MVs were first recognized by particle sizes (<1 μm), using 0.5-, 0.9-, and 3-μm standard microbeads (Biocytex, Marseille, France)18,19 and then detected for double labeling with an FITC-conjugated polyclonal VWF antibody (Abcam)27 and monoclonal antibodies against cell-specific markers of neurons, glial cells, platelets (CD41a), and endothelial cells (CD31; all from Abcam). Cell-specific markers of neuronal and glial cell MVs were collectively called BDMVs18 APC-conjugated Annexin V was used to detect phosphatidylserine (PS)-exposed MVs18 All buffers used for MV detection were filtered with a 0.1 μm filter (EDM Millipore, Billerica, MA).
Coagulation
TBI-induced coagulopathy was defined by 3 measurements: plasma levels of fibrinogen and the fibrinolytic product D-dimer, measured by commercial kits from Abcam and LifeSpan Biosciences, respectively (Seattle, WA). The anionic phospholipid-dependent clotting time was measured in the presence of MVs purified from TBI or sham mice and 0.02 U/mL coagulation factor Xa using the STA-Procoag-PPL kit (Diagnostica Stago, France).18,19,25
VWF measurements
VWF antigen (VWF:Ag) was measured using a mouse ELISA kit (Lifespan Biosciences). VWF adhesive activity was measured using a collagen-binding assay (VWF:CB).27 Briefly, 96-well plates were coated with 600 ng/well of human type III collagen (Sigma Aldrich) overnight at 4°C and blocked with 2.5% bovine serum albumin in PBS for 1 hour at room temperature. Platelet-poor plasma (PPP) from FPI and sham mice was diluted 30-fold with PBS containing 0.5% BSA and 0.05% Tween-20 (PBS-BSA-Tween-20) and incubated with collagen for 60 minutes at room temperature. The wells were washed with PBS-BSA-Tween-20 and incubated with an horseradish peroxidase-conjugated polyclonal VWF antibody (DAKO) for 1 hour, followed by 10 min of incubation with the substrate TMB at 37°C. VWF:CB reported as OD values before and after VWF:Ag was normalized with plasma from VWF−/− mice (kindly provided by Jose A. Lopez, Bloodworks Research Institute) to the value of noninjured mice. The normalization allowed us to measure intrinsic VWF adhesive activity after the elevated VWF:Ag in FPI mice was adjusted. For data validation, we also defined this intrinsic VWF activity by a calculated VWF:CB to VWF:Ag ratio. To specifically evaluate the effect of MV clearance on VWF:Ag and VWF:CB, mice were given 400 μg/kg lactadherin (Biomatik, Wilmington, DE) 30 minutes before FPI.25 Blood samples collected at baseline and at 3 and 6 hours after FPI were analyzed for levels of Annexin V+ MV, VWF-bound MVs, VWF:Ag, and VWF:CB.
To detect MV-bound and plasma VWF separately, PPP obtained by centrifuging citrated blood samples at 1500g for 20 minutes at 22°C was centrifuged again at 13 000g for 2 minutes to obtain cell-free plasma, which was further centrifuged at 100 000g for 60 minutes at 4°C to collect the supernatant as MV-free plasma (MVFP). The MV pellets from the ultracentrifugation were washed and resuspended in PBS at a volume equal to MVFP.18 MV and MPFP were solubilized with radioimmunoprecipitation assay buffer containing 1% sodium dodecyl sulfate, subjected to 4% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and probed for VWF using a polyclonal VWF antibody (Agilent, Santa Clara, CA).27
To measure VWF cleavage by ADAMTS-13, mouse PPP was solubilized and separated on sodium dodecyl sulfate-polyacrylamide gel electrophoresis under reducing conditions. After being transferred to nitrocellulose membrane, the uncleaved and ADAMTS-13-cleaved VWF were sequentially probed, first with a rabbit polyclonal antibody that primarily recognizes uncleaved VWF (Agilent, Santa Clara, CA) and then, after stripping the membrane, with a goat antibody (Bethyl Laboratories, Montgomery, TX). This antibody was made against a synthetic M1606VTGNPASDEIKRL peptide that is the C-terminal sequence of VWF after cleavage by ADAMTS-13 (M1606 is at the cleavage site) and recognizes only cleaved VWF, as we have previously reported.27 The rate of VWF cleavage was then quantified as the ratio of cleaved to uncleaved bands by densitometry.
Tissue histology
Mice were sacrificed 24 hours after FPI to collect the lungs, which were fixed with 4% formaldehyde overnight at room temperature, embedded, and sectioned. The sections were stained with hematoxylin and eosin to detect microvascular bleeding as previously described.18,19 We studied not only the brain (by Evans’ blue test) but also the lungs to demonstrate the systemic nature of TBI-induced vascular leakage.
Statistical analysis
Quantitative data were presented as percentages for categorical variables and as the mean ± SEM for continuous variables. They were analyzed using Sigma plot (V. 11.2, SYSTAT Software, San Jose, CA). Variations among groups were analyzed with paired t tests, 1-way or 1-way-repeated measures ANOVA, as specified for each dataset. For multiple group ANOVA, Bonferroni correction was performed to identify subgroup differences. The survival data were analyzed by Kaplan-Meier plot. A P value of < .05 was considered to be statistically significant.
Results
ADAMTS-13 prevented FPI-induced vascular leakage and coagulopathy
After FPI was induced by a physical impact of 1.9 atm, significant vascular leakage occurred at the impact site, in the surrounding tissue of the brain (Figure 1A-D), and in the lungs (Figure 1E-G). The pulmonary vascular leakage and bleeding were also detected in noninjured mice infused with purified BDMVs (supplemental Figure 1A). Consistent with our previous reports,18,19 FPI mice developed a hypercoagulable state that rapidly turned into consumptive coagulopathy defined by significantly reduced plasma fibrinogen (Figure 1H), increased plasma D-dimer (Figure 1I), and MV-induced shortening of clotting time (supplemental Figure 1B).18,19 Preconditioning mice with 200 μg/kg rADAMTS-13 reduced injury-induced vascular leakage in tissue surrounding the impact site (Figure 1A-D), pulmonary microvascular leakage (Figure 1E-G), and coagulopathy (Figure 1H-I), resulting in improved neurological function (Figure 2A) and survival (Figure 2B). At the dose tested, exogenous rADAMTS-13 did not prolong the tail bleeding time of recipient mice (supplemental Figure 1C).
Figure 1.

rADAMTS-13-preconditioning reduced FPI-induced vascular leakage and coagulopathy. (A-C) Topical and coronal views of the brains of C57BL/6J mice preconditioned with a single dose (200 μg/kg) of rADAMTS-13 or an equal volume of PBS before being subjected to 1.9 atm of FPI. Control mice received PBS and underwent sham surgery (representative images from 27 independent experiments). The top panels show surface areas of the impact site, and the coronal views show a depth of injury that is indicative of secondary injury. The reduction of vascular leakage is more visible in the coronal views. (D) Evans blue dye released from vessels into the interstitial space of the brain was measured in brain homogenates (n = 27; 1-way ANOVA). (E-G) Representative hematoxylin and eosin-stained lungs from mice that received the 3 treatments (arrow: microvascular bleeds; bar = 100 μm). Plasma levels of fibrinogen (H) and D-dimer (I) from mice in the above treatment groups (n = 27; 1-way ANOVA).
Figure 2.

ADMATS-13-preconditioning on TBI recovery and VWF reactivity. The modified Neurological Severity Scores (A; n = 33; 1-way ANOVA). (B) Kaplan-Meier survival plot of mice preconditioned with either rADAMTS-13 or PBS before FPI and sham mice (n = 33; P < .05 for FPI+rADAMTS-13 vs FPI+PBS, and for sham vs FPI+PBS, but not for FPI+rADAMTS-13 vs sham). Plasma VWF:Ag (C) and VWF:CB (D) of mice in the above groups (n = 33; 1-way ANOVA, *P < .01, **P < .001). (E) Plasma VWF-CB before (black bars) and after (white bars) adjustments for VWF:Ag (n = 18; paired t test, *P < .01, **P < .05). (F) PPP from mice receiving different treatments was probed for uncleaved (top) and cleaved (middle) VWF, using 2 specific antibodies. The bottom panel shows the densitometry ratio of cleaved to uncleaved VWF (n = 9, paired t test, *P < .01 and ** P < .05). (G) CD62 expression on platelets from mice of the above 3 treatment groups (n = 33, 1-way ANOVA, *P < .001).
ADAMTS-13 reduced FPI-induced VWF adhesive hyperactivity
The protective effect of rADAMTS-13 suggests that the VWF secreted during acute FPI was not sufficiently cleaved and was thus overly adhesive. Consistent with this notion, the levels and adhesive activity of circulating VWF were significantly but transiently increased, reaching peak levels at 3 hours post-FPI (Figure 2C-D). The rADAMTS-13 preconditioning prevented the increase in VWF:CB, but not the elevation of VWF:Ag. The VWF:CB remained higher in plasma from FPI mice than from sham mice after VWF:Ag was normalized to the baseline level of C57 BL/6J mice (Figure 2E), suggesting that the VWF in the FPI mice was intrinsically active. This notion was further validated by a higher VWF:CB-to-VWF:Ag ratio in FPI mice (supplemental Figure 2B). VWF cleavage by ADAMTS-13 was moderately higher in FPI mice receiving PBS and further increased in mice preconditioned with rADAMTS-13 (Figure 2F). Consistent with the enhanced VWF activity, CD62p expression was increased on platelets from FPI mice, and the increase was partially prevented in mice preconditioned with rADAMTS-13 (Figure 2G).
MV-bound VWF disrupted the integrity of the endothelial barrier
We have previously shown that anionic phospholipids exposed on the surface of BDMVs and extracellular mitochondria induce vascular permeability in vitro and in mouse models.18,19,22 The finding that rADAMTS-13 prevented the FPI-induced BBB disruption (Figure 1A-D) suggests a role of VWF in the MV-induced vascular permeability. We examined the effects of MVs and VWF on endothelial permeability in a transwell culture system. Plasma collected from mice 3 hours after FPI, but not that from sham mice induced significant leakage of cultured human umbilical cord endothelial cells (Figure 3A). This activity was almost exclusively found in the MV fraction of plasma, and it was blocked by a VWF antibody. In contrast, purified VWF, even at 100- to 300-fold of the mouse plasma concentration failed to induce the vascular leakage. When probed separately, VWF was detected in both the MV and MV-free fractions of plasma from FPI mice, but only in the MV-free fraction from sham mice (Figure 3B). Flow cytometry further determines that 11.8 ± 7.9%, 41.6 ± 12.3%, and 8.1 ± 3.7% of VWF-bound microvesicles were derived from the brain (NES+ and glial cell+), platelets, and endothelial cells, respectively (Figure 3C-E), suggesting that VWF activated platelets to produce microvesicles. Furthermore, lactadherin (400 μg/kg), which we have recently shown to promote the clearance of PS-expressing MVs,25 reduced the plasma levels not only of PS+ MVs (Figure 3F) but also of VWF-bound MVs (Figure 3G), reducing both plasma VWF:Ag and VWF:CB (Figure 3H).
Figure 3.

The activity of VWF-bound MVs from different cells. (A) MV-induced increase in endothelial permeability is blocked by the VWF antibody (n = 16; 1-way ANOVA, *P < .01). (B) VWF is detected in the MV and MV-free (MVFP) fractions of plasma from TBI but only in MVFP fraction from sham mice. (C-E) The levels of plasma VWF+ MVs from brain cells (glial cells and neurons), platelets (CD41a+), and endothelial cells (CD31+) in the acute phase of TBI (n = 27; 1-way ANOVA, *P < .01 vs sham). Plasma levels of Annexin V+ (F) and VWF-bound MVs (G) in FPI mice treated with lactadherin or PBS and in sham mice (n = 12; 1-way ANOVA, *P < .001). (H) VWF:Ag (top) and VWF:CB (bottom) of plasma from C57BL/6J mice that received either 400 μg/kg of lactadherin or an equal volume of PBS before being subjected to FPI. Control mice underwent sham surgery (n = 12; 1-way ANOVA, *P < .01).
ADAMTS-13 deficiency worsened the effect of FPI
To further delineate the role of ADAMTS-13 in reducing the ability of VWF-bound microvesicles to disrupt BBB and to enhance coagulation, we exposed ADAMTS-13−/− mice on CASK background to 1.9 atm FPI. These mice have active VWF at baseline (supplemental Figure 3) because of the lack of ADAMTS-13 cleavage, and they develop thrombotic thrombocytopenic purpura-like pathology after challenge with shigatoxin.23 As with the C57BL/6J mice, FPI acutely increased VWF:CB (Figure 4A) and VWF:Ag (Figure 4B) in ADAMTS-13−/− mice, and rADAMTS-13 selectively reduced VWF:CB with minimal influence on VWF:Ag. VWF:CB was greater in ADAMTS-13−/− mice than in C57 BL/6J mice subjected to FPI of a similar intensity, but the dynamic profiles of VWF adhesive activity in the first 24 hours post-FPI were similar in the 2 strains (Figure 4C). ADAMTS-13−/− mice preconditioned with rADAMTS-13 had reduced vascular leakage in the lungs (Figure 4D) and in the brain (supplemental Figure 4), lower plasma D-dimer (Figure 4E), and milder fibrinogen depletion (Figure 4F), leading to improved neurological function (Figure 4G) and survival (Figure 4H). We did not study VWF-deficient mice because their bleeding diathesis is a significant confounding factor that would be difficult to stratify in a study designed to evaluate TBI-induced coagulopathy.
Figure 4.

Effect of FPI on ADAMTS-13−/−mice. VWF:CB (A) and VWF:Ag (B) of ADAMTS-13−/−-CASA mice preconditioned with rADAMTS-13 or PBS before being subjected to FPI. Control mice underwent sham surgery (n = 27; 1-way repeated measures ANOVA, *P < .01, **P < .001). (C) The effect of rADAMTS-13 preconditioning on dynamic changes in VWF:CB of ADAMTS-13−/− and C57 BL/6J mice during the first 24 hours after FPI (n = 18; 1-way ANOVA, *P < .01 vs sham). (D) Evans blue dye extravasation of brain tissues from ADAMTS-13−/− mice preconditioned with either rADAMTS-13 or PBS before FPI (n = 27; 1-way ANOVA, *P < .01). Plasma levels of D-dimer (E) and fibrinogen (F) of ADAMTS-13−/− mice from the above groups (n = 27; 1-way ANOVA, *P < .01). (G) Modified Neurological Severity Scores (mNSS; n = 27; 1-way-repeated measures ANOVA, *P < .001, **P < .01, ***P < .05) and (H) Kaplan-Meier survival plot (n = 76) of ADAMTS-13−/−-CASA mice in the above treatment groups.
ADAMTS-13 was therapeutic
The finding that preconditioning mice with rADAMTS-13 prevented injury-induced vascular leakage and coagulopathy, improving the neurological function and survival not only of ADAMTS-13-deficient (Figure 4) but also of ADAMTS-13-efficient (Figures 1-2) mice strongly suggests the therapeutic potential of this metalloprotease for TBI. We tested this hypothesis by studying mice that received the same rADAMTS-13 dosing 30 minutes after FPI. This early dosing was chosen because TBI induced cerebral edema and coagulopathy within 60 minutes of injury.18,19,25 rADAMTS-13, which sustained its elevated plasma level for at least 6 hours post-TBI (supplemental Figure 5), significantly reduced VWF:CB (Figure 5A) without reducing plasma VWF:Ag (supplemental Figure 6). This rADAMTS-13-reduced VWF adhesive activity was reproduced by infusing TBI mice with a polyclonal VWF antibody (supplemental Figure 7). rADAMTS-13 also partially corrected FPI-induced fibrinogen depletion (Figure 5B), D-dimer production (Figure 5C), platelet activation (Figure 5D), and thrombocytopenia (supplemental Figure 8A), without increasing tail bleeding time (Figure 5E) or changing hematocrit (supplemental Figure 8B). These rADAMTS-13-treated mice developed less severe cerebral vascular leakage (Figure 5F) and improved their neurological function (Figure 5G) and survival (Figure 5H).
Figure 5.

rADAMTS-13 was therapeutic. rADAMTS-13 given after FPI reduced VWF:CB (A), partially restored plasma fibrinogen (B), reduced plasma D-dimer (C), reduced platelet CD62p expression (D), and reversed the shortened tail bleeding time (E) in mice receiving a single bolus dose of rADAMTS-13 at 200 μg/kg 30 min after FPI compared with those either receiving an equal volume of PBS 30 minutes after FPI or undergoing sham surgery (n = 27; 1-way ANOVA, *P < .01). rADAMTS-13 in the same treatment regimen reduced cerebral vascular leakage (F) (n = 27; 1-way ANOVA, *P < .001) and improved neurological function (G) (n = 27; 1-way ANOVA, *P < .001, **P < .01) and accumulative survival (H) in FPI mice.
Discussion
VWF deficiency has been well characterized for bleeding diathesis found in patients with von Willebrand disease. However, as an acute phase reactant, plasma VWF is significantly increased in many disease states, and the increase consistently predicts poor outcomes of those diseases, including TBI.12-14 The question is whether elevated plasma VWF serves as a marker for the activation of endothelial cells and platelets or plays a causal role for the development and propagation of TBI. We conducted a study to investigate the causal role of VWF in the TBI-induced disruption of endothelial integrity and coagulopathy. We made the following important and novel observations.
First, TBI induced a rapid VWF secretion that reached a plateau approximately 3 hours postinjury (Figure 2). More important, the acutely released VWF bound and activated platelets (Figure 2G) to produce VWF-bound MVs (Figure 3D). The MV-bound VWF tethered MVs to endothelial cells18 through its interaction with CD62p28 and integrin αvβ3,29 activating the endothelial cells, which further produced endothelial MVs (Figure 3E), released ultralarge and hyperadhesive VWF,21 and increased vascular permeability (Figure 3A). This VWF-mediated MV-endothelial cell interaction is particularly important in vivo because VWF tethers platelets to endothelial cells against the pulling force of flowing blood.21 In addition, activated platelets could release molecules that promote the transendothelial migration of MVs18 It is interesting that more than 10% of the VWF-bound MVs were from neurons and glial cells (Figure 3C), likely mediated by VWF interaction with integrins on those cells.29,30
Second, the finding that TBI induced the transient release of hyperadhesive VWF suggests that ADAMTS-13 may have either been inhibited or become insufficient after acute injury. The former is unlikely because VWF cleavage did increase after TBI without exogenous rADAMTS-13 (Figure 2F). Therefore, the baseline ADAMTS-13 activity was likely insufficient to cleave the hyperadhesive VWF that was suddenly and substantially released from activated endothelial cells and platelets. This notion is supported by the finding that exogenous rADAMTS-13, given either before or after TBI, protected mice against TBI-induced vascular leakage and consumptive coagulopathy to improve neurological function and survival. One limitation of the current study is its short follow-up period for post FPI survival (72 hours). Although it is consistent with the study’s focus on acute coagulopathy, which occurs within 30 minutes in this FPI model, the 72-hour monitoring period is relatively short for post-TBI neurological evaluation. We should be able to address this pitfall in a follow-up study focusing on whether rADAMTS-13 promotes cerebral tissue repair after TBI. Nevertheless, several lines of evidence suggest that rADAMTS-13 protected TBI mice by enhancing VWF proteolysis: rADAMTS-13 reduced VWF adhesive activity without affecting its secretion or antigen levels (Figures 2 and 5), VWF cleavage was substantially enhanced in mice receiving rADAMTS-13 (Figure 2F), and ADAMTS-13−/− mice developed worse vascular leakage and coagulopathy, which were partially corrected by exogenous rADAMTS-13 (Figure 4).
We also show that rADAMTS-13 given at identical dosage after TBI was equally effective in protecting endothelial cell integrity and reducing coagulopathy (Figure 5). This ability of rADAMTS-13 to protect TBI mice against coagulopathy is consistent with our previous reports that TBI-induced coagulopathy is developed from a hypercoagulable state.18,19 The finding that rADAMTS-13 reduced VWF adhesive activity but, even at 5 times the normal plasma concentration, did not increase tail-vein bleeding time (Figure 5E) suggests that the metalloprotease selectively cleaves activated VWF without affecting its basal hemostatic activity. This selective regulation of VWF activity was similarly observed in a study of rADAMTS-13 for stroke therapy.31 It is also consistent with previous reports that ADAMTS-13 cleaves plasma VWF only after prolonged incubation and in the presence of the protein-denaturing agents,9,10 but instantaneously cleaves ultralarge and hyperadhesive VWF fibrils anchored to the surface of endothelial cells.21 The finding that ADAMTS-13 becomes insufficient during acute injury also raises the question of whether patients with intrinsically low ADAMTS-13 activity are prone to TBI-induced cerebral injury and coagulopathy. Low plasma ADAMTS-13 has previously been associated with poor outcomes of ischemic stroke32 and disseminated intravascular coagulation.33,34 Disseminated intravascular coagulation is a well-defined condition of consumptive coagulopathy35 similar to TBI-induced coagulopathy.18,22
Third, lactadherin, which promotes the clearance of PS+ MVs (Figure 3F),25 also reduced the plasma level of VWF-bound MVs from different cell types (Figure 3G) and decreased both VWF antigen and adhesive activity (Figure 3H). These results indicate that MV-bound VWF activated platelets and endothelial cells to produce microvesicles; VWF-bound MVs remained procoagulant, capable of inducing consumptive coagulopathy in TBI mice; and PS expressed on MVs was not blocked by VWF and remained available for the lactadherin-mediated phagocytosis of MVs.22 However, the actual rates of removing VWF-bound and VWF-free MVs by lactadherin were not specifically measured and compared.
In summary, we have demonstrated that TBI induces the rapid and transient secretion of hyperadhesive VWF, which is not cleaved by ADAMTS-13 in a timely manner. The VWF secretion is likely triggered by BDMVs released from the injured brain, as we have previously shown.18 The VWF activates platelets and endothelial cells to produce more MVs to propagate the BDMV-induced vascular permeability and consumptive coagulopathy. These findings demonstrate that VWF not only serves as a marker of endothelial injury but also contributes to vascular leakage, coagulopathy, and neurological dysfunction induced by TBI. The early and transient release of activated VWF highlights the need for early interventions in severe TBI. Enhancing VWF cleavage by ADAMTS-13 or promoting MV clearance by lactadherin protects endothelial integrity and prevents coagulopathy, without increasing the risk of bleeding.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
This study is supported by National Institutes of Health, National Institute of Neurological Disorders and Stroke grant NS087296 (J.-f.D.), National Institutes of Health, National Heart, Lung, and Blood Institute grants HL119391 and HL125957 (J.-f.D.), Natural Science Foundation of China State Key Program Grant 81330029 (J.Z.), and research grants 81271361, 81271359, and 16PTSTJC00180 (J.Z.) and 81672399 (M.L.), and a grant from the Veterans Administration Research Service (P.T.).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: Y.W. designed and performed experiments, analyzed data, and wrote the manuscript; W.L. (second author) performed tissue histology, analyzed data, and wrote the manuscript; Y.Z. performed experiments, analyzed data, and wrote the manuscript; T.H., Z.Z., and W.L. (sixth author) performed experiments and analyzed data; M.W. performed tissue histology and analyzed data; J.Y. and K.H. performed experiments; P.T. generated experimental agents and wrote the manuscript; F.Z. and F.-D.S. wrote the manuscript; X.W. provided technical support for experiments, analyzed data, and wrote the manuscript; M.L., J.Z., and J.-f.D. developed hypotheses, designed experiments, analyzed data, and wrote the manuscript
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jing-fei Dong, Bloodworks Research Institute, 1551 Eastlake Avenue East, Seattle, WA; e-mail: jfdong@bloodworksnw.org; Jianning Zhang, Department of Neurosurgery, Tianjin Medical University General Hospital, Tianjin, China; e-mail: jianningzhang@hotmail.com; Min Li, Institute of Pathology, Lanzhou University School of Basic Medical Sciences, Lanzhou, China; e-mail: limin@lzu.edu.cn.
References
- 1.Sadler JE. Biochemistry and genetics of von Willebrand factor. Annu Rev Biochem. 1998;67:395-424. [DOI] [PubMed]
- 2.Wagner DD, Lawrence SO, Ohlsson-Wilhelm BM, Fay PJ, Marder VJ. Topology and order of formation of interchain disulfide bonds in von Willebrand factor. Blood. 1987;69(1):27-32. [PubMed] [Google Scholar]
- 3.Vischer UM, Wagner DD. von Willebrand factor proteolytic processing and multimerization precede the formation of Weibel-Palade bodies. Blood. 1994;83(12):3536-3544. [PubMed] [Google Scholar]
- 4.Mayadas TN, Wagner DD. In vitro multimerization of von Willebrand factor is triggered by low pH. Importance of the propolypeptide and free sulfhydryls. J Biol Chem. 1989;264(23):13497-13503. [PubMed] [Google Scholar]
- 5.Sporn LA, Marder VJ, Wagner DD. Inducible secretion of large, biologically potent von Willebrand factor multimers. Cell. 1986;46(2):185-190. [DOI] [PubMed] [Google Scholar]
- 6.Fowler WE, Fretto LJ, Hamilton KK, Erickson HP, McKee PA. Substructure of human von Willebrand factor. J Clin Invest. 1985;76(4):1491-1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moake JL, Chow TW. Increased von Willebrand factor (vWf) binding to platelets associated with impaired vWf breakdown in thrombotic thrombocytopenic purpura. J Clin Apher. 1998;13(3):126-132. [DOI] [PubMed] [Google Scholar]
- 8.Sporn LA, Marder VJ, Wagner DD. von Willebrand factor released from Weibel-Palade bodies binds more avidly to extracellular matrix than that secreted constitutively. Blood. 1987;69(5):1531-1534. [PubMed] [Google Scholar]
- 9.Furlan M, Robles R, Lämmle B. Partial purification and characterization of a protease from human plasma cleaving von Willebrand factor to fragments produced by in vivo proteolysis. Blood. 1996;87(10):4223-4234. [PubMed] [Google Scholar]
- 10.Tsai HM. Physiologic cleavage of von Willebrand factor by a plasma protease is dependent on its conformation and requires calcium ion. Blood. 1996;87(10):4235-4244. [PubMed] [Google Scholar]
- 11.Levy GG, Nichols WC, Lian EC, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413(6855):488-494. [DOI] [PubMed] [Google Scholar]
- 12.Yokota H, Naoe Y, Nakabayashi M, et al. Cerebral endothelial injury in severe head injury: the significance of measurements of serum thrombomodulin and the von Willebrand factor. J Neurotrauma. 2002;19(9):1007-1015. [DOI] [PubMed] [Google Scholar]
- 13.De Oliveira CO, Reimer AG, Da Rocha AB, et al. Plasma von Willebrand factor levels correlate with clinical outcome of severe traumatic brain injury. J Neurotrauma. 2007;24(8):1331-1338. [DOI] [PubMed] [Google Scholar]
- 14.Kutcher ME, Ferguson AR, Cohen MJ. A principal component analysis of coagulation after trauma. J Trauma Acute Care Surg. 2013;74(5):1223-1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu D, Mahmood A, Goussev A, Qu C, Zhang ZG, Chopp M. Delayed thrombosis after traumatic brain injury in rats. J Neurotrauma. 2004;21(12):1756-1766. [DOI] [PubMed] [Google Scholar]
- 16.Suidan GL, Brill A, De Meyer SF, et al. Endothelial Von Willebrand factor promotes blood-brain barrier flexibility and provides protection from hypoxia and seizures in mice. Arterioscler Thromb Vasc Biol. 2013;33(9):2112-2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aymé G, Adam F, Legendre P, et al. A novel single-domain antibody against von Willebrand factor a1 domain resolves leukocyte recruitment and vascular leakage during inflammation-brief report. Arterioscler Thromb Vasc Biol. 2017;37(9):1736-1740. [DOI] [PubMed] [Google Scholar]
- 18.Tian Y, Salsbery B, Wang M, et al. Brain-derived microparticles induce systemic coagulation in a murine model of traumatic brain injury. Blood. 2015;125(13):2151-2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao Z, Wang M, Tian Y, et al. Cardiolipin-mediated procoagulant activity of mitochondria contributes to traumatic brain injury-associated coagulopathy in mice. Blood. 2016;127(22):2763-2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaplan JE, Moon DG, Weston LK, et al. Platelets adhere to thrombin-treated endothelial cells in vitro. Am J Physiol. 1989;257(2 Pt 2):H423-H433. [DOI] [PubMed] [Google Scholar]
- 21.Dong JF, Moake JL, Nolasco L, et al. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood. 2002;100(12):4033-4039. [DOI] [PubMed] [Google Scholar]
- 22.Zhou Y, Cai W, Zhao Z, et al. Lactadherin promotes microvesicle clearance to prevent coagulopathy and improves survival of severe TBI mice. Blood. 2018;131(5):563-572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Motto DG, Chauhan AK, Zhu G, et al. Shigatoxin triggers thrombotic thrombocytopenic purpura in genetically susceptible ADAMTS13-deficient mice. J Clin Invest. 2005;115(10):2752-2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feys HB, Liu F, Dong N, et al. ADAMTS-13 plasma level determination uncovers antigen absence in acquired thrombotic thrombocytopenic purpura and ethnic differences. J Thromb Haemost. 2006;4(5):955-962. [DOI] [PubMed] [Google Scholar]
- 25.Zhou YCW, Zhao Z, Hilton T, et al. Lactadherin promotes microvesicle clearance to prevent coagulopathy and improves survival of mice subjected to severe traumatic brain injury. Blood. 2018;131(5):563-572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao W, Zhao Z, Yu G, et al. VEGI attenuates the inflammatory injury and disruption of blood-brain barrier partly by suppressing the TLR4/NF-κB signaling pathway in experimental traumatic brain injury. Brain Res. 2015;1622:230-239. [DOI] [PubMed] [Google Scholar]
- 27.Nascimbene A, Hilton T, Konkle BA, Moake JL, Frazier OH, Dong JF. von Willebrand factor proteolysis by ADAMTS-13 in patients on left ventricular assist device support. J Heart Lung Transplant. 2017;36(4):477-479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Padilla A, Moake JL, Bernardo A, et al. P-selectin anchors newly released ultralarge von Willebrand factor multimers to the endothelial cell surface. Blood. 2004;103(6):2150-2156. [DOI] [PubMed] [Google Scholar]
- 29.Huang J, Roth R, Heuser JE, Sadler JE. Integrin alpha(v)beta(3) on human endothelial cells binds von Willebrand factor strings under fluid shear stress. Blood. 2009;113(7):1589-1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu X, Reddy DS. Integrins as receptor targets for neurological disorders. Pharmacol Ther. 2012;134(1):68-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakano T, Irie K, Hayakawa K, et al. Delayed treatment with ADAMTS13 ameliorates cerebral ischemic injury without hemorrhagic complication. Brain Res. 2015;1624:330-335. [DOI] [PubMed] [Google Scholar]
- 32.Denorme F, Kraft P, Pareyn I, et al. Reduced ADAMTS13 levels in patients with acute and chronic cerebrovascular disease. PLoS One. 2017;12(6):e0179258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hyun J, Kim HK, Kim JE, et al. Correlation between plasma activity of ADAMTS-13 and coagulopathy, and prognosis in disseminated intravascular coagulation. Thromb Res. 2009;124(1):75-79. [DOI] [PubMed] [Google Scholar]
- 34.Habe K, Wada H, Ito-Habe N, et al. Plasma ADAMTS13, von Willebrand factor (VWF) and VWF propeptide profiles in patients with DIC and related diseases. Thromb Res. 2012;129(5):598-602. [DOI] [PubMed] [Google Scholar]
- 35.Hardaway RM, Williams CH, Vasquez Y. Disseminated intravascular coagulation in sepsis. Semin Thromb Hemost. 2001;27(6):577-583. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.