

Publisher's Note: There is a Blood Commentary on this article in this issue.
Key Points
During late erythropoiesis, exogenous glutamine, rather than TCA cycle intermediates, provides carbons for succinyl-CoA for heme synthesis.
Itaconate, a compound manufactured as part of the inflammatory response, inhibits heme synthesis in cells undergoing erythropoiesis.
Abstract
During erythroid differentiation, the erythron must remodel its protein constituents so that the mature red cell contains hemoglobin as the chief cytoplasmic protein component. For this, ∼109 molecules of heme must be synthesized, consuming 1010 molecules of succinyl-CoA. It has long been assumed that the source of succinyl-coenzyme A (CoA) for heme synthesis in all cell types is the tricarboxylic acid (TCA) cycle. Based upon the observation that 1 subunit of succinyl-CoA synthetase (SCS) physically interacts with the first enzyme of heme synthesis (5-aminolevulinate synthase 2, ALAS2) in erythroid cells, it has been posited that succinyl-CoA for ALA synthesis is provided by the adenosine triphosphate–dependent reverse SCS reaction. We have now demonstrated that this is not the manner by which developing erythroid cells provide succinyl-CoA for ALA synthesis. Instead, during late stages of erythropoiesis, cellular metabolism is remodeled so that glutamine is the precursor for ALA following deamination to α-ketoglutarate and conversion to succinyl-CoA by α-ketoglutarate dehydrogenase (KDH) without equilibration or passage through the TCA cycle. This may be facilitated by a direct interaction between ALAS2 and KDH. Succinate is not an effective precursor for heme, indicating that the SCS reverse reaction does not play a role in providing succinyl-CoA for heme synthesis. Inhibition of succinate dehydrogenase by itaconate, which has been shown in macrophages to dramatically increase the concentration of intracellular succinate, does not stimulate heme synthesis as might be anticipated, but actually inhibits hemoglobinization during late erythropoiesis.
Visual Abstract
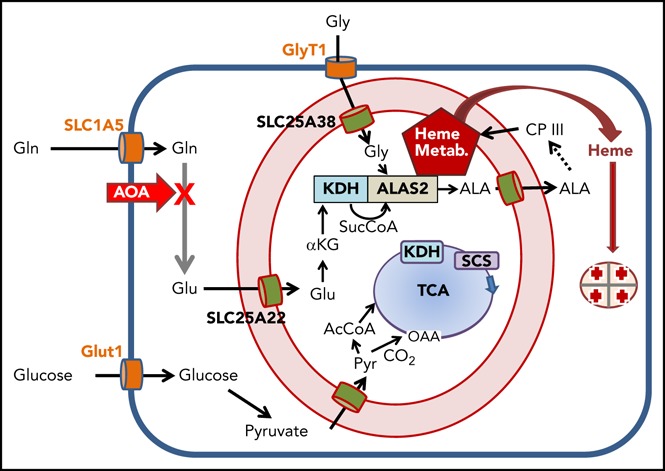
Introduction
The ability of a cell to maintain appropriate levels of key metabolites in response to changing organismal and environmental demands is essential for survival and growth. Monitoring and understanding regulatory schemes is challenging, but essential if one is to understand the metabolic basis for diseases as well as the potential impact of xenobiotics on cellular metabolism. All cell types have their own metabolic and regulatory needs, but those undergoing differentiation into a new cell type present unique challenges. This is clearly true for cells undergoing erythropoiesis where, starting with hematopoietic stem cells (HSCs), every cell division results in daughter cells that are distinct from their parent.1,2 Adding complexity to the overall process is that erythropoiesis occurs within the bone marrow in erythroblastic islands where red cell precursors exist in close association with so-called nurse macrophages.3,4
One of the hallmark events of terminal erythroid differentiation is the coordinated induction of heme and globin synthesis. Onset of heme synthesis occurs when the first pathway enzyme, the erythroid-specific 5-aminolevulinate synthase (ALAS2), is induced.5 In humans, this occurs in the basophilic erythroblast, reaching a maximum in the polychromatic/orthochromatic erythroblast, whereas in murine systems the initial induction occurs earlier, in the proerythroblast.6 GATA1-induced erythroid differentiation results in the upregulation of heme synthesis by induction of ALAS2, enhancement of the transcription of all heme pathway enzyme genes,5-7 and phosphorylation of the terminal pathway enzyme, ferrochelatase (FECH).8 In addition to transcriptional regulation, ALAS2 is also subject to translational regulation by iron via the iron-regulatory protein/iron-responsive element machinery9-12 and microRNAs.13 Posttranslational modification of the protein via the addition of the cofactor pyridoxal phosphate is essential.14 Hydroxylation of ALAS2 decreases the activity of ALAS2 and destabilizes it for proteasomal degradation,15 and posttranslational translocation of ALAS into the mitochondrion is inhibited by the binding of heme to the cytoplasmic precursor form of ALAS.16-18
Synthesis of heme in metazoan cells requires iron, glycine, and succinyl-coenzyme A (CoA) (supplemental Figure 1, available on the Blood Web site).2,19 The supply of iron for heme both in housekeeping activities and during erythroid differentiation has been well studied, and a reasonable view of the components involved in this process at both the organismal and the cellular level exists.20 Glycine is abundant in plasma (∼250 µM), and studies of glycine transporters are consistent with glycine being supplied from extracellular sources.21-23 In contrast, our knowledge of how succinyl-CoA is provided for heme synthesis during erythroid differentiation is not well understood. The assumption that the tricarboxylic acid (TCA) cycle supplies succinyl-CoA for erythroid heme synthesis has been made, but there is a paucity of data to support this view. It is possible that for nonerythroid cells, where only modest levels of heme are synthesized, there is sufficient capacity from the TCA cycle to supply succinyl-CoA.24 However, evoking this mechanism for erythroid cells would clearly place cells under strain as every molecule of heme generated requires 8 molecules of succinyl-CoA, and cells make 1.2 × 109 molecules of heme in a span of 2 to 3 days. It has been shown that ALAS2, but not the housekeeping ALAS (ALAS1), physically interacts with the adenosine triphosphate (ATP)-dependent succinyl-CoA synthetase (SCS) β-subunit SUCLA2.25,26 Because ALAS2 utilizes succinyl-CoA to synthesize ALA, and because SUCLA2 as a subunit of SCS may be involved in the ATP-dependent reverse reaction of SCS to generate succinyl-CoA from succinate, one hypothesis forwarded was that the ALAS2-SUCLA2 interaction exists to provide succinyl-CoA for ALA synthesis during erythropoiesis. However, although it is clear that an association between ALAS2 and SUCLA2 can exist, there are no data available to demonstrate what role, if any, this interaction plays in the supply of succinyl-CoA for ALAS2.
Data gathered in the current work from cells undergoing erythroid differentiation in vitro demonstrate that succinyl-CoA for ALA synthesis does not arise from succinate of the TCA cycle, but from α-ketoglutarate (KG) derived from glutamine and that during late erythropoiesis cellular metabolism is remodeled so that the major role of glutamine is to serve as a precursor for heme synthesis. This has significant implications to our understanding of cellular metabolism during erythropoiesis. Interestingly, we found that itaconate, a compound produced and excreted by lipopolysaccharide-induced macrophages,27 that is known to cause an increase in cellular succinate levels by inhibiting succinate dehydrogenase (SDH),27-29 inhibits heme synthesis and erythroid differentiation and that this inhibition is reversed by exogenously supplied ALA. A physiological role for macrophage-generated itaconate during erythropoiesis has not been identified, but data presented here suggest that it inhibits ALA synthesis in an as yet unidentified fashion.
Methods
13C labeling, cellular extraction, and liquid chromatography–mass spectrometry (LC/MS) characterization of murine erythroleukemia (MEL) cells undergoing erythroid differentiation are described in the supplemental materials. Assays of TCA cycle enzymes were done with mitochondria isolated from differentiating MEL cells as detailed in the supplemental materials. CD34+ cells were expanded and differentiated following StemCell protocols (StemCell Technologies Inc). Additional details are in the supplemental materials. Flow cytometry was carried out at the University of Utah Flow Cytometry Facility.
Results
Fate of 13C-labeled metabolic heme precursors during erythroid differentiation
Protoheme IX has a total of 34 carbons with 26 of these derived from succinyl-CoA and 8 carbons derived from glycine (Figure 1B-C). Assuming that glycine for heme synthesis is exogenously supplied,22,23 then 76% of the heme carbons originate from a source that produces succinyl-CoA (Figure 1A). During late erythropoiesis, elevation of heme synthesis creates a significantly increased demand for succinyl-CoA believed to come from the TCA cycle. Draining this cycle intermediate would require replenishment via anaplerotic supplementation.30 Both glucose and glutamine can replenish the TCA cycle by the conversion of pyruvate into oxaloacetate and glutamate into α-KG, respectively. In order to determine if either of these anaplerotic reactions provides carbon for heme synthesis during erythropoiesis, we employed uniformly (U) labeled 13C precursors and followed their incorporation into heme in differentiating MEL cells via LC/MS. MEL cells were induced to differentiate, and for a 24-hour period between 48 and 72 hours postinduction, the time of maximal heme synthesis in this system, the medium was supplemented with U-13C-labeled (1) glucose, (2) glutamine, or (3) diethylsuccinate (DES), the commonly employed cell-permeable analog of succinate.31 Heme was extracted and isolated from cells at 72-hours postinduction prior to analysis by LC/MS. A 24-hour window of exposure to U-13C-labeled metabolite was chosen in an effort to minimize cellular reshuffling of the label.
Figure 1.

Labeling patterns of ALA by succinate or glucose via the TCA cycle or glutamine. (A) The TCA cycle is shown with the carbons of intermediate compounds color coded to their original source. Carbons coming from glucose via acetyl-CoA are black. These carbons become the 2 carbons distal to the amino group of ALA. Carbons from exogenously provided succinate that transit the TCA cycle once are colored red and become the 2 carbons adjacent to the glycine-derived (green) carbon and amino group of ALA. Succinate that is directly converted to succinyl-CoA by the ATP-dependent SCS reverse reaction is colored yellow. All 4 succinate carbons that enter in this fashion are incorporated into ALA. Glutamine carbons are labeled in blue. Following decarboxylation to α-KG, all 4 atoms are incorporated into ALA. If these carbons were to pass through the TCA cycle, they would label ALA as is shown in red for succinate. (B-C) Ball and stick models of protoporphyrin heme IX are shown. In both panels, carbon atoms originating from glycine are in green. (B) Carbons originating from glucose via the TCA cycle as shown in panel A are black. Because of decarboxylations during porphyrin synthesis, only 10 glucose-derived carbons can contribute to the final heme, whereas up to 16 carbons from succinate molecules that transits the cycle (red) could potentially end up in heme. (C) Carbons originating from glutamine are in blue. It should be noted that if succinate is directly converted to succinyl-CoA by the ATP-driven reverse SCS reaction, then its labeling pattern would be identical to what is shown for the glutamine-derived carbons.
The data obtained revealed that 2.5% of heme carbons were labeled when U-13C DES was the sole source of isotope label, whereas 8.3% were labeled with U-13C glucose and 23% of heme carbons were labeled by U-13C glutamine. Because the amount of heme synthesized in the first 48 hours postinduction is not subject to labeling, parallel experiments where heme synthesis was inhibited at 48 hours by the aminolevulinate dehydratase (porphobilinogen synthase) inhibitor succinylacetone32 were carried out. These demonstrated that 30% of the heme present at 72 hours was synthesized prior to the 24-hour period of the 13C-labeling experiments. Thus, total U-13C-labeled DES, glucose, and glutamine contributions are 3.6, 12.0, and 33.0%, respectively.
Isotopomer data demonstrated that succinate supplied as U-13C DES provided significant label in protoporphyrin only up to a total of 5 carbons (M+5) with the greatest contribution above background level to be M+2, M+3, and M+4 (Figure 2A). These data points indicate that succinate is not directly converted to succinyl-CoA by reverse SCS and that it is also a poor source of carbon for ALAS2 even after a passage through the TCA cycle. U-13C from glucose had the highest degree of labeling around M+8 and provided labeling above background approaching M+16 (Figure 2B). Significant labeling was seen as low as M+2 but not as high as M+18. These data points demonstrate that the overall contribution of glucose to cellular heme synthesis during erythropoiesis (12%) is minor. Glutamine-supplied U-13C resulted in heme with a maximum incorporation around M+13 and significant label up to M+23 (Figure 2C). There is no significant labeling below M+4. These data points show that glutamine is a superior precursor of succinyl-CoA for ALAS2 than is glucose or succinate and that it is used directly rather than passing through the TCA cycle (see supplemental materials). The isotopomer data suggest that no single compound in the cell during erythropoiesis is the sole source of carbons for the succinyl-CoA that is used by ALAS2. However, it is clear that glutamine is the most effective and plays a significant role during elevated heme synthesis.
Figure 2.

Isotopic labeling of heme and assorted metabolites by 13C diethyl succinate, 13C glucose or 13C glutamine, demonstrates that glutamine is strongly used as a precursor for heme but succinate is not. (A-C) Bar graphs show the experimentally determined labeling of heme, isolated from differentiating MEL cells after treatment with the indicated labeled metabolite and show the distribution of 13C-provided mass (“M”) for isolated hemes. In these figures, M represents heme detected with no 13C incorporated into the 34 carbons of heme (carbons are all 12C): M+1 is heme with 1 atom of 13C incorporated; M+2 is heme with 2 13C carbons, etc. The green bars represent the control values from samples treated with unlabeled metabolite and show the natural distribution of 13C incorporation. (A) The source of 13C is succinate as supplied by DES (yellow bars). Carbons derived from DES are only poorly used in heme biosynthesis. (B) The source of 13C label is glucose (black bars). The bar graph shows that some carbons from labeled glucose are incorporated into heme. (C) The source of 13C is glutamine (blue bars), which is converted to α-KG by deamination prior to KDH-driven synthesis of succinyl-CoA for ALA synthesis. Glutamine is shown here to be a superior precursor for heme biosynthesis, and isolated hemes are found to contain up to 23 carbon atoms derived from labeled glutamine. (D) The distribution of label originating from 13C glucose, 13C glutamine, and 15N glutamine into glutamine and glutamate are shown. (E) The distribution of label originating from 13C glucose, 13C glutamine, and 15N glutamine into the TCA cycle intermediates malate and fumarate are shown. (F) The distribution of label originating from 13C glucose, 13C glutamine, and 15N glutamine into proline is shown. Each labeling experiment was carried out in triplicate.
Realizing that glucose may be metabolized to glutamate and that this may cause an apparent dilution of glutamine label going into heme during erythropoiesis, we examined the amount of glutamine and glutamate in the cell that were produced from U-13C labeled glucose during the experimental period (Figure 2D). The data indicate that glucose contributes ∼10% to the cellular glutamate and glutamine pools at the M+2 level even when medium-supplied glutamine is present. Presumably this results from transamination of TCA cycle-derived α-KG. Thus, the isotopomer-based data for heme carbons derived from glutamine represent an underestimation, because the available U-13C glutamine pool is significantly diluted by endogenous unlabeled glutamine.
As shown in Figure 2E, in differentiating MEL cells, glucose is converted into the TCA cycle intermediates fumarate and malate as the M+2 and M+3 isotopomers better than is glutamine. This is consistent with our model, where glutamine is preferentially converted into succinyl-CoA for use by ALAS2 and not for a generic replenishing of the TCA cycle. However, glutamate is the precursor to proline, and this is evident from 13C- and 15N-glutamine labeling of proline in differentiating MEL cells (Figure 2F). Not surprising, and in accord with our data presented above, glucose also contributes carbons to proline via TCA cycle-generated α-KG that is transaminated to glutamate for subsequent proline synthesis.
TCA cycle enzymes during erythropoiesis
If alterations in central metabolite flux occur upon initiation of erythropoiesis, one might expect a corresponding change in key enzyme activities. Thus, if succinyl-CoA for ALA synthesis is supplied by the TCA cycle, there would be an increase in TCA cycle enzymes. In contrast, for a model that has glutamine as the major source of carbon for ALA, it would be anticipated that there would be an increase in the enzyme α-ketoglutarate dehydrogenase (KDH), but not necessarily an increase in all other TCA cycle enzymes. KDH is composed of 3 types of subunits: oxoglutarate dehydrogenase (E1), dihydrolipoamide S-succinyltransferase (E2), and dihydrolipoamide dehydrogenase (E3).33,34 Examination of data in ErythronDB35,36 reveals that during adult definitive erythropoiesis messenger RNA (mRNA) levels for the KDH subunit E1 have a positive correlation with heme synthesis enzymes ALAS2 (0.92) and FECH (0.90), which both show significant increases during erythropoiesis. However, ALAS2 and FECH have negative correlations with many TCA cycle enzymes (SDHB, −0.49; SUCLA2, −0.55; ACLY, −0.57; and SDHD, −0.75), which generally remain constant or decrease. Realizing that mRNA levels may not always accurately reflect enzyme activities, we measured the activity of 6 TCA cycle enzymes in MEL cells (Figure 3A).
Figure 3.

Activity of TCA cycle enzymes during erythroid differentiation. (A) The activity of 6 TCA cycle enzymes was determined in mitochondrial lysates from undifferentiated and differentiating MEL cells. Activity of cells induced toward erythroid differentiation was normalized to the activity of uninduced cells. CS, citrate synthase; Acon, aconitase; IDH, isocitrate dehydrogenase. Data shown are averages of 5 determinations. Error bars represent ± standard deviation. *P < .05; **P < .01. (B) The induction of KDH subunit E1 during erythropoiesis. E1 levels as detected by western blot analysis in differentiating MEL cells. On top is shown the western blot gel photograph and below is a densitometry analysis of the western blot in which E1 protein levels were normalized to total protein per lane. “Days” represents days postinduction. (C) In vitro protein-protein interaction between KDH and His-tagged ALAS2 is shown. Anti-E1 western blot showing KDH subunit E1 interacts with ALAS2 immobilized on a cobalt resin column. (D) The inhibition of hemoglobinization in differentiating MEL cells by itaconate is shown. Differentiating MEL cultures were treated with 2.5 mM itaconate ± ALA (100 µM [+]) for 4 days before being analyzed for hemoglobin content. All conditions were run in triplicate. Hb, hemoglobin; IA, itaconate.
Only aconitase, isocitrate dehydrogenase, and KDH showed increases in cells undergoing erythroid differentiation relative to their activity in undifferentiated cells. Due to the significant increase in KDH enzyme activity as measured in vitro, further examination of KDH was warranted because it plays a central role in any model of heme synthesis. We verified that the ErythronDB mRNA increase of E1 was reflected in protein expression in differentiating MEL cells via western blot analysis (Figure 3B). There is a more than threefold increase in E1 protein. Thus, mRNA, protein, and enzyme activity measurements all demonstrate an increase in the E1 KDH subunit during erythropoiesis.
Because KDH can be a nucleating point for some TCA cycle enzymes,33 we wondered if it might interact with ALAS2. When purified, His-tagged, recombinant ALAS2 and untagged KDH purified from porcine hearts were mixed in solution and applied to a cobalt resin column, and the E1 component of KDH coeluted with ALAS2 (Figure 3C). This illustrates that in vitro the E1 component of KDH interacts with ALAS2.
In addition, in situ interactions between KDH and ALAS2 were probed with antibody pull-downs of mitochondria from differentiating MEL cells using flag-tagged ALAS2 as bait as described previously.37 These data points clearly demonstrated that ALAS2 interacts with the E1, E2, and E3 subunits of KDH (Table 1).
Table 1.
Affinity purification and MS results of ALAS2
| Murine protein | |||||
|---|---|---|---|---|---|
| Human input | Kdh E1 | Kdh E2 | Kdh E3 | Alas2 | Fumarate hydratase |
| ALAS2 (n = 2) | 20-45 | 10-23 | 10-20 | 90-347 | 4-10 |
| 15-33 | 7-13 | 6-10 | 22-34 | 4-8 | |
| 17-29% | 19-25% | 16-19% | 42-56% | 11-13% | |
| Empty vector (n = 2) | 0-2 | 6-7 | 0-4 | 0 | 3-6 |
| 0-2 | 2-3 | 0-3 | 0 | 2-3 | |
| 0-2% | 7-10% | 0-7% | 0% | 6-7% | |
Data shown top to bottom as spectral counts, number of unique peptides, and percent coverage, respectively.58
Impact of itaconate
Factors that influence the cellular supply of succinyl-CoA during erythropoiesis could clearly have an impact on heme synthesis. Itaconate, which is produced from the TCA cycle intermediate cis-aconitate by the enzyme IRG1, has been shown to disrupt the TCA cycle by blocking SDH resulting in increased cellular succinate.27-29,38-40 Given that itaconate causes accumulation of intracellular succinate, it could be imagined that it might stimulate porphyrin synthesis by increasing the amount of succinyl-CoA available (from reverse ATP-dependent SCS activity). In differentiating MEL cells, we monitored heme synthesis during erythroid differentiation41 (Figure 3D). Our data demonstrated that addition of 2.5 mM itaconate to cultures of differentiating cells did not result in an increase in heme synthesis, but instead caused an arrest in hemoglobinization. Significantly, this block could be reversed by adding ALA to the culture. These results are consistent with the finding from the isotopomeric studies that succinate is not the main source of succinyl-CoA for heme synthesis during late erythropoiesis.
Impact of metabolic inhibitors on erythroid heme synthesis
In order to further probe the source of precursors of α-KG and succinyl-CoA for heme synthesis, the impact of inhibitors that target enzymes potentially required for the synthesis of succinyl-CoA was examined in cells undergoing erythropoiesis. Aminooxyacetic acid (AOA), a broad-spectrum inhibitor of pyridoxal phosphate–dependent transaminases, has been shown to block early erythroid differentiation of CD34+ HSCs.42 AOA blocks glutamine deamination and thus the eventual conversion of glutamine into α-KG.43 This block can be bypassed by the addition of dimethyl-α-ketoglutarate (DMK), a cell-permeable analog of α-KG, to culture medium. Given that our data support the hypothesis that glutamine serves as source of carbons for heme synthesis by way of α-KG, we tested whether AOA impairs late erythroid differentiation by blocking heme synthesis. To do this, we examined the level of heme (as hemoglobin) produced by differentiating MEL cells treated with AOA.
MEL cells were selected because they are transformed murine spleen red cell precursors that are arrested at the proerythroblast stage. Upon DMSO-induced differentiation, there is an induction of heme biosynthesis.44,45 Addition of AOA at the initiation of erythroid differentiation reduced hemoglobin content by 76% as compared with untreated cells (Figure 4A). The addition of either DMK or ALA to AOA-treated differentiating MEL cell cultures rescued hemoglobinization (Figure 4B), whereas supplementing cultures with DES did not. These data points are consistent with the proposal that succinyl-CoA for heme synthesis during erythropoiesis arises from α-KG, and not succinate from the TCA cycle.
Figure 4.

Rescue of hemoglobinization in AOA treated differentiating MEL cells. ALA and DMK but not DES rescue hemoglobinization of AOA treated MEL cells at 72 hours. MEL cells were treated with AOA and/or metabolites at erythroid differentiation induction (A) and at 48 hours postinduction (B). Data shown are averages of 3 determinations. Error bars represent mean ± standard deviation. **P < .01. SA, succinylacetone.
In addition to MEL cells, we also examined human CD34+ cells to differentiate between cellular needs for glutamine prior to and during hemoglobinization. Human CD34+ cells are primary cells that represent a population of HSCs. When induced toward erythroid differentiation, they first pass through several early stages of erythropoiesis before becoming heme synthesizing basophilic erythroblasts.46 We monitored early erythroid differentiation at day 6, prior to significant elevation of ALAS2 and FECH that occurs at day 7 (supplemental Figure 2), by following expression of the surface receptors CD36, CD71 (transferrin), and CD235a (glycophorin A) and determined the impact that addition of DMK, DES, nucleosides, and/or ALA had on the differentiating CD34+ cells (Figure 5A). As previously reported,42 we found that the AOA-inhibited early erythroid differentiation could be overcome by addition of DMK to the medium (Figure 5B). This inhibition was not overcome by individual additions of DES, ALA, or nucleosides. These data points are consistent with the acknowledged multiple roles of glutamine (via α-KG) in cells undergoing the early stages of erythropoiesis.
Figure 5.
Erythroid differentiation of CD34+cells. (A) CD34+-enriched cells were induced to erythroid differentiation in the absence or presence of DMK, ALA, nucleosides, or AOA. (B) Cells were exposed to DMK, ALA, nucleosides, or DES in combination with AOA as indicated. After 6 days of differentiation and treatment in StemSpan media with erythroid expansion supplement, cells were collected and differentiation was assessed by flow cytometry detection of cell surface markers CD235a and CD71. Labels for each treatment type are shown at the top of the figures. For each treatment type, from top to bottom, graphs illustrate flow cytometric pseudocolor plots of cells stained using CD235a (x-axis) and CD71 (y-axis) antibodies, histograms of these cells stained for CD71, and histograms of these cells stained for CD235a. (C) Results from CD34+ cells that were grown for 6 days in StemSpan media with erythroid expansion supplement before the removal of defined growth factors/cytokines other than EPO as described in the text. Cellular heme levels are shown for cultures after 3 and 6 days in the presence of EPO with indicated compound. UN, untreated. The bar graph presents combined biological and technical replicates (n = 4-6), mean ± standard deviation.


For cells that are undergoing late erythropoiesis and beginning to hemoglobinize, we measured cellular heme content41 at days 3 and 6 following removal of cytokines/growth factors other than erythropoietin (EPO). For these cells, we found that AOA inhibited hemoglobinization and that this could be overcome by addition of DMK and ALA, but not DES, to the medium (Figure 5C; supplemental Figure 3). These data points indicate that a major role served by glutamine during hemoglobinization must be for heme synthesis because ALA alone overcame the AOA block during late stages of erythropoiesis. Interestingly, when media were supplemented with ALA, there was background fluorescence, likely due to the presence of unmetallated porphyrin, that was not seen in the DMK or DES supplemented samples.
Discussion
In going from a proerythroblast to mature erythrocyte, cells can repurpose amino acids from a wide variety of cytoplasmic proteins for synthesis of new globins chains, but each cell must provide ∼1010 molecules of succinyl-CoA and an equivalent amount of glycine for heme synthesis. Although plasma and mitochondrial membrane glycine transporters support the provisioning of glycine from the circulatory system during erythropoiesis,21-23 significant stores of succinyl-CoA do not exist and so this must be provided endogenously in a coordinated, real-time fashion. It is conceivable that in nonerythroid cells the TCA cycle can supply succinyl-CoA without compromising other metabolic pathways. However, given the level of heme synthesis during erythropoiesis, it is implausible that the TCA cycle would be able to function without robust anplerosis to resupply intermediates.
Although there have been proposals for the TCA cycle SUCLA2-dependent reverse SCS supply of succinyl-CoA for ALAS2,25,26 there exist no experimental data to support this hypothesis, and a number of observations are not consistent with this model. First, early studies with the particulate fraction from anemic chicken reticulocytes showed that α-KG served more efficiently than succinate as a precursor for ALA synthesis.19,47 Second, from an overall carbon economy perspective, utilizing TCA cycle intermediates for ALA production would require large-scale replenishment of the TCA cycle via anaplerotic reactions, not to mention the energetic costs of running the SCS reaction backward to generate succinyl-CoA for heme. Third, mice heterozygous for SUCLA2 knockout48 and human patients with SUCLA2 deficiency (MIM ID #612073) exhibit no signs of anemia, which one would anticipate if the ALAS2-SUCLA2 interaction is essential for the supply of succinyl-CoA for heme synthesis. Fourth, mRNA expression profiling during erythropoiesis shows a negative correlation (−0.7) between SUCLA2 and ALAS2 gene expression.35,36 Fifth, ALAS2 variants that have decreased interactions with SUCLA2 do not exhibit a uniform impact on ALAS2 activity. Some mutations result in diminished ALAS2 activity causing sideroblastic anemia,25,26 whereas others result in increased ALAS2 activity causing X-linked protoporphyria.49 Finally, we have demonstrated that succinate provided to differentiating erythroid precursor cells is a poor source of carbons for ALA synthesis when compared with glutamine.
What role SUCLA2 plays is unclear at present. Because it binds to both ALAS2 and FECH,37 it could be that during early stages of differentiation it stabilizes the apoprotein forms of ALAS2, which requires pyridoxyl phosphate as a cofactor,50 and FECH, which requires the presence of a [2Fe2S] cluster.51 Another possibility is that by SUCLA2 binding to ALAS2 and FECH it may prevent assembly of functional ATP-dependent SCS and, thereby, reduce conversion of succinyl-CoA into succinate when ALAS2 levels are high.
Our isotopomer-based analysis clearly demonstrated that glutamine is the preferred source of carbon for protoporphyrin synthesis (Figure 2C). Experimentally we determined that 13C-glutamine labeled 30% of the eligible total carbons in heme.
However, liquid LC/MS analysis reveals that only about one-third of the glutamine in the cell after 24 hours of incubation contains 13C label and that glucose contributes ∼10% of the cellular glutamate and glutamine pools. With these factors taken into consideration, we estimate that the experimentally determined magnitude of carbon flux from glutamine into heme is low by at least a factor of 2. A model where ALA is derived from glutamine via KDH rather than from succinate via SCS (Figure 6) is consistent with both the data presented herein and the published clinical observation that deficiency in thiamine, a required cofactor for KDH, results in a sideroblastic anemia52,53 similar to that found in X-linked sideroblastic anemia caused by deficiency in ALAS2 activity.
Figure 6.

Overview of cellular metabolism for heme synthesis during early and late erythropoiesis. A cartoon of a cell is shown where the plasma membrane is a heavy blue line, and the mitochondrial inner and outer membranes are red with intermembrane space in light pink. Plasma membrane transporters for glucose, glycine, and glutamine are orange; mitochondrial inner membrane transporters for glutamate, glycine, ALA, and pyruvate are green. The heme biosynthetic pathway is abbreviated as a dashed arrow, but is shown in its entirety in supplemental Figure 1. Heme, the end product, exits the mitochondrial matrix following its release from FECH, but the pathway for this is currently undetermined. The TCA cycle is in blue with the enzymes KDH and SCS see text) shown boxed in the figure. The site of inhibition by AOA is illustrated with a bold red X. Multiple glutamine transaminases exist within the cell, and all are inhibited by AOA. (A) Cellular metabolism for late erythropoiesis when large quantities of heme are necessary for hemoglobinization of the developing erythron. Inside the mitochondrion are shown the pathways of glutamate to ALA with KDH in complex with ALAS2 and the previously described heme metabolon (Heme Metab.)38 as a red pentagon. (B) Cellular metabolism during early erythropoiesis when heme for cellular functions is synthesized utilizing ALAS1. Under these conditions, the amounts of heme synthesized are relatively low, and the required succinyl-CoA can be provided by the TCA cycle.
Our data are consistent with a separation of glutamine for heme synthesis from a general anaplerotic replenishment of the TCA cycle in that we show glucose contributes to the TCA cycle intermediates during erythropoiesis more effectively than glutamine.
A glucose-derived label is found in malate and fumarate at higher levels than glutamine-derived malate and fumarate. This, along with the observation that 13C label from exogenously supplied glucose is found in significant amounts in cellular glutamate (Figure 2D), suggests that glucose may make contributions to heme via the cellular glutamate pool.
One interesting observation was that exposure of differentiating erythroid cells to itaconate, which is produced and secreted by immunoactivated macrophages and has been shown to increase intracellular succinate levels,27,28 did not stimulate heme synthesis as might be anticipated if succinate is a precursor for ALA synthesis. Instead, it inhibited hemoglobinization of these cells. This inhibition was rescued by addition of ALA to the culture medium, demonstrating that the blockage in hemoglobinization caused by itaconate occurs by inhibiting the availability of a necessary precursor or by directly inhibiting ALA production. This observation that itaconate decreases hemoglobinization in the developing erythron may be evidence for a link between anemia of inflammation and heme production. Itaconate is produced from the TCA cycle intermediate aconitate by the enzyme IRG1 and Irg1 as 1 of the most highly upregulated genes in macrophages under proinflammatory conditions28,38 Activated macrophages, which are present in erythroblastic islands along with erythroid precursor cells,54,55 excrete high concentrations of itaconate into the surrounding milieu. Thus, the action of itaconate on heme synthesis bears a resemblance to the action of hepcidin on heme synthesis during chronic infection.56 Both hepcidin and itaconate are antimicrobial compounds produced during the inflammatory response, and both inhibit heme synthesis, hepcidin by regulating iron and itaconate by regulating porphyrin synthesis. Thus, these may represent complementary systems that not only regulate overall heme synthesis but also prevent the accumulation of 2 toxic compounds: protoporphyrin and iron.
Glucose and glutamine metabolism via KDH have been shown to be essential for erythroid lineage commitment of CD34+ HSCs.43 Because glutamine is the most abundant amino acid in plasma, and plasma glucose levels in a healthy person range from 4 to 6 mM, both metabolites are readily available to developing erythroblasts. Knockdown of the glucose transporter Glut1 or the glutamine transporter ASCT2 in CD34+ stem cells decreased erythroid differentiation as measured by erythroid lineage-specific surface antigens. Oburoglu et al42 reported that glutamine is essential for nucleotide biosynthesis during erythropoiesis. Their conclusion that glutamine is required for nucleotide synthesis was based upon nucleoside rescue of CD34+ cells treated with 6-diazo-5-oxo-l-norleucine (DON). DON is a known inhibitor of glutamine utilizing enzymes, such as carbamoyl phosphate synthase, CTP synthase, NAD synthase, guanosine monophosphate synthetase, glutamine phosphoribosylpyrophosphate amidotransferase, formylglycinamide ribonucleotide amidotransferase, and asparagine synthase, which can all directly block nucleotide synthesis.57 This is aside from its ability to function as a glutaminase inhibitor. Because of DON’s targeted role in transaminase-independent inhibition of nucleotide synthesis, we focused on inhibition by AOA, which is more restricted to transaminases and not downstream pathways. Interestingly, we found that AOA inhibition of early erythropoiesis of CD34+ cells was not reversed by addition of nucleosides to the medium. Likewise, unlike DMK, ALA addition either alone or with nucleosides did not rescue the appearance of cell surface markers of erythroid maturation in CD34+ cell during early erythropoiesis. These data points demonstrate that glutamine plays a much greater role during early erythropoiesis than simply as a precursor to heme and nucleotide synthesis. The fact that either DMK or ALA rescues the AOA-mediated block of hemoglobinization in MEL cells and late-stage CD34+ cells supports an essential role for α-KG in hemoglobinization during late erythropoiesis. Glucose, which was present in all culture media, and DES did not rescue hemoglobinization in these cells, supporting the fact that their contributions alone to the production of succinyl-CoA via the TCA cycle are insufficient to support heme synthesis during erythropoiesis. However, it is clear from the 13C labeling, enzyme activity, cell culture data, and identification of the interaction between KDH and ALAS2 that KDH, in addition to being a component of the TCA cycle, serves a critical “moonlighting” role as primary supplier to succinyl-CoA for heme synthesis during erythropoietic hemoglobinization.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors gratefully acknowledge Tamara A. Dailey (deceased) for outstanding editorial assistance and critical discussions.
This work was supported by National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases grants DK111653 (A.E.M.), U54 DK110858, DK090257 (J.D.P.), DK096501 (H.A.D.), NIH Office of the Director grant S10 OD016232 (J.C.) and by the University of Utah Flow Cytometry Facility in addition to the National Institutes of Health, National Cancer Institute through Award Number 5P30CA042014-24. Research reported in this publication was supported by the National Center for Research Resources of the National Institutes of Health under Award Number 1S10RR026802-01.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: J.S.B. designed experiments, analyzed data and wrote the paper; J.R.M., J.A.M., and L.K.J. performed research and analyzed data; J.E.C., A.E.M., and J.D.P. analyzed data and edited the paper; and H.A.D. designed experiments, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Harry A Dailey Jr, Department of Microbiology, 230 Coverdell Center, University of Georgia, Athens, GA 30602; e-mail: hdailey@uga.edu.
REFERENCES
- 1.Hattangadi SM, Wong P, Zhang L, Flygare J, Lodish HF. From stem cell to red cell: regulation of erythropoiesis at multiple levels by multiple proteins, RNAs, and chromatin modifications. Blood. 2011;118(24):6258-6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132(4):631-644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bessis M. [Erythroblastic island, functional unity of bone marrow]. Rev Hematol. 1958;13(1):8-11. [PubMed] [Google Scholar]
- 4.Manwani D, Bieker JJ. The erythroblastic island. Curr Top Dev Biol. 2008;82:23-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dailey HA, Meissner PN. Erythroid heme biosynthesis and its disorders. Cold Spring Harb Perspect Med. 2013;3(4):a011676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.An X, Schulz VP, Li J, et al. Global transcriptome analyses of human and murine terminal erythroid differentiation. Blood. 2014;123(22):3466-3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Medlock AE, Dailey HA. Regulation of mammalian heme biosynthesis. In: Warren MJ, Smith AG, eds. Tetrapyrroles: Birth, Life and Death. New York, NY: Landes Bioscience and Springer Science + Business Media; 2009:116-127. [Google Scholar]
- 8.Chung J, Wittig JG, Ghamari A, et al. Erythropoietin signaling regulates heme biosynthesis. eLife. 2017;6:e24767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhasker CR, Burgiel G, Neupert B, Emery-Goodman A, Kühn LC, May BK. The putative iron-responsive element in the human erythroid 5-aminolevulinate synthase mRNA mediates translational control. J Biol Chem. 1993;268(17):12699-12705. [PubMed] [Google Scholar]
- 10.Melefors O, Goossen B, Johansson HE, Stripecke R, Gray NK, Hentze MW. Translational control of 5-aminolevulinate synthase mRNA by iron-responsive elements in erythroid cells. J Biol Chem. 1993;268(8):5974-5978. [PubMed] [Google Scholar]
- 11.Schranzhofer M, Schifrer M, Cabrera JA, et al. Remodeling the regulation of iron metabolism during erythroid differentiation to ensure efficient heme biosynthesis. Blood. 2006;107(10):4159-4167. [DOI] [PubMed] [Google Scholar]
- 12.Ye H, Rouault TA. Human iron-sulfur cluster assembly, cellular iron homeostasis, and disease. Biochemistry. 2010;49(24):4945-4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doyle F, Tenenbaum SA. Trans-regulation of RNA-binding protein motifs by microRNA. Front Genet. 2014;5:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kardon JR, Yien YY, Huston NC, et al. Mitochondrial ClpX activates a key enzyme for heme biosynthesis and erythropoiesis. Cell. 2015;161(4):858-867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abu-Farha M, Niles J, Willmore WG. Erythroid-specific 5-aminolevulinate synthase protein is stabilized by low oxygen and proteasomal inhibition. Biochem Cell Biol. 2005;83(5):620-630. [DOI] [PubMed] [Google Scholar]
- 16.Dailey TA, Woodruff JH, Dailey HA. Examination of mitochondrial protein targeting of haem synthetic enzymes: in vivo identification of three functional haem-responsive motifs in 5-aminolaevulinate synthase. Biochem J. 2005;386(Pt 2):381-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lathrop JT, Timko MP. Regulation by heme of mitochondrial protein transport through a conserved amino acid motif. Science. 1993;259(5094):522-525. [DOI] [PubMed] [Google Scholar]
- 18.Yamauchi K, Hayashi N, Kikuchi G. Translocation of delta-aminolevulinate synthase from the cytosol to the mitochondria and its regulation by hemin in the rat liver. J Biol Chem. 1980;255(4):1746-1751. [PubMed] [Google Scholar]
- 19.Gibson KD, Laver WG, Neuberger A. Initial stages in the biosynthesis of porphyrins. 2. The formation of delta-aminolaevulic acid from glycine and succinyl-coenzyme A by particles from chicken erythrocytes. Biochem J. 1958;70(1):71-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen C, Paw BH. Cellular and mitochondrial iron homeostasis in vertebrates. Biochim Biophys Acta. 2012;1823(9):1459-1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fernández-Murray JP, Prykhozhij SV, Dufay JN, et al. Glycine and folate ameliorate models of congenital sideroblastic anemia. PLoS Genet. 2016;12(1):e1005783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garcia-Santos D, Schranzhofer M, Bergeron R, Sheftel AD, Ponka P. Extracellular glycine is necessary for optimal hemoglobinization of erythroid cells. Haematologica. 2017;102(8):1314-1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schranzhofer MBR, Ponka P. Glycine transporter 1 plays a crucial role in hemoglobinization [abstract]. Blood. 118(21). Abstract 345. [Google Scholar]
- 24.Bishop DF, Tchaikovskii V, Hoffbrand AV, Fraser ME, Margolis S. X-linked sideroblastic anemia due to carboxyl-terminal ALAS2 mutations that cause loss of binding to the β-subunit of succinyl-CoA synthetase (SUCLA2). J Biol Chem. 2012;287(34):28943-28955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Furuyama K, Sassa S. Interaction between succinyl CoA synthetase and the heme-biosynthetic enzyme ALAS-E is disrupted in sideroblastic anemia. J Clin Invest. 2000;105(6):757-764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Labbe RF, Kurumada T, Onisawa J. The role of succinyl-CoA synthetase in the control of heme biosynthesis. Biochim Biophys Acta. 1965;111(2):403-415. [DOI] [PubMed] [Google Scholar]
- 27.Michelucci A, Cordes T, Ghelfi J, et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci USA. 2013;110(19):7820-7825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cordes T, Wallace M, Michelucci A, et al. Immunoresponsive gene 1 and itaconate inhibit succinate dehydrogenase to modulate intracellular succinate levels. J Biol Chem. 2016;291(27):14274-14284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lampropoulou V, Sergushichev A, Bambouskova M, et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. 2016;24(1):158-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Owen OE, Kalhan SC, Hanson RW. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem. 2002;277(34):30409-30412. [DOI] [PubMed] [Google Scholar]
- 31.Zacharias NM, Chan HR, Sailasuta N, Ross BD, Bhattacharya P. Real-time molecular imaging of tricarboxylic acid cycle metabolism in vivo by hyperpolarized 1-(13)C diethyl succinate. J Am Chem Soc. 2012;134(2):934-943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ebert PS, Hess RA, Frykholm BC, Tschudy DP. Succinylacetone, a potent inhibitor of heme biosynthesis: effect on cell growth, heme content and delta-aminolevulinic acid dehydratase activity of malignant murine erythroleukemia cells. Biochem Biophys Res Commun. 1979;88(4):1382-1390. [DOI] [PubMed] [Google Scholar]
- 33.Lyubarev AE, Kurganov BI. Supramolecular organization of tricarboxylic acid cycle enzymes. Biosystems. 1989;22(2):91-102. [DOI] [PubMed] [Google Scholar]
- 34.Porpáczy Z, Sümegi B, Alkonyi I. Association between the alpha-ketoglutarate dehydrogenase complex and succinate thiokinase. Biochim Biophys Acta. 1983;749(2):172-179. [DOI] [PubMed] [Google Scholar]
- 35.Greenfest-Allen E, Malik J, Palis J, Stoeckert CJ Jr. Stat and interferon genes identified by network analysis differentially regulate primitive and definitive erythropoiesis. BMC Syst Biol. 2013;7:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kingsley PD, Greenfest-Allen E, Frame JM, et al. Ontogeny of erythroid gene expression. Blood. 2013;121(6):e5-e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Medlock AE, Shiferaw MT, Marcero JR, et al. Identification of the mitochondrial heme metabolism complex. PLoS One. 2015;10(8):e0135896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luan HH, Medzhitov R. Food fight: role of itaconate and other metabolites in antimicrobial defense. Cell Metab. 2016;24(3):379-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cordes T, Michelucci A, Hiller K. Itaconic acid: the surprising role of an industrial compound as a mammalian antimicrobial metabolite. Annu Rev Nutr. 2015;35:451-473. [DOI] [PubMed] [Google Scholar]
- 40.Németh B, Doczi J, Csete D, et al. Abolition of mitochondrial substrate-level phosphorylation by itaconic acid produced by LPS-induced Irg1 expression in cells of murine macrophage lineage. FASEB J. 2016;30(1):286-300. [DOI] [PubMed] [Google Scholar]
- 41.Marcero JR, Piel Iii RB, Burch JS, Dailey HA. Rapid and sensitive quantitation of heme in hemoglobinized cells. Biotechniques. 2016;61(2):83-91. [DOI] [PubMed] [Google Scholar]
- 42.Oburoglu L, Tardito S, Fritz V, et al. Glucose and glutamine metabolism regulate human hematopoietic stem cell lineage specification [published correction appears in Cell Stem Cell. 2014;15(5):666-668]. Cell Stem Cell. 2014;15(2):169-184. [DOI] [PubMed] [Google Scholar]
- 43.Katt WP, Cerione RA. Glutaminase regulation in cancer cells: a druggable chain of events. Drug Discov Today. 2014;19(4):450-457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Conder LH, Woodard SI, Dailey HA. Multiple mechanisms for the regulation of haem synthesis during erythroid cell differentiation. Possible role for coproporphyrinogen oxidase. Biochem J. 1991;275(Pt 2):321-326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lake-Bullock H, Dailey HA. Biphasic ordered induction of heme synthesis in differentiating murine erythroleukemia cells: role of erythroid 5-aminolevulinate synthase. Mol Cell Biol. 1993;13(11):7122-7132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neildez-Nguyen TM, Wajcman H, Marden MC, et al. Human erythroid cells produced ex vivo at large scale differentiate into red blood cells in vivo. Nat Biotechnol. 2002;20(5):467-472. [DOI] [PubMed] [Google Scholar]
- 47.Laver WG, Neuberger A, Udenfriend S. Initial stages in the biosynthesis of porphyrins. I. The formation of delta-am-inolaevulic acid by particles obtained from chicken erythrocytes. Biochem J. 1958;70(1):4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kacso G, Ravasz D, Doczi J, et al. Two transgenic mouse models for β-subunit components of succinate-CoA ligase yielding pleiotropic metabolic alterations. Biochem J. 2016;473(20):3463-3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bishop DF, Tchaikovskii V, Nazarenko I, Desnick RJ. Molecular expression and characterization of erythroid-specific 5-aminolevulinate synthase gain-of-function mutations causing X-linked protoporphyria. Mol Med. 2013;19(1):18-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Neuberger A. Aspects of the metabolism of glycine and of porphyrins. Biochem J. 1961;78:1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dailey HA, Finnegan MG, Johnson MK. Human ferrochelatase is an iron-sulfur protein. Biochemistry. 1994;33(2):403-407. [DOI] [PubMed] [Google Scholar]
- 52.Diaz GA, Banikazemi M, Oishi K, Desnick RJ, Gelb BD. Mutations in a new gene encoding a thiamine transporter cause thiamine-responsive megaloblastic anaemia syndrome. Nat Genet. 1999;22(3):309-312. [DOI] [PubMed] [Google Scholar]
- 53.Fleming JC, Tartaglini E, Steinkamp MP, Schorderet DF, Cohen N, Neufeld EJ. The gene mutated in thiamine-responsive anaemia with diabetes and deafness (TRMA) encodes a functional thiamine transporter. Nat Genet. 1999;22(3):305-308. [DOI] [PubMed] [Google Scholar]
- 54.Alam MZ, Devalaraja S, Haldar M. The heme connection: linking erythrocytes and macrophage biology. Front Immunol. 2017;8:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Soares MP, Hamza I. Macrophages and iron metabolism. Immunity. 2016;44(3):492-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nemeth E, Valore EV, Territo M, Schiller G, Lichtenstein A, Ganz T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood. 2003;101(7):2461-2463. [DOI] [PubMed] [Google Scholar]
- 57.Cervantes-Madrid D, Romero Y, Duenas-Gonzalez A. Reviving lonidamine and 6-diazo-5-oxo-L-norleucine to be used in combination for metabolic cancer therapy. Biomed Res Int. 2015;2015:690492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stehling O, Vashisht AA, Mascarenhas J, et al. MMS19 assembles iron-sulfur proteins required for DNA metabolism and genomic integrity. Science. 2012;337(6091):195-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.