

Key Points
Inhibition of the ibrutinib-resistant C481S BTK mutant can be achieved by the reversible BTK inhibitor GDC-0853.
BTK inhibitors, which lack ITK inhibition, preserve natural killer cell–mediated cellular cytotoxicity to CD20 directed monoclonal antibodies.
Abstract
The clinical success of ibrutinib validates Bruton tyrosine kinase (BTK) inhibition as an effective strategy for treating hematologic malignancies, including chronic lymphocytic leukemia (CLL). Despite ibrutinib’s ability to produce durable remissions in patients, acquired resistance can develop, mostly commonly by mutation of C481 of BTK in the ibrutinib binding site. Here, we characterize a novel BTK inhibitor, GDC-0853, to evaluate its preclinical efficacy in ibrutinib-naive and ibrutinib-resistant CLL. GDC-0853 is unique among reported BTK inhibitors in that it does not rely upon covalent reaction with C481 to stabilize its occupancy within BTK’s adenosine triphosphate binding site. As with ibrutinib, GDC-0853 potently reduces B-cell receptor signaling, viability, NF-κB–dependent transcription, activation, and migration in treatment naïve CLL cells. We found that GDC-0853 also inhibits the most commonly reported ibrutinib-resistant BTK mutant (C481S) both in a biochemical enzyme activity assay and in a stably transfected 293T cell line and maintains cytotoxicity against patient CLL cells harboring C481S BTK mutations. Additionally, GDC-0853 does not inhibit endothelial growth factor receptor or ITK, 2 alternative targets of ibrutinib that are likely responsible for some adverse events and may reduce the efficacy of ibrutinib-antibody combinations, respectively. Our results using GDC-0853 indicate that noncovalent, selective BTK inhibition may be effective in CLL either as monotherapy or in combination with therapeutic antibodies, especially among the emerging population of patients with acquired resistance to ibrutinib therapy.
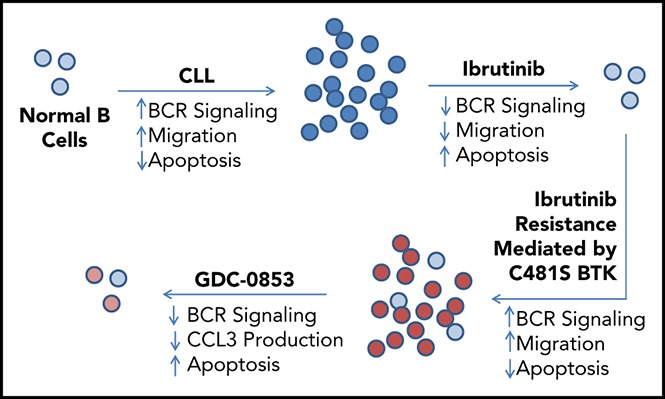
Visual Abstract
Introduction
Chronic lymphocytic leukemia (CLL) is the most prevalent adult leukemia, with an estimated annual incidence >18 000 in the United States.1 Treatment of CLL has changed dramatically in recent years with the introduction of therapies that target B-cell receptor (BCR) signaling, a pathway known to enhance proliferation, immune function, and survival of malignant B lymphocytes.2-5 Among the components of the BCR pathway, Bruton tyrosine kinase (BTK) has emerged as an effective therapeutic target in a number of B-cell malignancies including CLL.6,7 In addition to its role in the BCR pathway, BTK propagates signaling cascades originating from toll-like receptors (TLRs), chemokine receptors, and a variety of survival inducing cytokine receptors.8-11 Mouse models that overexpress BTK in B cells show increased mortality because of systemic autoimmune disease.12 B lymphocytes from these mice demonstrate hyperresponsiveness to BCR stimulation, increased NF-κB activity, and resistance to Fas-mediated apoptosis.12 Conversely, XID mice, which lack BTK kinase activity because of a point mutation in the pleckstrin homology domain of BTK, exhibit slower rates of CLL development and have significantly improved survival when crossed with the Eμ-TCL1 (TCL1) murine CLL model compared with the parental TCL1 strain.6 Taken together, these observations implicate BTK as an important driver of CLL disease progression.
Ibrutinib is a first-in-class irreversible BTK inhibitor developed for the treatment of B-cell malignancies and is currently approved for the treatment of CLL, relapsed mantle cell lymphoma, marginal zone lymphoma, and Waldenström macroglobulinemia. Ibrutinib irreversibly inhibits BTK kinase activity by covalently reacting with the C481 amino acid residue in the adenosine triphosphate binding site.13-16 Ibrutinib has been extraordinarily successful in CLL therapy, including in patients with high-risk cytogenetic abnormalities including del(17)(p13.1).17 Randomized phase 3 trials have also shown a survival advantage with ibrutinib treatment over standard therapies for both treatment-naïve and relapsed/refractory CLL.18,19 The preclinical work and clinical success of ibrutinib validates BTK inhibition as an effective strategy for treating many low-grade hematologic malignancies.
Despite ibrutinib’s activity in CLL, acquired resistance to ibrutinib does develop in a subset of heavily pretreated patients and is most commonly mediated by mutation of BTK cysteine-481, the amino acid of BTK with which ibrutinib irreversibly reacts, to serine.20,21 C481S BTK mutations have been reported to diminish ibrutinib’s potency 500-fold and prevent its covalent binding, rendering it unable to effect irreversible inhibition of BTK.20,22 This same pattern of resistance has also been seen with acalabrutinib, a second-generation irreversible BTK inhibitor,23 although the incidence of resistance mutations associated with this more selective agent requires further investigation. The observation that acquired resistance to irreversible BTK inhibitors is facilitated via a mutation of ibrutinib’s BTK-binding site whereas other irreversible targets of ibrutinib are not mutated suggests that BTK is of extreme importance to disease progression. Maintaining BTK inhibition in the context of ibrutinib resistance may therefore provide further clinical benefit.
Ibrutinib’s irreversible inhibition of alternative targets other than BTK (eg, endothelial growth factor receptor [EGFR], ITK, TEC, BMX, BLK, HER2, HER4, and JAK3) has been suggested to be associated with adverse treatment-related events and immune modulation. Ibrutinib-associated adverse events include rash, diarrhea, atrial fibrillation, and thrombocytopenia.17,19 Patients who discontinue ibrutinib as the result of treatment-related adverse events have been found to outnumber those who stop treatment because of disease progression.24,25 EGFR inhibition has been previously linked to rash and diarrhea26,27 and is proposed to mediate these adverse events in patients who experience them on ibrutinib. Inhibition of ITK by ibrutinib modulates T-cell populations28 and has been shown to suppress antibody-dependent cell-mediated cytotoxicity (ADCC) by natural killer (NK) cells in response to therapeutic anti-CD20 antibodies in vitro.29,30 Combinations of ibrutinib with antibody therapies have not yet been shown to be more effective than ibrutinib alone.31,32 Development of a therapeutic agent that inhibits multiple BTK variants without inhibiting alternative targets such as EGFR and ITK may offer a potential therapeutic advantage.
Herein, we characterize GDC-0853, a reversible small molecule drug that is extremely selective for inhibition of BTK.33 GDC-0853 binds to BTK without covalently reacting with C481.33-35 Because of the noncovalent mechanism of inhibition and novel BTK-binding mode of GDC-0853 and other similar inhibitors,36 we expected that GDC-0853 would maintain efficacy in situations of ibrutinib resistance mediated by C481S BTK mutations. Recent work has shown that noncovalent inhibitors similar in structure to GDC-0853 are able to fully retain potent inhibition of C481S BTK mutant enzyme in biochemical and cellular systems.36 We attempt to extend these findings to patient-derived CLL B cells and other models to further validate the efficacy of noncovalent inhibition on C481S BTK. We further hypothesize that, because of its high degree of specificity for BTK, that GDC-0853 lacks ITK and EGFR inhibition, thereby diminishing alternative on-target effects of ibrutinib. This work supports the clinical trial design and first-in-human phase 1 study of the BTK inhibitor GDC-0853 in relapsed or refractory B-cell non-Hodgkin lymphoma and CLL in which patients that had relapsed on ibrutinib and were identified to have the C481S mutation were enrolled.37
Methods
Subject population and lymphocyte isolation
Blood samples were obtained from patients with CLL at our institution who consented to an institutional review board–approved tissue procurement protocol or who were enrolled in institutional review board–approved clinical trials of ibrutinib in CLL. Primary CLL cells were either used fresh or were viably cryopreserved and used at later dates. Human T cells were obtained from healthy volunteers. All patients gave written informed consent in accordance with the Declaration of Helsinki. Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood through Ficoll density gradient centrifugation. Specific leukocyte populations were negatively selected using Rosette-sep isolation from whole blood or Easy-sep negative selection from PBMC (STEMCELL Technologies).
Cell culture and drug treatments
RPMI-1640 media supplemented with 100 U/mL penicillin, 100 µg/mL streptomycin, and 10% fetal bovine serum were used for all cell cultures. To specifically inhibit the irreversible targets of ibrutinib, treatment with this agent occurred for 1 hour followed by washout in which cells were pelleted and resuspended in fresh RPMI-1640 medium. Cells treated with dimethyl sulfoxide (DMSO) or GDC-0853 (obtained under a material transfer agreement with Genentech) were similarly pelleted and then resuspended in 10% fetal bovine serum RPMI-1640 medium containing DMSO or GDC-0853. Experiments that occurred over several days included daily addition of drug and medium replacement.
Immunoblotting
Proteins were detected using the following Cell Signaling antibodies: anti-phospho-BTK (Y223, catalog #5082), anti-BTK (catalog #8547), anti-phospho-PLCγ2 (Y1217, catalog #3871), anti-PLCγ2 (catalog #3872), anti-phospho-AKT (S473, catalog #9271), anti-AKT (catalog #4685), anti-phospho-ERK (T202/Y204, catalog #9101), anti-ERK (catalog #4695), anti-phospho-IκBα (S32, catalog #2859), anti-IκBα (catalog#4812), anti-phospho-EGFR (Y1068, catalog #2234), anti-EGFR (catalog #54359), anti-NFAT (catalog #4389), anti-GAPDH (catalog #5179), anti-Actin (catalog. #3700), and anti-Lamin (catalog #13435). Blots were probed with primary antibodies and horseradish peroxidase–conjugated secondary antibodies (Santa Cruz Biotechnologies) and then visualized with SuperSignal chemiluminescent substrate (Thermo Fisher Scientific) on X-ray film. Densitometry analysis was performed using AlphaView software.
CLL cells treated with GDC-0853 or ibrutinib were stimulated through their BCR by spinning onto a 6-well plate coated with 10 µg/mL anti-immunoglobulin M (anti-IgM) antibody (Jackson Laboratories). Following 15 minutes of stimulation, cells lysates were collected and analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Healthy human T cells were treated with BTK inhibitor and stimulated for 45 minutes with a combination of 2 µg/mL plate bound anti-CD3 and 10 µg/mL soluble anti-CD28 (eBiosciences). A549 (ATCC CCL-185) lung cancer cells were incubated with BTK inhibitor for 1 hour before lysis and SDS-PAGE analysis as described previously. Nuclear and cytoplasmic lysates were collected using Thermo Scientific NE-PER nuclear and cytoplasmic extraction reagents (catalog #78833) and analyzed by SDS-PAGE.
Cytotoxicity analysis
HS5 (ATCC CRL-11882) stromal cells were stably transfected with green fluorescent protein (GFP) allowing us to selectively gate on our GFP-negative CLL population in addition to gates based upon cell size. GFP-HS5 stromal cells were plated and permitted to reach 50% to 75% confluence before being cocultured with 1 × 106 cells/mL primary CLL. Both populations were then treated with drug and viability was measured at 48 hours. Samples with DMSO-treated viabilities below 40% at 48 hours were omitted from analysis because of their rapid rate of ex vivo apoptosis and unpredictable behavior in vitro. Viability was defined as the percentage of PBMC staining negative for 7-AAD (Life Technologies). Viability of C481S BTK-mutated patient CLL cells was determined by annexin V and propidium iodine staining (eBioscience). Viability was defined as the percentage of cells staining dually negative for both annexin V and propidium iodine. All viability measurements were acquired with a Beckman Coulter FC3000 flow cytometer; Kaluza software was used for analysis.
CpG-induced activation
Following BTK inhibitor treatment, CLL cells were stimulated with 3.2 µg/mL class A oligodeoxynucleotide CpG (Eurofins MWG Operon). Subsequent expression of CD86 was determined by mean fluorescence intensity after 48 hours with a CD86-PE–conjugated antibody (BD Biosciences). Staining intensity of CD86-PE was measured using a Beckman Coulter FC3000 flow cytometer and Kaluza software was used for analysis.
CXCL12-induced migration
Following 1 µM BTK inhibitor treatment of 1 hour, CLL cells were placed into an 8.0-μm transwell insert (Sigma-Aldrich) resting in media containing 200 ng/mL CXCL12 (R&D Systems). After incubating for 4 hours, the inserts were removed and the number of cells migrating through the transwell insert toward CXCL12 were counted by flow cytometry and normalized to input controls. Kaluza software was used for analysis.
Real-time PCR
CLL cells were treated with 1 µM BTK inhibitor with or without stimulation with 10 µg/mL anti-IgM antibody (Jackson Laboratories) for 72 hours before Trizol lysis and RNA purification via an RNeasy isolation kit (Qiagen). Lysates were analyzed for messenger RNA expression via reverse transcription polymerase chain reaction (RT-PCR) using the following primers: BCL2 (catalog #Hs00608023_m1), MCL1 (catalog #Hs01050896_m1), MYC (catalog #Hs00153408_m1), OCT2 (catalog #Hs00922172_m1), and TBP (catalog #Hs00427620_m1) (Applied Biosystems). RNA expression was measured using a Viia7 Real-Time PCR system and normalized against TBP expression.
Kinase assay
A total of 160 ng human recombinant wild-type and C481S BTK (SignalChem) were incubated with DMSO or 1 µM GDC-0853 for 30 minutes. Recombinant protein was then combined with 50 µM adenosine triphosphate and 5 µg poly (4:1, Glu:Tyr) peptide for 30 minutes at room temperature in 1× reaction buffer (Promega catalog #V6930) to allow for phosphorylation of the peptide substrate. ADP-glo kinase reagent and kinase detection reagent (Promega catalog #V6930) were then used to quench and quantify the reaction, respectively. Luminescence was measured using a DTX880 plate reader.
CCL3 ELISA
A total of 1 × 106 B cells from patients with CLL and C481S BTK mutations were treated with 1 µM BTK inhibitor and stimulated with 10 µg/mL soluble anti-IgM (Jackson Laboratories) for 24 hours. Supernatants were then collected and CCL3 production was measured using a human CCL3/MIP-1 α Quantikine ELISA kit (R&D Systems) according to the manufacturer’s instructions.
NK cell–mediated ADCC
Effector NK cells were isolated from Leukopaks obtained through the American Red Cross and incubated with target CLL cells loaded with radioactive Cr51 at an effector to target ratio of 25:1. Following treatment of purified NK cells with DMSO, 1 µM GDC-0853, or 1 µM ibrutinib for 1 hour, CLL cells were incubated with trastuzumab, alemtuzumab, rituximab, ofatumumab, or obinutuzumab at a concentration of 10 µg/mL and cocultured with NK cells to allow for lysis. After 4 hours of coculture, supernatant was collected and measured for radiation using a PerkinElmer Wizard2 γ counter. β decay measurements were scaled according to a no-NK cell coculture group with baseline CLL lysis and a detergent-treated CLL group with complete lysis.
Statistical analysis
Mixed effects models were used to assess changes in BTK, PLCγ2, AKT, ERK activation, and cytotoxicity between GDC-0853 vs DMSO to account for correlations among observations from the same patient. For RT-PCR data, models were fit using the ΔCT values to reduce skewness and stabilize variances; fold changes were estimated from the models. P values for the primary comparisons (GDC-0853 vs untreated) were adjusted for multiple comparisons using Holm’s procedure. Similarly, differences in migration, CD86 expression, and BTK inhibition (via kinase assay) and CCL3 production were assessed using mixed effects models; data were log-transformed to reduce skewness. Finally, differences in cytotoxicity between GDC-0853 and ibrutinib in combination with other antibodies were assessed directly using interaction contrasts. P values were again adjusted for multiple comparisons using Holm’s method. All analyses were performed using SAS/STAT software, Version 9.4 (SAS Institute Inc., Cary, NC).
Results
GDC-0853 inhibits BCR signaling
Inhibition of BTK and subsequent downstream BCR signaling is thought to be the most important mechanism by which ibrutinib exerts its therapeutic benefit. To investigate pharmacologically effective concentrations of GDC-0853, we assessed its ability to inhibit BCR signaling over a range of concentrations. We found that CLL cells treated with GDC-0853 in vitro before BCR stimulation demonstrate reduced levels of BTK phosphorylation and diminished activation of downstream targets including PLCγ2, AKT, and ERK (Figure 1A). In the representative immunoblot, 10 nM GDC-0853 is sufficient to abrogate activation of both BTK (Y223) and AKT (S473), whereas PLCγ2 (Y1217) and ERK (T202/Y204) demonstrate modest inhibition at concentrations ranging from 10 nM to 1 µM. Using immunoblot densitometry analysis, we quantified BCR pathway activation in 5 CLL patients treated with GDC-0853 or ibrutinib. As expected, we found significant decreases in the mean phosphorylation of BTK (78.6%, P = .001), PLCγ2 (43.7%, P = .023), AKT (60.1%, P = .033), and ERK (85.8%, P < .001) following treatment with 1 µM GDC-0853 (Figure 1B-E). Pharmacokinetic data obtained from a clinical trial of GDC-0853 established a mean maximal plasma concentration ranging from 0.354 to 2.10 µM (100 mg and 400 mg daily dose, respectively) on day 15 of the study, well above the concentrations observed in the present study that were necessary for effective BTK inhibition.37
Figure 1.

BCR signaling is inhibited by GDC-0853 in CLL patient cells. (A) Representative immunoblot demonstrating the effect of GDC-0853 on BCR signaling. (B-E) Quantification of BCR signaling from 5 patients with CLL using immunoblot densitometry analysis. *P ≤ .05; ***P ≤ .001. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
GDC-0853 induces modest cytotoxicity and overcomes stromal protection
CLL cells were treated with GDC-0853 for 48 hours and assessed for viability to determine whether this inhibitor mediates direct cytotoxicity. A total of 1 µM GDC-0853 induced a modest but statistically significant mean viability reduction of 9.8% normalized to DMSO treatment (P = .031) (Figure 2), similar to the effect observed with 1 µM ibrutinib. Cytotoxicity was also analyzed following coculture with the HS5 stromal cell line, a model used to recapitulate the protective bone marrow microenvironment that is not dependent on BCR signaling. GDC-0853 was found to induce a slight increase in cytotoxicity (15.3%, P < .001) in the presence of HS5 stroma. Furthermore, the viability following treatment with GDC-0853 was not found to significantly differ in the presence or absence of stromal coculture (P = .234), suggesting that GDC-0853 may abrogate the survival benefit from the bone marrow microenvironment in addition to its direct, albeit modest, cytotoxicity.
Figure 2.

GDC-0853 is modestly cytotoxic to CLL B cells and limits stroma-induced survival. Viability of CLL cells with or without stromal support following 48 hours of BTK inhibitor treatment (N = 9). Cells were treated with 1 µM BTK inhibitor and measured for viability by flow cytometry. Results were normalized to untreated CLL cells in medium. *P ≤ .05; ***P ≤ .001. n.s., not significant.
GDC-0853 inhibits NF-κB–dependent transcription, reduces activation, and impairs migration
Multiple B-cell pathways signal through BTK leading to subsequent intracellular calcium release, activation of NF-κB, and changes in gene transcription.38 To determine the influence of GDC-0853 on NF-κB–dependent transcription, we treated CLL cells with 1 µM BTK inhibitor for 72 hours and stimulated with anti-IgM to induce BCR signaling. We found that the mean transcript levels of BCL2, MCL1, MYC, and OCT2 significantly decreased in anti-IgM stimulated CLL cells following treatment with 1 µM GDC-0853 (46%, 44%, 62%, and 31% compared with DMSO-treated conditions, respectively; P < .01 in all comparisons) (Figure 3). The effect of GDC-0853 on gene transcription was found to be comparable to that of ibrutinib in all cases.
Figure 3.

GDC-0853 represses NF-κB–dependent gene transcription. CLL patient cells were treated with 1 µM BTK inhibitor for 72 hours with or without anti-IgM stimulation and assessed for gene expression changes of NF-κB targets by RT-PCR (N = 8). **P ≤ .01; ***P ≤ .001.
To determine whether GDC-0853 could prevent B-cell activation through TLR9 signaling, we treated primary CLL cells with CpG deoxynucleotides, which activate TLR9, and then measured expression of the activation marker CD86. We found a mean 39.4% reduction in CD86 expression among patient samples treated with 1 µM GDC-0853 when compared with those treated with DMSO (P = .024) (Figure 4A), similar to what we observed with ibrutinib.
Figure 4.

GDC-0853 abrogates important cellular functions including CLL activation and migration. (A) Activation of CLL cells 48 hours following 3.2 µM CpG stimulation as determined by CD86 expression via flow cytometry (N = 22). MFI results were normalized to untreated paired samples. (B) Migration of CLL cells toward the chemokine CXCL12 in the presence of DMSO, 1 µM GDC-0853, or 1 µM ibrutinib (N = 11). *P ≤ .05; **P < .01. MFI, mean fluorescence intensity.
Chemokine-mediated migration of malignant CLL lymphocytes to protective lymphoid microenvironments is also a BTK-dependent process. Both GDC-0853 and ibrutinib were found to prevent CXCR4 mediated migration of CLL cells toward its ligand CXCL12 (Figure 4B), with GDC-0853 showing a 51% reduction in migration compared with DMSO (P = .003).
GDC-0853 does not inhibit cellular EGFR or ITK
To assess the cellular specificity of GDC-0853 for BTK, 2 alternative targets of ibrutinib, EGFR and ITK, were tested for inhibition. Although ibrutinib was found to decrease the constitutive activation of EGFR in the A549 cell line, GDC-0853 spared EGFR phosphorylation up to 10 µM (Figure 5A), confirming the results of prior biochemical assays34,35 in a cellular system.
Figure 5.

GDC-0853 lacks inhibition of EGFR and ITK in cells. (A) The A549 lung cancer cell line was treated with varying concentrations of BTK inhibitor to determine the effect on EGFR activation. (B) T-cell activation in healthy donor T cells following treatment with 1 µM BTK inhibitor before T-cell receptor stimulation with anti-CD3 and anti-CD28.
ITK propagates T-cell receptor stimulation leading to phosphorylation of IKBα, nuclear translocation of the transcription factor NFAT, and subsequent changes in gene expression.38 Because of its inhibition of ITK, ibrutinib was found to inhibit IKBα phosphorylation and NFAT nuclear localization in healthy human T cells. GDC-0853, however, which does not inhibit ITK enzyme,34,35 preserved T-cell receptor signaling allowing for the activation of IKBα and NFAT nuclear localization confirming that GDC-0853 lacks ITK inhibition in a cellular system and does not affect T-cell receptor activation (Figure 5B).
GDC-0853 preserves NK cell–mediated ADCC
CD20 antibody therapy with rituximab or ofatumumab when combined with chemotherapy has been used to prolong patient survival in previously untreated CLL.39,40 However, in vitro data suggest that ITK inhibition by ibrutinib antagonizes the efficacy of NK cell–mediated ADCC.30 Knowing that GDC-0853 does not inhibit ITK in biochemical assays or in cells, we hypothesized that GDC-0853 would be effective in combination with anti-CD20 antibody therapies. Indeed, we found that combinations of GDC-0853 with antibody therapies were significantly more effective at inducing NK cell–mediated lysis of target CLL cells than combinations of ibrutinib with antibody (Figure 6). Combining GDC-0853 with obinutuzumab, the most pharmacologically active anti-CD20 antibody in CLL, produced 128% greater cytotoxicity compared with ibrutinib (P = .01). The differences in cytotoxicity between GDC-0853 and ibrutinib in combination with the less active anti-CD20 antibodies rituximab (31.8% vs 4.0%, P < .001) and ofatumumab (28.5% vs 0.2%, P < .001) were even more pronounced.
Figure 6.

GDC-0853 preserves NK cell–mediated ADCC in response to anti-CD20 antibodies. NK cells treated with 1 µM BTK inhibitor were cocultured with CLL cells loaded with radioactive chromium in the presence of alemtuzumab, rituximab, ofatumumab, or obinutuzumab at a concentration of 10 µg/mL. Supernatant fluids were collected after 4 hours and measured for radioactivity to determine the efficacy of CLL cell lysis by NK cell–mediated ADCC (N = 4). **P ≤ .01; ***P ≤ .001.
GDC-0853 demonstrates equivalent inhibition of wild-type and C481S-mutated BTK
The most common acquired means of ibrutinib resistance occurs via a cysteine to serine mutation of BTK’s ibrutinib binding site (C481S) that prevents ibrutinib’s irreversible inhibition and results in a reduced apparent potency.20 Based on data36 for similar inhibitors with a common mode of action as GDC-0853 and recently published data35 for GDC-0853, we confirmed that, as a noncovalent inhibitor, GDC-0853 maintains equivalent efficacy against both wild-type and C481S-mutant BTK. Using a biochemical BTK kinase assay, we sought to determine the efficacy of GDC-0853 against both wild-type and C481S BTK. A total of 1 µM of GDC-0853 effectively inhibited wild-type BTK and the C481S BTK variant (Figure 7A). The relative inhibition of wild-type and C481S BTK by GDC-0853 were similar at 96.6% and 94.1%, respectively (P < .001 for both comparisons). These results are in agreement with previous results35 for GDC-0853.
Figure 7.

GDC-0853 inhibits both wild-type and C481S-mutated BTK variants. (A) Inhibition of wild-type and C481S recombinant BTK protein by 1 µM GDC-0853 as measured by a biochemical kinase activity assay (N = 3). (B) The HEK 293T cell line was stably transfected with either wild-type or C481S-mutated BTK. Following BTK inhibitor treatment, these cells were immunoblotted to determine the phosphorylation status of BTK (Y223). (C) Primary CLL cells from patients expressing the C481S BTK mutation were treated with DMSO, 1 µM GDC-0853, or 1 µM ibrutinib for 48 hours and assessed for viability (N = 9). (D) CLL cells from patients acquiring C481S BTK mutations during the course of ibrutinib therapy were treated with GDC-0853 to determine its ability to inhibit BCR-mediated production of CCL3 (N = 8). *P ≤ .05; **P ≤ .01.
GDC-0853 and ibrutinib were tested in HEK 293T cell lines expressing either wild-type or C481S BTK to determine their ability to inhibit BTK activation. Although effective against wild-type BTK, ibrutinib did not reduce the phosphorylation of C481S-mutated BTK. GDC-0853, however, was able to inhibit both wild-type and C481S-mutated BTK (Figure 7B). The results in this cell system are also in agreement with previous results35 for GDC-0853.
We further validated the efficacy of GDC-0853 against C481S-mutated BTK by testing its cytotoxicity in CLL patient samples harboring this mutation. We found a 10.3% normalized decrease in mean viability for C481S BTK patient samples treated with GDC-0853 (P = .030) after 72 hours compared with a 2.9% decrease in viability following treatment with ibrutinib (P = .661) (Figure 7C). Additionally, the cytokine CCL3, which is produced by B cells following BCR stimulation, was decreased by GDC-0853 in CLL cells from patients with C481S BTK mutations (Figure 7D). GDC-0853 was found to decrease CCL3 expression by 70% compared with DMSO-treated cells (P = .003) and 59% compared with ibrutinib-treated cells (P = .015), indicating that GDC-0853 more effectively inhibits BCR signaling in CLL cells with C481S BTK than ibrutinib.
Discussion
Mutation of the C481 amino acid of BTK to which ibrutinib irreversibly, covalently, and irreversibly reacts is the most common and important mechanism of resistance to ibrutinib in CLL. As the clinical usage of ibrutinib continues to increase in patients with heavily pretreated CLL, so too does the incidence of acquired resistance to this covalent BTK inhibitor. Follow-up on previously untreated cohorts of CLL patients receiving ibrutinib is shorter so the frequency of resistance in this population is less well defined. Noncovalent inhibition of wild-type and C481S BTK by GDC-0853 validates that reversible inhibitors are capable of producing sustained BTK inhibition that can overcome the ibrutinib-resistant mutation. Developing potent BTK inhibitors such as GDC-0853, which can circumvent acquired mutations in patients, is therefore of high importance. Additionally, the inhibition of targets other than BTK likely causes some of the adverse events seen with ibrutinib, such as the diarrhea and rash that may be associated with EGFR inhibition. As well, in vitro data suggest that inhibition of ITK reduces the efficacy of therapeutic antibodies when given in combination with ibrutinib. Because GDC-0853 lacks EGFR and ITK inhibition, we expect fewer off-target drug-related toxicities and the potential for more effective combination therapies. Here, we have profiled GDC-0853, a noncovalent and highly selective BTK inhibitor with the ability to inhibit ibrutinib-resistant patient clones and to enable more effective combinations of BTK inhibitor with anti-CD20 antibody therapies.
Among patients treated with ibrutinib, 19.1% discontinue treatment at or before 4 years of therapy because of CLL progression and >80% of these patients have been found to possess BTK mutations.25 Data also exist that demonstrate that BTK mutations facilitate resistance to ibrutinib in Waldenström macroglobulinemia41 and mantle cell lymphoma.42 Because of its noncovalent mechanism of action, GDC-0853 was expected retain efficacy against C481S mutant BTK variants. Indeed, GDC-0853 demonstrates preclinical efficacy in biochemical assays, cell lines, and primary CLL cells harboring C481S-mutated BTK. Thus, our work demonstrates that BTK inhibition can be maintained in ibrutinib-resistant CLL cells by using a reversible inhibitor that does not rely on reaction with the C481 amino acid of BTK. Given that patients who develop acquired resistance to ibrutinib have few available therapeutic options, the need for effective therapies in this population is great. Our data provide evidence that BTK inhibition can still be achieved even in the context of ibrutinib resistance. Although GDC-0853 is currently not being actively pursued for development in CLL, our results are an exciting proof of principle that noncovalent BTK inhibitors such as GDC-0853 can overcome acquired resistance to ibrutinib in patients. The topic of ibrutinib resistance is relatively new and the role that BTK mutations serve in the development of progressive CLL disease leaves many questions unanswered.43 Further investigation of reversible BTK inhibitors for use in acquired resistance to ibrutinib is justified.
The addition of anti-CD20 monoclonal antibodies to chemotherapy is known to improve patient survival when compared with chemotherapy alone, making the combination of a BTK inhibitor with an anti-CD20 monoclonal antibody appealing. Ibrutinib in combination with clinical antibodies used in CLL has been effective, but it is unclear whether the combination is superior to single-agent ibrutinib therapy. By blocking BTK signaling and promoting CLL clearance through immune effector cell activation, combinations of antibody therapy with ITK-sparing BTK inhibitors, such as GDC-0853 and other irreversible selective BTK inhibitors (acalabrutinib, ONO-4059, BGB3111), may be more synergistic than similar combinations with ibrutinib. Although BTK inhibition prevents CXCL12-mediated upregulation of CD20 in CLL, GDC-0853 would still be expected to outperform ibrutinib in combination with anti-CD20 targeting antibodies in vivo because of its preservation of T- and NK-cell activity.44 Determining this will require trials directly comparing these combinations.
GDC-0853 and potentially other selective reversible BTK inhibitors currently in development represent a novel treatment strategy for CLL patients who have C481S mutations. This molecule performs comparably to ibrutinib in terms of its ability to mitigate BCR signaling, CLL survival, activation, migration, and its ability to repress the transcription of disease drivers including MYC. As such, these data justify further investigation of GDC-0853 and related agents for the treatment of C481S mutated CLL.
Acknowledgments
GDC-0853 was kindly provided by Genentech.
This research was supported by the National Institutes of Health, National Cancer Institute (K23 CA178183, R01 CA197870, and R35 CA197734).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: S.D.R. designed and conducted experiments, analyzed data, generated figures, interpreted results, and wrote the manuscript; W.B.Y. is a coinventor of GDC-0853, planned the project, designed experiments, and led the research efforts at Genentech; A.R.J. designed experiments, analyzed data, and interpreted results; L.L. analyzed clinical pharmacology data; D.G. provided necessary reagents; E.M.M., C.A.F., C.C., R.M., and L.S. generated data; A.L. performed the statistical analysis; A.J.J. supervised research; J.C.B. and J.A.W. planned the project, designed the experiments, supervised the research, analyzed data, interpreted the results, and obtained funding for the research work; and all authors approved, reviewed, and modified drafts of the manuscript and approved the final version.
Conflict-of-interest disclosure: A.R.J., L.L., and W.B.Y. are current employees of Genentech Inc. The remaining authors declare no competing financial interests.
Correspondence: Jennifer A. Woyach, Department of Internal Medicine, Division of Hematology, Comprehensive Cancer Center, The Ohio State University, 4th Floor CCC Building, 410 W. 12th Ave; e-mail: jennifer.woyach@osumc.edu.
References
- 1.American Cancer Society. Cancer Facts & Figures 2016. Atlanta: American Cancer Society; 2016. [Google Scholar]
- 2.Chiorazzi N, Ferrarini M. B cell chronic lymphocytic leukemia: lessons learned from studies of the B cell antigen receptor. Annu Rev Immunol. 2003;21(1):841-894. [DOI] [PubMed] [Google Scholar]
- 3.Packham G, Stevenson F. The role of the B-cell receptor in the pathogenesis of chronic lymphocytic leukaemia. Semin Cancer Biol. 2010;20(6):391-399. [DOI] [PubMed] [Google Scholar]
- 4.Woyach JA, Johnson AJ, Byrd JC. The B-cell receptor signaling pathway as a therapeutic target in CLL. Blood. 2012;120(6):1175-1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jain N, O’Brien S. Targeted therapies for CLL: practical issues with the changing treatment paradigm. Blood Rev. 2016;30(3):233-244. [DOI] [PubMed] [Google Scholar]
- 6.Woyach JA, Bojnik E, Ruppert AS, et al. Bruton’s tyrosine kinase (BTK) function is important to the development and expansion of chronic lymphocytic leukemia (CLL). Blood. 2014;123(8):1207-1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buggy JJ, Elias L. Bruton tyrosine kinase (BTK) and its role in B-cell malignancy. Int Rev Immunol. 2012;31(2):119-132. [DOI] [PubMed] [Google Scholar]
- 8.Lee KG, Xu S, Wong ET, Tergaonkar V, Lam KP. Bruton’s tyrosine kinase separately regulates NFkappaB p65RelA activation and cytokine interleukin (IL)-10/IL-12 production in TLR9-stimulated B Cells. J Biol Chem. 2008;283(17):11189-11198. [DOI] [PubMed] [Google Scholar]
- 9.Lougaris V, Baronio M, Vitali M, et al. Bruton tyrosine kinase mediates TLR9-dependent human dendritic cell activation. J Allergy Clin Immunol. 2014;133(6):1644-50.e4. [DOI] [PubMed] [Google Scholar]
- 10.de Gorter DJ, Beuling EA, Kersseboom R, et al. Bruton’s tyrosine kinase and phospholipase Cgamma2 mediate chemokine-controlled B cell migration and homing. Immunity. 2007;26(1):93-104. [DOI] [PubMed] [Google Scholar]
- 11.Chen SS, Chang BY, Chang S, et al. BTK inhibition results in impaired CXCR4 chemokine receptor surface expression, signaling and function in chronic lymphocytic leukemia. Leukemia. 2016;30(4):833-843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kil LP, de Bruijn MJ, van Nimwegen M, et al. Btk levels set the threshold for B-cell activation and negative selection of autoreactive B cells in mice. Blood. 2012;119(16):3744-3756. [DOI] [PubMed] [Google Scholar]
- 13.Honigberg LA, Smith AM, Sirisawad M, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci USA. 2010;107(29):13075-13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herman SE, Gordon AL, Hertlein E, et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood. 2011;117(23):6287-6296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herman SE, Mustafa RZ, Gyamfi JA, et al. Ibrutinib inhibits BCR and NF-κB signaling and reduces tumor proliferation in tissue-resident cells of patients with CLL. Blood. 2014;123(21):3286-3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y, Zhang LL, Champlin RE, Wang ML. Targeting Bruton’s tyrosine kinase with ibrutinib in B-cell malignancies. Clin Pharmacol Ther. 2015;97(5):455-468. [DOI] [PubMed] [Google Scholar]
- 17.Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Byrd JC, Brown JR, O’Brien S, et al. ; RESONATE Investigators. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371(3):213-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burger JA, Tedeschi A, Barr PM, et al. ; RESONATE-2 Investigators. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med. 2015;373(25):2425-2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woyach JA, Furman RR, Liu TM, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370(24):2286-2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maddocks KJ, Ruppert AS, Lozanski G, et al. Etiology of ibrutinib therapy discontinuation and outcomes in patients with chronic lymphocytic leukemia. JAMA Oncol. 2015;1(1):80-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furman RR, Cheng S, Lu P, et al. Ibrutinib resistance in chronic lymphocytic leukemia [published correction in N Engl J Med. 2014;370(26):2547]. N Engl J Med. 2014;370(24):2352-2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Byrd JC, Harrington B, O’Brien S, et al. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374(4):323-332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parikh SA, Chaffee KR, Call TG, et al. Ibrutinib therapy for chronic lymphocytic leukemia (CLL): an analysis of a large cohort of patients treated in routine clinical practice [abstract]. Blood. 2015;126(23). Abstract 2935. [Google Scholar]
- 25.Woyach JA, Ruppert AS, Guinn D, et al. BTKC481S-mediated resistance to ibrutinib in chronic lymphocytic leukemia. J Clin Oncol. 2017;35(13):1437-1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harandi A, Zaidi AS, Stocker AM, et al. Clinical efficacy and toxicity of anti-EGFR therapy in common cancers. J Oncol 2009;2009:567486. [DOI] [PMC free article] [PubMed]
- 27.Lynch TJ Jr, Kim ES, Eaby B, Garey J, West DP, Lacouture ME. Epidermal growth factor receptor inhibitor-associated cutaneous toxicities: an evolving paradigm in clinical management. Oncologist. 2007;12(5):610-621. [DOI] [PubMed] [Google Scholar]
- 28.Dubovsky JA, Beckwith KA, Natarajan G, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood. 2013;122(15):2539-2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khurana D, Arneson LN, Schoon RA, Dick CJ, Leibson PJ. Differential regulation of human NK cell-mediated cytotoxicity by the tyrosine kinase Itk. J Immunol. 2007;178(6):3575-3582. [DOI] [PubMed] [Google Scholar]
- 30.Da Roit F, Engelberts PJ, Taylor RP, et al. Ibrutinib interferes with the cell-mediated anti-tumor activities of therapeutic CD20 antibodies: implications for combination therapy. Haematologica. 2015;100(1):77-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burger JA, Keating MJ, Wierda WG, et al. Safety and activity of ibrutinib plus rituximab for patients with high-risk chronic lymphocytic leukaemia: a single-arm, phase 2 study. Lancet Oncol. 2014;15(10):1090-1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jaglowski SM, Jones JA, Nagar V, et al. Safety and activity of BTK inhibitor ibrutinib combined with ofatumumab in chronic lymphocytic leukemia: a phase 1b/2 study. Blood. 2015;126(7):842-850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crawford JJ, Johnson AR, Misner DL, et al. Discovery of GDC-0853: a potent, selective, and noncovalent Bruton's tyrosine kinase inhibitor in early clinical development. J Med Chem. 2018;61(6):2227-2245. [DOI] [PubMed] [Google Scholar]
- 34.Erickson RI, Schutt LK, Tarrant JM, et al. Bruton’s tyrosine kinase small molecule inhibitors induce a distinct pancreatic toxicity in rats. J Pharmacol Exp Ther. 2017;360(1):226-238. [DOI] [PubMed] [Google Scholar]
- 35.Crawford JJ, Johnson AR, Misner DL, et al. Discovery of GDC-0853: a potent, selective, and noncovalent Bruton’s tyrosine kinase inhibitor in early clinical development. J Med Chem. 2018;61(6):2227-2245. [DOI] [PubMed] [Google Scholar]
- 36.Johnson AR, Kohli PB, Katewa A, et al. Battling Btk mutants with noncovalent inhibitors that overcome Cys481 and Thr474 mutations. ACS Chem Biol. 2016;11(10):2897-2907. [DOI] [PubMed] [Google Scholar]
- 37.Byrd JC, Smith S, Wagner-Johnston N, et al. First-in-human phase 1 study of the BTK inhibitor GDC-0853 in relapsed or refractory B-cell NHL and CLL. Oncotarget. 2018;9(6):13023-13035. [DOI] [PMC free article] [PubMed]
- 38.Blomberg KEM, Boucheron N, Lindvall JM, et al. Transcriptional signatures of Itk-deficient CD3+, CD4+ and CD8+ T-cells. BMC Genomics. 2009;10(1):233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fischer K, Bahlo J, Fink AM, et al. Long-term remissions after FCR chemoimmunotherapy in previously untreated patients with CLL: updated results of the CLL8 trial. Blood. 2016;127(2):208-215. [DOI] [PubMed] [Google Scholar]
- 40.Goede V, Fischer K, Busch R, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med. 2014;370(12):1101-1110. [DOI] [PubMed] [Google Scholar]
- 41.Xu L, Tsakmaklis N, Yang G, et al. Acquired mutations associated with ibrutinib resistance in Waldenström macroglobulinemia. Blood. 2017;129(18):2519-2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stephens DM, Spurgeon SE. Ibrutinib in mantle cell lymphoma patients: glass half full? Evidence and opinion. Ther Adv Hematol. 2015;6(5):242-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lampson BL, Brown JR. Are BTK and PLCG2 mutations necessary and sufficient for ibrutinib resistance in chronic lymphocytic leukemia? Expert Rev Hematol. 2018;11(3):185-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pavlasova G, Borsky M, Seda V, et al. Ibrutinib inhibits CD20 upregulation on CLL B cells mediated by the CXCR4/SDF-1 axis. Blood. 2016;128(12):1609-1613. [DOI] [PMC free article] [PubMed] [Google Scholar]