

Key Points
Blood and its components activated the iRhom2/ADAM17-dependent release of the proinflammatory cytokine TNF-α from macrophages.
The iRhom2/ADAM17/TNF-α pathway emerged as a potential new target to prevent bone resorption following a joint bleed in mice.
Abstract
Hemophilic arthropathy (HA) is a debilitating degenerative joint disease that is a major manifestation of the bleeding disorder hemophilia A. HA typically begins with hemophilic synovitis that resembles inflammatory arthritides, such as rheumatoid arthritis, and frequently results in bone loss in patients. A major cause of rheumatoid arthritis is inappropriate release of the proinflammatory cytokine tumor necrosis factor-α (TNF-α) by the TNF-α convertase (TACE; also referred to as ADAM17) and its regulator, iRhom2. Therefore, we hypothesized that iRhom2/ADAM17-dependent shedding of TNF-α also has a pivotal role in mediating HA. Here, we show that addition of blood or its components to macrophages activates iRhom2/ADAM17-dependent TNF-α shedding, providing the premise to study the activation of this pathway by blood in the joint in vivo. For this, we turned to hemophilic FVIII-deficient mice (F8−/− mice), which develop a hemarthrosis following needle puncture injury with synovial inflammation and significant osteopenia adjacent to the affected joint. We found that needle puncture–induced bleeding leads to increased TNF-α levels in the affected joint of F8−/− mice. Moreover, inactivation of TNF-α or iRhom2 in F8−/− mice reduced the osteopenia and synovial inflammation that develops in this mouse model for HA. Taken together, our results suggest that blood entering the joint activates the iRhom2/ADAM17/TNF-α pathway, thereby contributing to osteopenia and synovitis in mice. Therefore, this proinflammatory signaling pathway could emerge as an attractive new target to prevent osteoporosis and joint damage in HA patients.
Visual Abstract
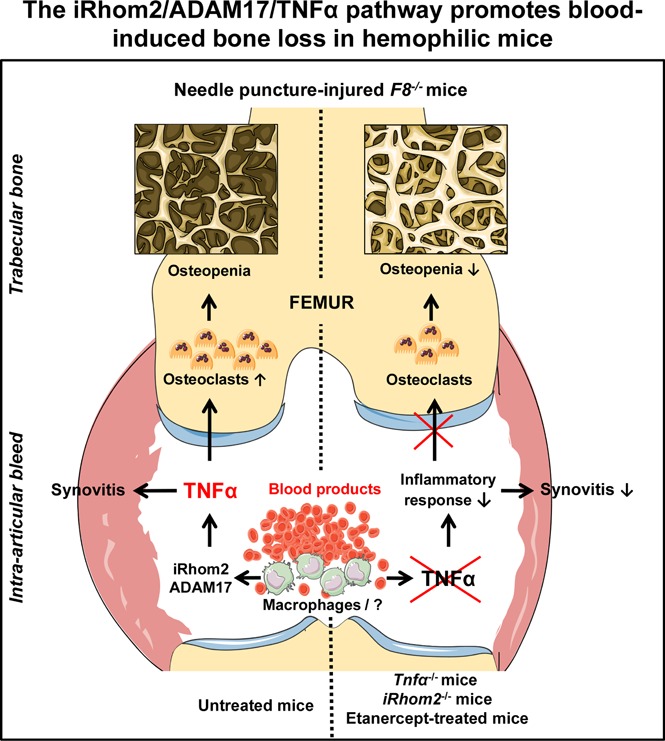
Introduction
A major manifestation of hemophilia A, an X-linked bleeding disorder, is hemophilic arthropathy (HA), a serious joint disease caused by intra-articular bleeding.1-3 HA typically begins with hemophilic synovitis (HS), a synovial hyperplasia with inflamed synovium,4 followed by cartilage destruction and bone resorption, resulting in osteopenia. Depending on the severity of the symptoms that develop, HA can have a devastating impact on patients.1,2 Current efforts to prevent HA are mainly focused on management of acute bleeds and optimizing the schedule for prophylactic factor replacement,5 which has significant advantages over episodic factor replacement following bleeding.6-9 However, breakthrough bleeds occur even on the best prophylaxis protocols,10 and HA remains a persistent problem and challenge for hemophilia patients. Therefore, improving the understanding of its pathogenesis and devising novel treatments remain major unmet needs in this area.5,11
Patients suffering from hemophilia frequently develop osteopenia or osteoporosis,12-18 raising questions about whether the underlying mechanism might be related to the presence of blood in the joint. Bleeding episodes are proposed to cause toxicity for articular chondrocytes by overloading them with iron,19 resulting in damage to articular cartilage. Moreover, repetitive joint bleeds elicit synovial hypertrophy, potentially caused by inflammation in the joint and its surrounding tissues.20-27 The proinflammatory cytokine tumor necrosis factor-α (TNF-α) is a major target for treatment of inflammatory arthritis28,29 and has been implicated in bone resorption.30-32 TNF-α is synthesized as a membrane-anchored precursor that is released by the TNF-α convertase (TACE; also referred to as ADAM17).33-36 We have recently uncovered a crucial role for ADAM17 and its upstream regulator, iRhom2, in the pathogenesis of inflammatory arthritis.37-39 Because inflammatory arthritides are caused, at least in part, by inappropriate activation of iRhom2/ADAM17/TNF-α and because the initial stages of synovitis in HA have been compared with rheumatoid arthritis (RA),20,40,41 we were interested in determining whether this signaling pathway could also have a role in HA. We explored this hypothesis using in vitro cell-based assays and a mouse model for HA to test whether inactivation of iRhom2 or TNF-α, or treatment with the anti–TNF-α biologic etanercept, would affect the development of HA and osteopenia.
Methods
Reagents
Reagents were from Sigma-Aldrich, unless indicated otherwise. The TNF ELISA kit was from Becton Dickinson Biosciences Pharmigen (San Diego, CA) (murine: OptEIA #555268; human: OptEIA #550610), the Hemoglobin Colorimetric assay kit (#700540) was from Cayman Chemical (Ann Arbor, MI), and anti-Mac2 antibodies (CL8942B) were from Cedarlane (Burlington, Canada). ELISA assays for the inflammatory marker Calprotectin (CAL35-K01) were from Eagle Biosciences (Nashua, NH). Etanercept is the generic name for Enbrel (NDC 58406-435-01), a fusion between the human p75TNFR and IgG-Fc.
Mice
Factor VIII-deficient (F8−/−) female mice (B6;129S4-F8tm1kaz/J; The Jackson Laboratory, Bar Harbor, ME) were crossed with wild-type (WT; C57BL/6J), TNF-α−/− (B6;129S-TNF-tm1Gkl/J; The Jackson Laboratory), or iRhom2−/− (C57BL/6J) male mice37 to generate male F8−/−/TNF-α−/− or F8−/−/iRhom2−/− mice.
Human subjects
Patients with anterior cruciate ligament (ACL) injury were 14 to 48 years old (mean, 25.4 years). Samples were taken 4 to 97 days after injury (mean, 45 days; Hospital for Special Surgery Institutional Review Board protocol #2014-042; all patients provided written informed consent). Calprotectin was measured in plasma from healthy controls or hemophilia patients with HA (diagnosed clinically by physical examination; patients with hemophilia A, hemophilia B, or type 3 von Willebrand disease; all with history of multiple hemarthroses [>20 joint bleeds]; age 19-72 years; 33% on continuous prophylaxis, 14% on intermittent activity-related prophylaxis, 55% on episodic treatment only with history of 2-10 bleeds per year during childhood) or disease controls with other bleeding disorders but no history of hemarthrosis (mild hemophilia A and B, n = 19; types 1 and 2 von Willebrand disease, n = 24; mild factor XI deficiency, n = 3; mild factor VII deficiency, n = 2; mild factor XIII deficiency, n = 2; mild factor I deficiency, n = 1). Plasma samples were from a biorepository collected through the Waste Protocol (ie, samples leftover after blood draw; Institutional Review Board Protocol #HS16-0169). No consent was necessary for these samples without identifiers. All samples were stored at −80°C.
Cell culture
Mouse bone marrow–derived macrophages (BMDMs) were isolated from C57BL/6J mice, as described,35 grown in Dulbecco’s modified Eagle medium (Thermo Fisher, Waltham, MA), 20% fetal bovine serum (Atlanta Biologicals, Flowery Branch, GA), and 10 ng/mL murine macrophage colony-stimulating factor (PeproTech, Rocky Hill, NJ). RAW264.7 murine macrophages (RL-2278; American Type Culture Collection, Manassas, VA) were grown in RPMI 1640 (Thermo Fisher) and 10% fetal bovine serum.
Mouse blood, macrophage treatments, and TNF-α ELISA
Blood isolated by cardiac puncture from TNF-α−/− mice was either washed twice in 50 mL of RPMI 1640 to isolate fresh blood cells by centrifugation, or it was immediately adjusted to 7.5 mM EDTA and centrifuged to separate plasma and cells (10 000g, 10 minutes, Eppendorf 5702 centrifuge). EDTA-treated plasma and cells were frozen at −80°C, thawed, and added undiluted to RAW264.7 cells (50 000 cells per well, 96-well plates). Fresh blood cells were resuspended at 2 × 109 cells per mL in RPMI 1640 and added to RAW264.7 cells or mouse BMDMs (0.5 × 106 cells per well; 24-well plate, differential blood counts showed mainly red blood cells [RBCs; mean 9 × 106 vs white blood cells 1.4 × 103/μL] and almost complete depletion of platelets. Hemin (Sigma-Aldrich), prepared at 1 mg/mL in dimethyl sulfoxide (Sigma-Aldrich), was used at 20 µM. Mouse Toll-like receptor 1-9 (TLR1-9) agonists (tlrl-kit1mw; InvivoGen, San Diego, CA) were added to RAW264.7 cells (TLR1/2: Pam3CSK4, 0.1 μg/mL; TLR2: keyhole limpet hemocyanin, 108 cells per mL; TLR3: polyinosinic-polycytidylic acid, 10 ng/mL and polyinosinic-polycytidylic acid low molecular weight, 10 μg/mL; TLR4: lipopolysaccharide from Escherichia coli, 10 ng/mL; TLR5: flagellin from Salmonella typhimurium, 0.1 μg/mL; TLR6/2: FLS-1, 0.5 μg/mL; TLR7: ssRNA40, 2.5 μg/mL; TLR9: ODN 1826, 2.5 μg/mL). TNF-α shedding from RAW264.7 cells (1- or 2-hour stimulation) or BMDMs (4-hour stimulation) was quantified by enzyme-linked immunosorbent assay (ELISA).
TNF-α in mouse blood and intra-articular hemorrhagic material
Twenty-four hours after needle puncture, blood from F8−/− mice was collected by cardiac puncture. Serum was isolated by centrifugation (15 minutes at 10 600g; Eppendorf, Westbury, NY). Intra-articular hemorrhagic material was harvested from the joint capsule, weighed (AB54-S balance; Mettler Toledo, Columbus, OH), and ground with a small pestle in phosphate-buffered saline (PBS). Equal amounts of coagulated F8−/− blood (1 hour at 37°C) ground in PBS served as control. TNF-α in serum, heparinated blood, intra-articular hemorrhagic material, or ground coagulated blood was quantified by ELISA and normalized to the weight of the collected material.
TNF-α and hemoglobin in synovial fluid from patients with ACL injury
Synovial fluid from patients with ACL injury was spun at 2100g for 10 minutes (Legend XTR; Sorvall, Langenselbold, Germany) to remove cells and debris. TNF-α and hemoglobin were quantified by ELISA after 1:1 dilution in PBS.
Faxitron
Radiograph images were generated 2 and 12 weeks after hemarthrosis (Specimen DR software version 3.1.0; Faxitron, Tucson, AZ).
HA mouse model
Male 8-12-week-old F8−/− mice were anesthetized, hair was removed from both knees with clippers, and the right knee capsule was punctured with a 30-G needle below the patella. The punctured joints were visually examined for hemorrhage.42,43 Etanercept treatment (10 mg/kg) was by subcutaneous injection 1 and 7 days after puncture. All procedures were approved by the Hospital for Special Surgery, Weill Cornell Medicine, or Rush University Medical Center Institutional Animal Care and Use Committee.
Microcomputed tomography
For trabecular bone, 1.35 mm of the distal femur, starting 100 µm from the growth plate, was scanned by microcomputed tomography (micro-CT) (µCT 35; SCANCO Medical, Brüttisellen, Switzerland); for cortical bone, a 1.4-mm portion of the middiaphysis was used. Scans were on femurs in 70% ethanol using 6-µm voxel size, 55 KVp, 0.36° rotation step (180° angular range), and a 400-millisecond exposure per view. SCANCO Biomedical µCT software (HP DECwindows Motif for Open VMS Version 1.6) was used for 3-dimensional reconstruction, and volumes were segmented using a global threshold of 0.4 g/c. Cortical area fraction (cortical area/total area), cortical area, and cortical thickness were calculated for cortical bone. Bone volume fraction, trabecular thickness, number of trabeculi, and separation of trabeculi were calculated for trabecular bone.
Histology and histomorphometry
Knees were fixed in 4% paraformaldehyde overnight, decalcified (10 days, 20% EDTA, pH 7.4), and embedded in paraffin. Sections (7 μm) were stained with hematoxylin and eosin or for tartrate-resistant acid phosphatase (TRAP) with sodium tartrate (50 mM), Naphthol AS-BI phosphate, and 0.02% Fast Green for counterstaining. TRAP+ cells were counted in the secondary spongiosa using a Nikon Labophot microscope (Nikon, Tokyo, Japan) at 200× magnification (2 sections per animal, 5 areas per section), and the osteoclast surface was calculated as described.44,45 Anti-Mac2 antibodies were used to label macrophages, with hematoxylin counterstaining. Mac2+ cells in the synovium were counted in 2 sections per knee (3 or 4 areas/section) to calculate the number of Mac2+ cells per square millimeter. Histopathological synovitis scoring was performed in a blinded manner using a modified Krenn score (see supplemental Figure 1 for details, available on the Blood Web site).46-48
RNA isolation, reverse-transcription PCR, and real-time qPCR
Long bones.
Tibiae were isolated 2 weeks after hemarthrosis, cleaned, and stored in RNAlater. Total RNA was isolated using TRIzol Reagent (Invitrogen, Carlsbad, CA). RNA samples were processed using an RNeasy Mini Kit (QIAGEN, Valencia, CA). Total RNA (600 ng per sample) was reverse transcribed into cDNA using 2.5 mM deoxynucleotide triphosphates, RNasin Plus (Promega, Madison, WI), and M-MuLV Reverse Transcriptase and Random Primer 9 (NEB, Ipswich, MA). Real-time quantitative polymerase chain reaction (qPCR) was performed on a StepOnePlus (Applied Biosystems, Foster City, CA) using SYBR Green (Thermo Scientific) and gapdh or trap primers (QuantiTect assay; QIAGEN).
Soft tissue.
Joint soft tissue was obtained 3, 7, 14, and 30 days after injury or from intact control mice and stored in RNAlater. Total RNA for each of 3 samples/group was from 2 or 3 pooled joint tissues and purified using an RNeasy Micro Kit. RNA was reverse transcribed using random primers (High-Capacity cDNA Reverse Transcription Kit; Life Technologies, Carlsbad, CA). Real-time PCR was performed using TaqMan custom-ordered probes and primers (ABI ViiA 7 Real-Time PCR System; Life Technologies).
Gene-expression analysis
The relative change in Trap and TNF-α genes compared with reference genes (gapdh for Trap and Ipo8 and Ubc for TNF-α) was determined using the 2ΔΔCT method.49
Statistical analysis
The distribution of continuous variables was assessed using the Shapiro-Wilk test. Approximately normally distributed variables are presented as mean ± standard error, whereas nonnormally distributed variables are presented as median and interquartile range. Pairwise comparisons were assessed using a Student t test or a Wilcoxon rank-sum test, as appropriate. Bonferroni correction was used to adjust for multiple comparisons.
Results
Blood and blood degradation products activate iRhom2-dependent release of TNF-α from macrophages
To test whether blood could trigger the production of TNF-α in myeloid cells, we cocultured mouse RAW264.7 myeloid cells and freshly isolated RBCs or frozen/thawed plasma or blood cells (see “Methods”). RBCs and frozen/thawed blood extract promoted significantly increased TNF-α levels after 1 and 2 hours compared with untreated controls (Figure 1A-B). Incubation of RAW264.7 cells with 20 µM hemin, a major component of RBCs, also triggered a significant induction of TNF-α at 2 hours (Figure 1C). In similar experiments with primary WT BMDMs or BMDMs lacking iRhom2, a crucial regulator of the TNF-α convertase ADAM17 in myeloid cells,37-39,50,51 we found that RBCs or 20 µM hemin stimulated significantly increased TNF-α release from WT BMDMs but very little from iRhom2−/− BMDMs (Figure 1D-E).
Figure 1.

Coculture of mouse macrophages with fresh blood or blood degradation products. RBCs (4 × 109/mL) (1 hour, 38 ± 38 pg/mL; 2 hours, 146 ± 25 pg/mL) (A) and blood extract from frozen and thawed blood (1 hour, 104 ± 21 pg/mL; 2 hours, 167 ± 23 pg/mL) (B) promoted significantly increased TNF-α shedding from RAW 264.7 cells compared with their controls. (C) Incubation of RAW 264.7 cells with 20 µM hemin also stimulated the release of TNF-α (45 ± 5 pg/mL compared with dimethyl sulfoxide–treated cells, 1 ± 0.8 pg/mL). Incubation with RBC for 4 hours (4 × 109/mL) (D) or with 20 µM hemin for 2 hours (E) stimulated the shedding of TNF-α from WT BMDMs (RBCs, 185 ±56 pg/mL; hemin, 80 ± 8 pg/mL), but not from BMDMs lacking iRhom2 (iRhom2−/−; RBCs, 21 ± 7 pg/mL; hemin, 30 ± 6 pg/mL). (F) Activation of different TLRs on RAW 264.7 cells for 2 hours also triggered the production of TNF-α from macrophages (1424 ± 445 pg/mL for TLR1/2-Pam3CSK4 [0.1 μg/mL]; 1008 ± 191 pg/mL for TLR2, keyhole limpet hemocyanin (108 cells per mL); 1787 ± 671 pg/mL for TLR4, lipopolysaccharide from E. coli (10 ng/mL); 714 ± 278 pg/mL for TLR6/2, FLS-1 (0.5 μg/mL); 506 ± 23 pg/mL for TLR7-ssRNA40 (2.5 μg/mL); 2601 ± 1185 pg/mL for TLR9, ODN1826 [2.5 μg/mL]). The mean ± SEM of 3 independent experiments in triplicate are shown. *P > .05 vs untreated control (A,D), vs EDTA-treated control (B), or vs DMSO-treated control (C,E).
Cells in blood can release damage-associated molecular patterns (DAMPs), such as phospholipids and nucleic acids, as they disintegrate.52,53 Because DAMPs could activate pattern recognition receptors (TLRs) on macrophages, and because TLR4-dependent production of TNF-α in BMDMs is iRhom2 dependent,37 we tested whether different TLR agonists could induce TNF-α release from macrophages. We found that activation of TLR1/2, TLR4, TLR6, TLR7, and TLR9 for 2 hours all increased TNF-α release over untreated controls (Figure 1F). Taken together, these findings suggest that blood and blood degradation products can induce proinflammatory conditions by activating TNF-α release from BMDMs, which depends on iRhom2.
Hemarthrosis triggers TNF-α production in mice and humans
To assess the potential relevance of TNF-α in HA in vivo, we turned to a mouse model for HA. In this model, F8−/− mice receive a needle puncture injury to the knee joint.42,43 This induces intra-articular bleeding, which progresses to a maximum hemarthrosis within 3 days. When we measured TNF-α levels in serum or heparin-treated blood from F8−/− mice, no TNF-α was detected (Figure 2A). However, a significant TNF-α accumulation was found in the intra-articular hemorrhagic material from the punctured knees of F8−/− mice but not in similar amounts of clotted control blood (Figure 2A; see “Methods”). TNF-α gene expression in soft joint tissues from F8−/− mice at different times after needle puncture showed a strong upregulation after 3 days, which then receded, but nevertheless remained increased at 7, 14, or 30 days after hemarthrosis (Figure 2B). This suggests that intra-articular bleeding triggers pronounced, but fleeting, local TNF-α production in the affected joint, presumably from activated synovial macrophages or immune cells in the blood that entered the joint.
Figure 2.

Hemarthrosis promotes TNF-α production. (A) TNF-α was measured by ELISA in serum isolated from F8−/− mice (5.5 ± 0.2 pg/mL; n = 7) 2 days after needle-puncture injury or in intra-articular hemorrhagic material isolated from the right knee of the same F8−/− mice (83 ± 34 pg/mL; n = 6). As control, TNF-α was also measured in heparin-treated blood (0.6 ± 0.6 pg/mL; n = 6) and in clotted blood (1.2 ± 1.2 pg/mL; n = 6) from F8−/− mice. (B) Measurement of TNF-α mRNA expression by qPCR following needle-puncture injury of F8−/− mice showed significantly increased expression at 3, 7, 14, and 30 days after the injury (13-, 5-, 6.4-, and 3.6-fold increase, respectively, compared with control; n = 3 independent experiments for each group and time point). For each experiment and time point, 2 or 3 joint tissues were pooled. (C) Joint exudates from patients following ACL injury showed a correlation between the hemoglobin concentration (an indicator of lysed blood in the joint fluid) and the levels of TNF-α. *P < .05 vs control.
To test whether joint bleeds also trigger TNF-α production in humans, we obtained joint fluid aspirates from patients following ACL injury, which typically results in hemarthrosis of varying severity. An analysis of these samples, which were taken up to 12 weeks after the injury, uncovered a significant correlation between the concentration of hemoglobin (an approximate indicator of the joint bleed severity) and TNF-α levels (Figure 2C). These findings suggest that intra-articular bleeding also promotes TNF-α production in humans, thereby establishing a proinflammatory milieu in the joint.
Inactivation of TNF-α or iRhom2 reduces macrophage invasion and synovitis following hemarthrosis
When the joints of F8−/− animals were analyzed histopathologically 2 weeks after needle puncture, substantial synovial thickening with inflammation was observed in the punctured knee (Figure 3B), but not in the unpunctured control (Figure 3A). Treatment of punctured F8−/− mice with etanercept to block TNF-α, or genetic inactivation of TNF-α or iRhom2, reduced synovial inflammation after 2 weeks (Figure 3C-F; see supplemental Figure 1 for analysis at 12 weeks and a table with inflammation scores at 2 and 12 weeks). Moreover, histopathological analysis showed significantly fewer Mac2+ macrophages in F8−/−/TNF-α−/− and F8−/−/iRhom2−/− mice compared with F8−/− mice (supplemental Figure 2), suggesting that iRhom2-dependent TNF-α release promotes macrophage invasion into the synovium. Etanercept did not protect from recruitment of synovial macrophages, perhaps because its levels in the joint space were not sufficient to fully inactivate TNF-α.
Figure 3.

Histopathological analysis of the knee joints 2 weeks after needle injury. Unpunctured control joints of F8−/− mice (A) and severe thickening of the synovium with inflammation in the punctured, but untreated, joints of F8−/− animals (B). Representative hematoxylin and eosin–stained sections of joints from needle-punctured F8−/− animals treated with the anti–TNF-α biologic etanercept (C) or from F8−/−TNF-α−/− (D) or F8−/−iRhom2−/− mice (E). (F) Evaluation of synovitis using a modified Krenn score indicated high levels in untreated needle-punctured F8−/− mice (8.5 ± 0.2; n = 6), with significantly reduced levels in etanercept-treated F8−/− mice (5.3 ± 1.4; n = 7), F8−/−/TNF-α−/− mice (6.8 ± 0.6 n = 6), and F8−/−/ iRhom2−/− mice (6.2 ± 0.4; n = 9). The images shown are representative for the average synovial inflammation observed in each treatment group; yellow arrows denote the analyzed synovial area. Scale bars, 100 µm. Please see supplemental Figure 1 for details. *P < .05 vs untreated F8−/− mice. F, femur; m, meniscus; T, tibia.
Targeting TNF-α or iRhom2 protects from osteopenia after hemarthrosis
Next, we used micro-CT to assess how the hemarthrosis affects the trabecular and cortical bone adjacent to the needle-punctured joint or the contralateral control joint at 2 or 12 weeks after injury. At 2 weeks, we noted severe osteopenia in the trabecular bone adjacent to the hemarthrosis, but not next to the unpunctured control knee, in F8−/− mice (Figure 4A). Treatment of needle-punctured F8−/− mice with etanercept, or the absence of TNF-α or iRhom2, protected from the unilateral trabecular bone loss (Figure 4A). Quantification by micro-CT corroborated that treatment with etanercept, or TNF-α or iRhom2 deficiency, offered significant protection from the trabecular bone loss seen in untreated F8−/− mice (Figure 4B-C). At 12 weeks, there was still significant bone loss in needle-punctured F8−/− mice, although to a lesser degree than at 2 weeks (∼30%), providing evidence for self-repair. The bone loss was prevented by etanercept and by inactivation of iRhom2 and was reduced in the absence of TNF-α (supplemental Figure 3). In addition, we noted thickened cortical bone underneath the blood-filled suprapatellar pouch using Faxitron (supplemental Figure 4) and micro-CT (supplemental Figure 5). Treatment with etanercept or inactivation of TNF-α largely prevented the cortical thickening, whereas iRhom2 deficiency had no significant effect.
Figure 4.

Evaluation of the development of osteopenia in the trabecular bone adjacent to the hemarthrosis, but not in the control knee, of F8−/−mice. (A) Micro-CT analysis of trabecular bone showed significant bone loss next to the hemarthrosis, but not next to the control joint, of F8−/− mice. The bone erosion was reduced by treatment with etanercept or in TNF-α–knockout or iRhom2-knockout mice. The micro-CT images shown are representative for the average sample of each treatment group. Quantification of the bone volume to total volume fraction (BV/TV) (B) and trabecular number (C) is shown as the percentage reduction compared with the unpunctured control knee (75% ± 4% and 39% ± 4%, respectively, for untreated F8−/− mice [n = 8]; 51% ± 6% and 11% ± 4%, respectively, for etanercept-treated F8−/− mice [n = 5]; 48% ± 6% and 15% ± 4% for F8−/−/TNF-α−/−mice [n = 7]; 49% ± 5.7% and 18% ± 3% for F8−/−/iRhom2−/− mice [n = 10]). *P ≤ .005, **P ≤ .001 vs untreated F8−/− mice.
The increase in TRAP+ osteoclasts depends on TNF-α or iRhom2
To explore the possible cause for the localized trabecular osteopenia adjacent to the hemarthrosis, we performed staining for TRAP, a marker for osteoclast activation. TRAP staining (red) of F8−/− joints, combined with Fast Green staining of trabecular bone (green), demonstrated a substantial increase in TRAP+ osteoclasts on the surface of trabecular bone next to the hemarthrosis (Figure 5A), consistent with previous studies.54 Treatment with etanercept or deficiency of TNF-α or iRhom2 strongly protected from bone resorption, as shown by quantification of the osteoclast surface compared with the total bone surface (Figure 5B). Moreover, all 3 treatment arms led to decreased TRAP gene expression in the distal femur adjacent to the hemarthrosis, as quantified by qPCR (Figure 5C).
Figure 5.

Evaluation of the osteoclastic phenotype by staining for TRAP and evaluation of its gene expression. (A) Fast Green staining of trabecular bone (green) and TRAP staining (red, see arrows) of the knee joints of F8−/− mice that were not subjected to needle puncture (unpunctured), or were punctured and were left untreated or treated with etanercept, or were punctured with additional genetic inactivation of TNF-α or iRhom2 (left panels: original magnification ×40; right panels: original magnification ×200). The micrographs shown are representative for the average sample of each treatment group. (B) Quantification of the osteoclast surface compared with the total bone surface (Oc. S/BS) in unpunctured versus punctured joints (11% ± 1% and 26% ± 4%, respectively, for F8−/− mice [n = 6]; 10% ± 1% and 10% ± 1%, respectively, for etanercept-treated F8−/− mice [n = 7], 11.6% ± 0.7% and 11.5% ± 1%, respectively, for F8−/−/TNF-α−/− mice [n = 7]; 8.4% ± 1% and 9.8% ± 1%, respectively, for F8−/−/ iRhom2−/− mice [n = 9]). (C) qPCR analysis of TRAP gene expression (fold change) compared with the unpunctured joint (3.3-fold for F8−/− mice [n = 5]; 0.8-fold for etanercept-treated F8−/− mice [n = 3]; 1.4-fold for F8−/−/TNF-α−/− mice [n = 4]; and 1.7-fold for F8−/−/iRhom2−/− mice [n = 4]). *P < .05 vs unpunctured untreated, **P ≤ .05 vs punctured untreated.
Increase in the inflammation marker calprotectin in HA patients
Taken together, our results suggest that activation of the proinflammatory iRhom2/ADAM17/TNF-α pathway could contribute to osteopenia and osteoporosis in HA patients. Therefore, we measured the levels of calprotectin, a TNF-α–dependent marker for detection of residual inflammation,55,56 in serum of needle-punctured F8−/− mice. We found increased calprotectin in F8−/− mice at 2 and 12 weeks after needle puncture injury (Figure 6A), suggesting that this could be a suitable serum biomarker for hemarthrosis. We also found increased calprotectin in serum from HA patients compared with healthy controls and patients suffering from bleeding disorders that do not cause hemarthrosis (Figure 6B), consistent with the notion that hemarthrosis can elicit inflammation in HA patients.
Figure 6.

Increased production of calprotectin, a biomarker for inflammation, in F8−/−punctured mice and HA patients. (A) The inflammation marker calprotectin was higher in serum of F8−/− punctured mice at 2 weeks (58 ± 23 pg/mL; n = 3) and 12 weeks (21 ±7 pg/mL; n = 7) compared with unpunctured F8−/− mice, where calprotectin was not detectable (0 ± 0 pg/mL; n = 3). (B) Calprotectin levels in hemophilia A patients were 57 ± 4 pg/mL (n = 40), in healthy controls they were 32 ± 3 pg/mL (n = 23) and in disease controls they were 38 ± 2 pg/mL (n = 36) (see “Methods” for details). *P < .001.
Discussion
This study was prompted by the observation that the inflammatory stages of HA have some features in common with inflammatory arthritides, such as RA.20 In mice, the proinflammatory iRhom2/ADAM17/TNF-α pathway contributes to the development of inflammatory arthritis.39 To explore whether intra-articular bleeding could activate this pathway and, thereby, contribute to the pathogenesis of HA, we used a combination of in vitro studies with mouse myeloid cells exposed to blood and blood extracts and a mouse model for HA.
Our observation that blood cells or frozen/thawed blood and hemin, as well as the stimulation of various Toll-like pattern recognition receptors, promote the production of TNF-α from myeloid cells provides strong support to the notion that TNF-α signaling might be dysregulated in HA. Our findings highlight the pleiotropic effects of blood in the joint, which can presumably activate several pathways that promote production and release of TNF-α, including the activation of TLRs by DAMPs released by cells in the bleed.52,53 The release of the soluble proinflammatory TNF-α depends on ADAM1733-35 and its upstream regulator, iRhom2.37-39,50,51 Our findings in a mouse model of HA support the concept that this pathway is also activated by intra-articular bleeding in mice in vivo. Moreover, hemostatically normal patients who had suffered an intra-articular bleed through an ACL injury had significantly increased TNF-α in joint fluid that correlated with hemoglobin concentrations, a surrogate for the amount of blood that entered the joint. These results are the first, to our knowledge, to explore how bleeding into the joint promotes TNF-α production. Our findings suggest that blood and blood degradation products promote a proinflammatory milieu by activating the iRhom2/ADAM17-dependent release of TNF-α from macrophages. The transient production of TNF-α can, in turn, elicit long-lasting proinflammatory changes in synovial fibroblasts57 and in the chromatin of target cells (together with type I interferons), which changes the response to inflammatory stimuli.58 Following a hemarthrosis, the highly increased expression of TNF-α, which remained elevated for 30 days postinjury, can presumably promote long-lasting changes, such as osteopenia, although the underlying mechanism of osteoclast activation remains to be established. In addition, hemophilia patients can suffer from repeated bleeds, which could further exacerbate their joint inflammation. These findings may potentially be relevant for other conditions, such as following a hemorrhagic stroke or fever, in which bleeding could activate immune cells to produce TNF-α.
Inactivation of TNF-α or iRhom2 prevents synovial thickening and inflammation in a mouse model for inflammatory arthritis.39 Interestingly, deficiency in TNF-α or iRhom2 also reduced the degree of synovial inflammation in the HA model. Moreover, the unilateral osteopenia in the trabecular bone adjacent to the hemarthrosis in F8−/− mice could be largely prevented by etanercept or by inactivation of TNF-α or iRhom2. This finding is particularly interesting in light of previous studies demonstrating that HA is associated with osteoporosis in patients12,17,59-63 and in hemophilic mice,64-66 because it suggests that the local release of TNF-α from the hemarthrosis has a role in promoting osteopenia in vivo. The lack of osteopenia in the contralateral control femur demonstrates that, in this mouse model of HA, TNF-α acts locally and not systemically. Perhaps the TNF-α produced in the joint space can enter the trabecular bone through channels connecting these 2 locations, which have also been implicated in the development of subchondral bone damage in RA and osteoarthritis.67 The underlying mechanism most likely involves a TNF-α–dependent activation of osteoclastogenesis,68,69 leading to osteopenia and bone resorption. Moreover, anti–TNF-α treatment also helps to prevent bone resorption in patients with RA and inflammatory bowel disease30,31,70,71 and in inflammatory arthritis models in mice.72,73 Interestingly, unilateral osteopenia adjacent to the injured knee can occur in patients following ACL injury,67,74 raising questions about a possible role for TNF-α in this process. In light of the significant variability in the development of HA in hemophiliacs with similar severities of factor deficiency,4 it will be interesting to determine whether polymorphisms in the iRhom2/ADAM17/TNF-α pathway or other inflammatory cytokines might contribute to this variability. Finally, it should be noted that factor VIII has been implicated in systemic bone remodeling through modulation of the thrombin/protease activated receptor 1 pathway.75
In addition to the osteopenia in trabecular bone, we observed local cortical thickening of the femur immediately under the blood-filled suprapatellar pouch.64,65 This localized cortical thickening depended on TNF-α but not on iRhom2. Perhaps membrane-anchored TNF-α is important for cortical thickening, which would be blocked by etanercept and removed in TNF-α−/− mice but would still be present in iRhom2−/− mice, which lack mature ADAM17 on myeloid cells.37-39 Alternatively, inactivation of iRhom2/ADAM17 could affect other substrates on BMDMs that counteract the effect of inactivating TNF-α. These results also suggest that cortical bone responds differently to increased TNF-α than does trabecular bone. Although the relevance of this observation to human HA remains to be established, the differential response of cortical and trabecular bone to TNF-α (ie, thickening vs resorption) could provide an opportunity to explore the underlying bone biology. Finally, it should be noted that IL-1β induced chondrocyte damage in cocultures in vitro, whereas TNF-α did not,76 further supporting the notion that different cell types have distinct responses to TNF-α.
Identification of biomarkers for HA is considered an important goal,77 so our observation that the inflammation marker calprotectin55,56 was significantly upregulated in HA mice and HA patients supports the concept that joint bleeds, especially repetitive bleeds, can promote a systemic proinflammatory state.78 Because none of these samples were obtained around the time of acute hemarthrosis, our data suggest that calprotectin, whose expression is TNF-α dependent, is a marker of persistent inflammation, as seen in RA and also in F8−/− mice at 12 weeks after injury. The increased bone resorption in HA mice is consistent with the significant increase in the bone resorption biomarker CTX-1 in HA patients.78 Taken together, these findings suggest that intra-articular bleeding promotes bone resorption. In principle, regular prophylactic factor replacement therapy is considered the gold standard to prevent HA, although it does not completely prevent joint disease.65 Previous studies have found increased proinflammatory cytokines, such as interleukin-6 (IL-6), IL-1β, KC, and MCP-1, in hemarthroses79 and increased IL-1, IL-6, and TNF-α in cultured synovial tissue from hemophilia patients. Moreover, anti–IL-6R and anti–TNF-α antagonists protect against HA when given with factor replacement.25 Finally, osteoprotegerin, RANK, and RANKL, as well as angiogenesis, are thought to contribute to the inflammatory process and pathogenesis in HA.80-82 That the bone resorption in mouse HA depends on iRhom2/TNF-α raises the possibility that bone resorption in HA patients could be prevented by anti–TNF-α biologics.
In summary, this study established that blood and blood degradation products elicit the production and release of TNF-α from macrophages in vitro. This requires the presence of iRhom2 to support the shedding of TNF-α by the TNF-α convertase (ADAM17). Moreover, TNF-α expression was increased in the joint soft tissue of mice following needle-puncture injury and in the synovial fluid of patients suffering from hemarthrosis caused by an ACL injury. In mice, the activation of osteoclasts and the resulting osteopenia in the trabecular bone adjacent to the hemarthrosis were prevented by genetic inactivation of TNF-α or iRhom2 or by treatment with etanercept. Previous studies have reported an increase in osteoporosis in HA patients,12,17,59-63 yet little is known about the underlying mechanism. Our results provide new insights into the role of the iRhom2/TNF-α pathway in bone resorption and synovial inflammation in F8−/− mice, thereby uncovering this signaling axis as a potential new target for prevention of osteoporosis and joint damage in HA patients.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Elaine Warner for serving as Bayer Healthcare Liaison for this project and for many insightful suggestions and comments and Hansjoerg Dürr for valuable discussions that first stimulated our interest in this area and for critically reading the manuscript. They are grateful to Lyudmila Lukashova, Maria Jiao, and Arthur Guiraud for excellent technical assistance and to Thorsten Maretzky, Gisela Weskamp, and Xiaoping Qing for advice and guidance.
This work was supported by the Bayer Hemophilia Awards Program and Bayer Healthcare, USA (C.P.B. and N.H.). S.M. and the Laboratory of Comparative Pathology Memorial Sloan Kettering Cancer Center, The Rockefeller University, Weill Cornell Medicine are supported in part by Cancer Center Support Grant P30CA008748 from the National Institutes of Health, National Cancer Institute.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: C.H. performed experiments and wrote and edited the manuscript; N.H. and X.S. provided TNF-α qPCR analysis in Figure 2; N.H. provided conceptual insights and editing; T.P. and S.M. performed histopathological analyses; C.C. and S.R. provided ACL injury samples; S.A. provided samples from hemophilia patients and controls in Figure 6; J.S. performed statistical analyses; D.L. performed experiments; D.M. and T.W.M. provided iRhom2-knockout mice and edited the manuscript; A.S. provided intellectual input and editing; and J.E.S. and C.P.B. supervised the project and wrote and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Carl P. Blobel, Hospital for Special Surgery, 535 East 70th St, New York, NY 10021; e-mail: blobelc@hss.edu.
REFERENCES
- 1.Simpson ML, Valentino LA. Management of joint bleeding in hemophilia. Expert Rev Hematol. 2012;5(4):459-468. [DOI] [PubMed] [Google Scholar]
- 2.Stephensen D, Rodriguez-Merchan EC. Orthopaedic co-morbidities in the elderly haemophilia population: a review. Haemophilia. 2013;19(2):166-173. [DOI] [PubMed] [Google Scholar]
- 3.Peyvandi F, Garagiola I, Young G. The past and future of haemophilia: diagnosis, treatments, and its complications. Lancet. 2016;388(10040):187-197. [DOI] [PubMed] [Google Scholar]
- 4.van Vulpen LFD, Mastbergen SC, Lafeber FPJG, Schutgens REG. Differential effects of bleeds on the development of arthropathy - basic and applied issues. Haemophilia. 2017;23(4):521-527. [DOI] [PubMed] [Google Scholar]
- 5.Hanley J, McKernan A, Creagh MD, et al. ; Musculoskeletal Working Party of the UKHCDO. Guidelines for the management of acute joint bleeds and chronic synovitis in haemophilia: A United Kingdom Haemophilia Centre Doctors’ Organisation (UKHCDO) guideline. Haemophilia. 2017;23(4):511-520. [DOI] [PubMed] [Google Scholar]
- 6.Rodriguez-Merchan EC. Prevention of the musculoskeletal complications of hemophilia. Adv Prev Med. 2012;2012:201271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aznar JA, Marco A, Jiménez-Yuste V, et al. ; Spanish Haemophilia Epidemiological Study Working Group. Is on-demand treatment effective in patients with severe haemophilia? Haemophilia. 2012;18(5):738-742. [DOI] [PubMed] [Google Scholar]
- 8.Khawaji M, Astermark J, Berntorp E. Lifelong prophylaxis in a large cohort of adult patients with severe haemophilia: a beneficial effect on orthopaedic outcome and quality of life. Eur J Haematol. 2012;88(4):329-335. [DOI] [PubMed] [Google Scholar]
- 9.Manco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357(6):535-544. [DOI] [PubMed] [Google Scholar]
- 10.Fischer K, Steen Carlsson K, Petrini P, et al. Intermediate-dose versus high-dose prophylaxis for severe hemophilia: comparing outcome and costs since the 1970s. Blood. 2013;122(7):1129-1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Srivastava A, Brewer AK, Mauser-Bunschoten EP, et al. ; Treatment Guidelines Working Group on Behalf of The World Federation Of Hemophilia. Guidelines for the management of hemophilia. Haemophilia. 2013;19(1):e1-e47. [DOI] [PubMed] [Google Scholar]
- 12.Lee A, Boyd SK, Kline G, Poon MC. Premature changes in trabecular and cortical microarchitecture result in decreased bone strength in hemophilia. Blood. 2015;125(13):2160-2163. [DOI] [PubMed] [Google Scholar]
- 13.Kempton CL, Antoniucci DM, Rodriguez-Merchan EC. Bone health in persons with haemophilia. Haemophilia. 2015;21(5):568-577. [DOI] [PubMed] [Google Scholar]
- 14.Anagnostis P, Karras S, Paschou SA, Goulis DG. Haemophilia A and B as a cause for secondary osteoporosis and increased fracture risk. Blood Coagul Fibrinolysis. 2015;26(6):599-603. [DOI] [PubMed] [Google Scholar]
- 15.Wells AJ, McLaughlin P, Simmonds JV, et al. A case-control study assessing bone mineral density in severe haemophilia A in the UK. Haemophilia. 2015;21(1):109-115. [DOI] [PubMed] [Google Scholar]
- 16.Paschou SA, Anagnostis P, Karras S, et al. Bone mineral density in men and children with haemophilia A and B: a systematic review and meta-analysis. Osteoporos Int. 2014;25(10):2399-2407. [DOI] [PubMed] [Google Scholar]
- 17.Kempton CL, Antun A, Antoniucci DM, et al. Bone density in haemophilia: a single institutional cross-sectional study. Haemophilia. 2014;20(1):121-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghosh K, Shetty S. Bone health in persons with haemophilia: a review. Eur J Haematol. 2012;89(2):95-102. [DOI] [PubMed] [Google Scholar]
- 19.Valentino LA, Hakobyan N, Enockson C, et al. Exploring the biological basis of haemophilic joint disease: experimental studies. Haemophilia. 2012;18(3):310-318. [DOI] [PubMed] [Google Scholar]
- 20.Valentino LA. Blood-induced joint disease: the pathophysiology of hemophilic arthropathy. J Thromb Haemost. 2010;8(9):1895-1902. [DOI] [PubMed] [Google Scholar]
- 21.Forsyth AL, Rivard GE, Valentino LA, et al. Consequences of intra-articular bleeding in haemophilia: science to clinical practice and beyond. Haemophilia. 2012;18(Suppl 4):112-119. [DOI] [PubMed] [Google Scholar]
- 22.Haxaire C, Blobel CP. With blood in the joint - what happens next? Could activation of a pro-inflammatory signalling axis leading to iRhom2/TNFα-convertase-dependent release of TNFα contribute to haemophilic arthropathy? Haemophilia. 2014;20(Suppl 4):11-14. [DOI] [PubMed] [Google Scholar]
- 23.Nieuwenhuizen L, Roosendaal G, Mastbergen SC, et al. Antiplasmin, but not amiloride, prevents synovitis and cartilage damage following hemarthrosis in hemophilic mice. J Thromb Haemost. 2014;12(2):237-245. [DOI] [PubMed] [Google Scholar]
- 24.Melchiorre D, Linari S, Innocenti M, et al. Ultrasound detects joint damage and bleeding in haemophilic arthropathy: a proposal of a score. Haemophilia. 2011;17(1):112-117. [DOI] [PubMed] [Google Scholar]
- 25.Narkbunnam N, Sun J, Hu G, et al. IL-6 receptor antagonist as adjunctive therapy with clotting factor replacement to protect against bleeding-induced arthropathy in hemophilia. J Thromb Haemost. 2013;11(5):881-893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Meegeren ME, Roosendaal G, Coeleveld K, Nieuwenhuizen L, Mastbergen SC, Lafeber FP. A single intra-articular injection with IL-4 plus IL-10 ameliorates blood-induced cartilage degeneration in haemophilic mice. Br J Haematol. 2013;160(4):515-520. [DOI] [PubMed] [Google Scholar]
- 27.Pulles AE, Mastbergen SC, Schutgens RE, Lafeber FP, van Vulpen LF. Pathophysiology of hemophilic arthropathy and potential targets for therapy. Pharmacol Res. 2017;115:192-199. [DOI] [PubMed] [Google Scholar]
- 28.Feldmann M, Maini RN. Lasker Clinical Medical Research Award. TNF defined as a therapeutic target for rheumatoid arthritis and other autoimmune diseases. Nat Med. 2003;9(10):1245-1250. [DOI] [PubMed] [Google Scholar]
- 29.Feldmann M, Maini RN. Anti-TNF therapy, from rationale to standard of care: what lessons has it taught us? J Immunol. 2010;185(2):791-794. [DOI] [PubMed] [Google Scholar]
- 30.Sakthiswary R, Das S. The effects of TNF-α antagonist therapy on bone metabolism in rheumatoid arthritis: a systematic review. Curr Drug Targets. 2013;14(13):1552-1557. [DOI] [PubMed] [Google Scholar]
- 31.Zhao B. TNF and bone remodeling. Curr Osteoporos Rep. 2017;15(3):126-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bertolini DR, Nedwin GE, Bringman TS, Smith DD, Mundy GR. Stimulation of bone resorption and inhibition of bone formation in vitro by human tumour necrosis factors. Nature. 1986;319(6053):516-518. [DOI] [PubMed] [Google Scholar]
- 33.Moss ML, Jin S-LC, Milla ME, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha [published correction appears in Nature 1997;386(6626):738]. Nature. 1997;385(6618):733-736. [DOI] [PubMed] [Google Scholar]
- 34.Black RA, Rauch CT, Kozlosky CJ, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385(6618):729-733. [DOI] [PubMed] [Google Scholar]
- 35.Horiuchi K, Kimura T, Miyamoto T, et al. Cutting edge: TNF-alpha-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. J Immunol. 2007;179(5):2686-2689. [DOI] [PubMed] [Google Scholar]
- 36.Peschon JJ, Slack JL, Reddy P, et al. An essential role for ectodomain shedding in mammalian development. Science. 1998;282(5392):1281-1284. [DOI] [PubMed] [Google Scholar]
- 37.McIlwain DR, Lang PA, Maretzky T, et al. iRhom2 regulation of TACE controls TNF-mediated protection against Listeria and responses to LPS. Science. 2012;335(6065):229-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adrain C, Zettl M, Christova Y, Taylor N, Freeman M. Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science. 2012;335(6065):225-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Issuree PD, Maretzky T, McIlwain DR, et al. iRHOM2 is a critical pathogenic mediator of inflammatory arthritis. J Clin Invest. 2013;123(2):928-932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lafeber FP, Miossec P, Valentino LA. Physiopathology of haemophilic arthropathy. Haemophilia. 2008;14(Suppl 4):3-9. [DOI] [PubMed] [Google Scholar]
- 41.Blobel CP, Haxaire C, Kalliolias GD, DiCarlo E, Salmon J, Srivastava A. Blood-induced arthropathy in hemophilia: mechanisms and heterogeneity. Semin Thromb Hemost. 2015;41(8):832-837. [DOI] [PubMed] [Google Scholar]
- 42.Hakobyan N, Enockson C, Cole AA, Sumner DR, Valentino LA. Experimental haemophilic arthropathy in a mouse model of a massive haemarthrosis: gross, radiological and histological changes. Haemophilia. 2008;14(4):804-809. [DOI] [PubMed] [Google Scholar]
- 43.Hakobyan N, Valentino LA, Cong L, et al. Haemarthrosis model in mice: BSS - Bleeding Severity Score assessment system. Haemophilia. 2016;22(5):790-798. [DOI] [PubMed] [Google Scholar]
- 44.Parfitt AM, Drezner MK, Glorieux FH, et al. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 1987;2(6):595-610. [DOI] [PubMed] [Google Scholar]
- 45.Parfitt AM. Bone histomorphometry: standardization of nomenclature, symbols and units. Summary of proposed system. Bone Miner. 1988;4(1):1-5. [PubMed] [Google Scholar]
- 46.Matsuzaki T, Alvarez-Garcia O, Mokuda S, et al. FoxO transcription factors modulate autophagy and proteoglycan 4 in cartilage homeostasis and osteoarthritis. Sci Transl Med. 2018;10(428):eaan0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matsukura Y, Muneta T, Tsuji K, et al. Mouse synovial mesenchymal stem cells increase in yield with knee inflammation. J Orthop Res. 2015;33(2):246-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Krenn V, Morawietz L, Burmester GR, et al. Synovitis score: discrimination between chronic low-grade and high-grade synovitis. Histopathology. 2006;49(4):358-364. [DOI] [PubMed] [Google Scholar]
- 49.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402-408. [DOI] [PubMed] [Google Scholar]
- 50.Li X, Maretzky T, Weskamp G, et al. iRhoms 1 and 2 are essential upstream regulators of ADAM17-dependent EGFR signaling. Proc Natl Acad Sci USA. 2015;112(19):6080-6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maretzky T, McIlwain DR, Issuree PD, et al. iRhom2 controls the substrate selectivity of stimulated ADAM17-dependent ectodomain shedding. Proc Natl Acad Sci USA. 2013;110(28):11433-11438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goulopoulou S, McCarthy CG, Webb RC. Toll-like Receptors in the vascular system: sensing the dangers within. Pharmacol Rev. 2016;68(1):142-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schaefer L. Complexity of danger: the diverse nature of damage-associated molecular patterns. J Biol Chem. 2014;289(51):35237-35245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Recht M, Liel MS, Turner RT, Klein RF, Taylor JA. The bone disease associated with factor VIII deficiency in mice is secondary to increased bone resorption. Haemophilia. 2013;19(6):908-912. [DOI] [PubMed] [Google Scholar]
- 55.Ometto F, Friso L, Astorri D, et al. Calprotectin in rheumatic diseases. Exp Biol Med (Maywood). 2017;242(8):859-873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Abildtrup M, Kingsley GH, Scott DL. Calprotectin as a biomarker for rheumatoid arthritis: a systematic review. J Rheumatol. 2015;42(5):760-770. [DOI] [PubMed] [Google Scholar]
- 57.Lee A, Qiao Y, Grigoriev G, et al. Tumor necrosis factor α induces sustained signaling and a prolonged and unremitting inflammatory response in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2013;65(4):928-938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Park SH, Kang K, Giannopoulou E, et al. Type I interferons and the cytokine TNF cooperatively reprogram the macrophage epigenome to promote inflammatory activation. Nat Immunol. 2017;18(10):1104-1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gerstner G, Damiano ML, Tom A, et al. Prevalence and risk factors associated with decreased bone mineral density in patients with haemophilia. Haemophilia. 2009;15(2):559-565. [DOI] [PubMed] [Google Scholar]
- 60.Wallny TA, Scholz DT, Oldenburg J, et al. Osteoporosis in haemophilia - an underestimated comorbidity? Haemophilia. 2007;13(1):79-84. [DOI] [PubMed] [Google Scholar]
- 61.Barnes C, Wong P, Egan B, et al. Reduced bone density among children with severe hemophilia. Pediatrics. 2004;114(2):e177-e181. [DOI] [PubMed] [Google Scholar]
- 62.Kovacs CS. Hemophilia, low bone mass, and osteopenia/osteoporosis. Transfus Apheresis Sci. 2008;38(1):33-40. [DOI] [PubMed] [Google Scholar]
- 63.Anagnostis P, Vakalopoulou S, Slavakis A, et al. Reduced bone mineral density in patients with haemophilia A and B in Northern Greece. Thromb Haemost. 2012;107(3):545-551. [DOI] [PubMed] [Google Scholar]
- 64.Lau AG, Sun J, Hannah WB, et al. Joint bleeding in factor VIII deficient mice causes an acute loss of trabecular bone and calcification of joint soft tissues which is prevented with aggressive factor replacement. Haemophilia. 2014;20(5):716-722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sun J, Hua B, Livingston EW, et al. Abnormal joint and bone wound healing in hemophilia mice is improved by extending factor IX activity after hemarthrosis. Blood. 2017;129(15):2161-2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liel MS, Greenberg DL, Recht M, Vanek C, Klein RF, Taylor JA. Decreased bone density and bone strength in a mouse model of severe factor VIII deficiency. Br J Haematol. 2012;158(1):140-143. [DOI] [PubMed] [Google Scholar]
- 67.Binks DA, Gravallese EM, Bergin D, et al. Role of vascular channels as a novel mechanism for subchondral bone damage at cruciate ligament entheses in osteoarthritis and inflammatory arthritis. Ann Rheum Dis. 2015;74(1):196-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Azuma Y, Kaji K, Katogi R, Takeshita S, Kudo A. Tumor necrosis factor-alpha induces differentiation of and bone resorption by osteoclasts. J Biol Chem. 2000;275(7):4858-4864. [DOI] [PubMed] [Google Scholar]
- 69.Miller CH, Smith SM, Elguindy M, et al. RBP-J-regulated miR-182 promotes TNF-α-induced osteoclastogenesis. J Immunol. 2016;196(12):4977-4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dimitroulas T, Nikas SN, Trontzas P, Kitas GD. Biologic therapies and systemic bone loss in rheumatoid arthritis. Autoimmun Rev. 2013;12(10):958-966. [DOI] [PubMed] [Google Scholar]
- 71.Zerbini CAF, Clark P, Mendez-Sanchez L, et al. ; IOF Chronic Inflammation and Bone Structure (CIBS) Working Group. Biologic therapies and bone loss in rheumatoid arthritis. Osteoporos Int. 2017;28(2):429-446. [DOI] [PubMed] [Google Scholar]
- 72.Coste E, Greig IR, Mollat P, et al. Identification of small molecule inhibitors of RANKL and TNF signalling as anti-inflammatory and antiresorptive agents in mice. Ann Rheum Dis. 2015;74(1):220-226. [DOI] [PubMed] [Google Scholar]
- 73.Saidenberg-Kermanac’h N, Corrado A, Lemeiter D, deVernejoul MC, Boissier MC, Cohen-Solal ME. TNF-alpha antibodies and osteoprotegerin decrease systemic bone loss associated with inflammation through distinct mechanisms in collagen-induced arthritis. Bone. 2004;35(5):1200-1207. [DOI] [PubMed] [Google Scholar]
- 74.van Meer BL, Waarsing JH, van Eijsden WA, et al. Bone mineral density changes in the knee following anterior cruciate ligament rupture. Osteoarthritis Cartilage. 2014;22(1):154-161. [DOI] [PubMed] [Google Scholar]
- 75.Aronovich A, Nur Y, Shezen E, et al. A novel role for factor VIII and thrombin/PAR1 in regulating hematopoiesis and its interplay with the bone structure. Blood. 2013;122(15):2562-2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.van Vulpen LF, Schutgens RE, Coeleveld K, et al. IL-1β, in contrast to TNF-α, is pivotal in blood-induced cartilage damage and is a potential target for therapy. Blood. 2015;126(19):2239-2246. [DOI] [PubMed] [Google Scholar]
- 77.van Vulpen LF, van Meegeren ME, Roosendaal G, et al. Biochemical markers of joint tissue damage increase shortly after a joint bleed; an explorative human and canine in vivo study. Osteoarthritis Cartilage. 2015;23(1):63-69. [DOI] [PubMed] [Google Scholar]
- 78.Hua B, Olsen EHN, Sun S, et al. Serological biomarkers detect active joint destruction and inflammation in patients with haemophilic arthropathy. Haemophilia. 2017;23(4):e294-e300. [DOI] [PubMed] [Google Scholar]
- 79.Øvlisen K, Kristensen AT, Jensen AL, Tranholm M. IL-1 beta, IL-6, KC and MCP-1 are elevated in synovial fluid from haemophilic mice with experimentally induced haemarthrosis. Haemophilia. 2009;15(3):802-810. [DOI] [PubMed] [Google Scholar]
- 80.Melchiorre D, Manetti M, Matucci-Cerinic M. Pathophysiology of hemophilic arthropathy. J Clin Med. 2017;6(7):E63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Acharya SS, Kaplan RN, Macdonald D, Fabiyi OT, DiMichele D, Lyden D. Neoangiogenesis contributes to the development of hemophilic synovitis. Blood. 2011;117(8):2484-2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bhat V, Olmer M, Joshi S, et al. Vascular remodeling underlies rebleeding in hemophilic arthropathy. Am J Hematol. 2015;90(11):1027-1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.