

Abstract
Background
Despite a high nonresponse rate, predictors of response to anti–tumor necrosis factor (anti-TNF) therapy in ulcerative colitis (UC) remain limited. We aim to determine clinical and genetic predictors of primary nonresponse (PNR) and durable response (DR) to anti-TNF therapy in a large prospective UC cohort.
Methods
Using the Illumina Immunochip, candidate polymorphisms associated with clinical outcomes of PNR and DR were separately evaluated and combined into weighted genetic risk scores. Combined genetic and clinical multivariable models for PNR and DR were compared with clinical predictive models using area under the receiver operating characteristic (AUROC) curves. Models were internally (DR) or externally (PNR) validated. Multivariable logistic regression was utilized to assess the association of genetic risk scores with infliximab levels and antibodies.
Results
Of 231 patients, 28 (12%) experienced PNR and 120 (52%) experienced DR. There was no significant difference in clinical features between primary nonresponders and responders. Eight alleles were associated with PNR. A combined clinical-genetic model (AUROC, 0.87) more accurately predicted PNR compared with a clinical-only model (AUROC, 0.57; P < 0.0001). In an external cohort of 131 patients, increasing tertiles of PNR genetic risk score correlated with increased risk of PNR (P = 0.052). Twelve candidate loci were associated with DR. Genetic risk score quartiles for DR demonstrated a strong dose-response relationship in predicting treatment duration. Genetic risk scores for PNR and DR were not associated with infliximab levels or antibody formation.
Conclusion
Genetic polymorphisms enhance prediction of PNR and DR to anti-TNF therapy in patients with UC.
Keywords: inflammatory bowel disease, ulcerative colitis, biologics, genomics, epidemiology
INTRODUCTION
Anti–tumor necrosis factor (anti-TNF) therapies have revolutionized our ability to achieve clinical remission and endoscopic healing in patients with moderate to severe ulcerative colitis.1–3 However, up to 30% of patients are primary nonresponders to these medications.4, 5 Primary nonresponse to anti-TNF therapy has serious implications on disease course, including predicting low likelihood of response to other treatments within the same therapy class and a greater need for surgery.5 In addition, approximately 15% of patients who do initially respond to therapy subsequently lose response annually, such that just over half of the patients initiated on therapy remain on the drug at the end of 2 years.6
To date, there are few tools to accurately predict primary nonresponse and durable response to anti-TNF therapy, primarily as the mechanisms remain incompletely understood. Clinical risk factors that have been associated with primary nonresponse include severe disease,4, 7 age,8, 9 duration of colitis, disease extent,10 elevated C-reactive protein levels,7, 11, 12 and lower baseline albumin levels.5, 13 However, the predictive value of clinical risk factors remains poorly replicated and inadequate, thereby limiting utility in current practice. A few prior studies have attempted to use genetic markers to predict response to anti-TNF therapy in ulcerative colitis,14, 15 premised on the hypothesis that single nucleotide polymorphisms (SNPs) related to the pathogenesis of disease or mechanisms of action of anti-TNFs determine likelihood of response. Limited, small prior studies examining individual candidate SNPs have examined the association with IL-13 receptor (IL13Rα2), IL-23 receptor (IL23R), TNF-receptor I (TNFRI), IgG Fc receptor IIIa (FcYRIIIa), neonatal Fc receptor (VNTR2/VNTR3), apoptosis-related genes (Fas ligands, caspase 9), and MAP kinases.16–24 These candidate gene studies have not been widely replicated, and a targeted approach may miss other relevant polymorphisms. In addition, many prior studies failed to differentiate between primary and secondary nonresponders, 2 events with a recognized distinct biologic basis.
Understanding predictors of primary nonresponse and durable response to anti-TNF therapy is a key unmet need and gains urgency with availability of therapies with distinct mechanisms of action that have broadly similar efficacy as anti-TNF therapies. A priori prediction of outcomes will help clinicians guide a personalized therapeutic approach to inflammatory bowel disease in choosing among multiple therapeutic classes with distinct mechanisms of action and avoiding unnecessary exposure to therapies that are unlikely to be of benefit. In a large prospective cohort of patients with ulcerative colitis, we aimed to identify clinical parameters and genetic markers that predict primary nonresponse and durable response to anti-TNF therapy among both inflammatory bowel disease (IBD) risk alleles and nonrisk alleles implicated in immune response and to validate our findings.
METHODS
Population
The Prospective Registry in IBD Study at Massachusetts General Hospital (PRISM) is an ongoing prospective registry of adult patients with Crohn’s disease (CD) and ulcerative colitis (UC) who receive care at the Massachusetts General Hospital Crohn’s and Colitis Center for inflammatory bowel disease.25, 26 All patients age ≥18 years with inflammatory bowel disease are offered voluntary enrollment without exclusion. However, this study included only Caucasian patients. From this population, for this study, we included all patients meeting the following criteria: (1) confirmed diagnosis of UC according to standard criteria; (2) initiation of first anti-TNF agent with full clinical documentation within our health care system; (3) follow-up duration of at least 12 weeks to identify whether the patient experiences primary nonresponse and 24 months to determine if a patient experiences durable response; and (4) genotyping with the Illumina Immunochip.
Genotyping
Patients included in the study were genotyped using the Illumina Immunochip-v1 on the Illumina Bead Express platform at the Broad Institute (Cambridge, MA, USA). The Immunochip is an Illumina genotyping platform containing 196524 polymorphisms (718 small insertion deletions, 195 806 SNPs), with dense coverage of known major immune and inflammatory disease loci.
Outcome and Covariates
Patients were characterized into clinical outcomes of primary nonresponse, nondurable response, and durable response through detailed chart review of gastroenterologist documentation from clinic office visits or hospitalizations, colonoscopy and/or sigmoidoscopy reports, radiologic imaging, and operative notes. Patients were categorized as primary nonresponders if no clinical response was noted within 12 weeks of initiating an anti-TNF therapy or if the patient required alteration in therapeutic approach for ongoing clinical activity within this time (addition of a corticosteroid or another class of medication, UC-related hospitalization, or surgery). Patients who discontinued anti-TNF therapy within 12 weeks of initiation due to adverse drug effects or intolerance were not included as primary nonresponders. Patients were categorized as durable responders by maintenance of response to anti-TNF therapy for at least 24 months after initiation. Patients who required anti-TNF dose escalation/optimization to maintain response were included in the durable response outcome if remission was maintained. Covariate clinical features, including duration of disease at anti-TNF initiation, age of diagnosis, sex, smoking status, and extent of disease, were collected via medical record review. Laboratory parameters within 1 month prior to initiation of anti-TNF therapy, including albumin, erythrocyte sedimentation rate, and C-reactive protein, were also collected for the subset of patients for whom these were available.
As part of routine clinical care at our center, many patients who lost response to infliximab underwent testing for infliximab trough levels and antibodies either at Prometheus laboratories or Mayo clinical laboratories. Where available, this information was retrieved from the medical record. If a patient had more than 1 set of antibodies and levels drawn, the first trough level was used for analysis. Infliximab trough levels were categorized as therapeutic (≥5 μg/mL) or subtherapeutic (<5 μg/mL), based on current guidelines.27 Antibodies were categorized as present or absent. Anti-TNF trough and antibody levels were only obtained for the subpopulation with infliximab exposure, as more than 80% of patients in our cohort were treated with infliximab, and until recently, drug levels and antibodies were only commercially available for this anti-TNF drug.
Genetic and Clinical Risk Score Development
Two separate genetic association analyses for PNR and DR were performed using Plink v1.07.28 For each analysis, immunochip single nucleotide polymorphisms (SNPs) that met the Hardy-Weinberg equilibrium threshold of P > 0.001, genotyping call rate >99%, and genotyping success rate >80% were included in analysis. For the 201 IBD-associated alleles, a genetic association for inclusion in PNR or DR genetic risk scores was set as significant if P < 0.05. For other Immunochip loci, in this hypothesis-generating study, a threshold of P < 1 × 10–6 was used. Using significant alleles from each genetic association analysis, separate weighted genetic risk scores for PNR and DR were calculated as a the cumulative sum of the product of the log-odds ratio and allele burden for each of the risk SNPs. Genetic risk scores for PNR and DR were then entered into multivariable logistic regression models, with relevant clinical covariates determined using a priori knowledge.
Discrimination of the Genetic and Combined Models
Calibration of the combined genetic/clinical models was tested using logistic regression with the Hosmer and Lemeshow goodness-of-fit test. To test discrimination, each combined genetic/clinical multivariable model was compared with the clinical predictive model and the genetic risk score using area under the receiver operating characteristic (AUROC) curves and likelihood ratio tests.
External Validation of the Primary Nonresponse Genetic Risk Score
External validation of the primary nonresponse genetic risk score was then performed in an independent cohort of 131 Caucasian adult patients (age ≥18 years) with UC recruited at the IBD Center at Cedars-Sinai Medical Center who underwent genotyping with the same Illumina Immunochip-v1 platform.29 Primary nonresponse was defined in this cohort as lack of response after 8 weeks of initiation of anti-TNF therapy. Logistic regression was utilized to determine the association of PNR genetic risk score tertile derived in the primary cohort, with primary nonresponse in the validation cohort.
Internal Validation of the Durable Response Genetic Risk Score
External validation of the durable response genetic risk score was not performed due to significant differences in the definition for durable response between the primary and external cohorts. Consequently, we performed internal validation by bootstrapping with 10 000-fold replications. We separately performed internal validation in our cohort through a Kaplan-Meier analysis, with time to cessation of anti-TNF therapy as a time-to-event analysis.
Assessment of the Association Between Genetic Risk Score and Infliximab Trough Levels and Antibodies
Among the 190 individuals who were exposed to infliximab, 84 (44%) underwent infliximab level and antibody testing, of which 79 were confirmed to have levels drawn at trough. We performed univariate and multivariable logistic regression to determine the association of genetic risk scores for PNR and DR with therapeutic trough level and antibodies.
All statistical analyses were performed using SAS Studio (Cary, NC, USA).
Ethical Considerations
The study was approved by the Institutional Review Board of Partners Healthcare.
RESULTS
Study Cohort
Five-hundred thirty-nine patients with an enrollment diagnosis of ulcerative colitis underwent genotyping on the Illumina Immunochip at our center. After excluding those with no exposure to anti-TNF therapy (n = 229), those with a diagnosis of CD or inflammatory bowel disease – unclassified (n = 41), those who were postcolectomy at anti-TNF initiation (n = 14), or those with insufficient follow-up documentation (n = 24), we had a final cohort of 231 patients with UC included in this analysis. The mean age at diagnosis of UC of included patients was 29.5 years, with a mean disease duration of 8.4 years at the time of anti-TNF initiation. More than 90% of patients had pancolitis (56.3%) or left-sided colitis (35.1%). Just over half (50.8%) of patients were women. Eighty-two percent of patients were initiated on infliximab as their first anti-TNF agent.
Of 231 patients, 28 (12%) experienced PNR, 120 (52%) experienced DR, and the remainder experienced nondurable response. There was no significant difference in baseline clinical features of primary nonresponders as compared with responders (Table 1). The mean disease duration of primary nonresponders was 7.0 years, as compared with 8.6 years for responders (P = 0.29). Pancolitis was observed in 57.1% of primary nonresponders, as compared with 56.2% of responders. Among the subset of patients with laboratory parameters within 1 month prior to initiation of anti-TNF therapy, the mean serum albumin levels were 4.2 g/dL (SD, 0.4; n = 13) among primary nonresponders and 4.0 g/dL (SD, 0.6; n = 118) among responders (P = 0.34). The median serum ESRs were 12.0 mm/h (IQR, 2–28; n = 10) in primary nonresponders and 18.0 mm/h (IQR, 12–37; n = 105) in responders (P = 0.16). The median serum C-reative proteins (CRPs) were 2.2 mg/dL (IQR, 0.2–14.7; n = 11) in primary nonresponders and 7.4 mg/dL (IQR, 1.2–26.1; n = 105) in responders (P = 0.28).
Table 1:
Comparison of Characteristics of Patients With Ulcerative Colitis Initiating Anti-TNF With and Without Primary Nonresponse
| Primary Nonresponders (n = 28) | Responders (n = 203) | P | |
|---|---|---|---|
| Age at diagnosis (SD), y | 29.6 (11.7) | 29.5 (11.8) | 0.99 |
| Disease duration (SD), y | 7.0 (6.2) | 8.6 (7.6) | 0.29 |
| Female, No. (%) | 14 (50.0) | 103 (50.7) | 0.94 |
| Disease extent, No. (%) | 0.74 | ||
| Proctitis | 1 (3.6) | 15 (7.4) | |
| Left-sided colitis | 11 (39.3) | 70 (34.5) | |
| Pancolitis | 16 (57.1) | 114 (56.2) | |
| Unknown | 0 (0.0) | 4 (2.0) | |
| First anti-TNF therapy, No. (%) | 0.58 | ||
| Infliximab | 21 (75.0) | 169 (83.3) | |
| Adalimumab | 5 (17.9) | 27 (13.3) | |
| Golimumab | 1 (3.6) | 2 (1.0) | |
| Certolizumab | 1 (3.6) | 5 (2.4) | |
| Tobacco use, No. (%) | 0.66 | ||
| Current smoker | 1 (3.6) | 12 (5.9) | |
| Former smoker | 9 (32.1) | 49 (24.1) | |
| Never-smoker | 18 (64.3) | 137 (67.5) | |
| Unknown | 0 (0.0) | 5 (2.5) |
Predictors of Primary Nonresponse
On genetic association analysis, 7 IBD-associated alleles and 1 non-IBD-associated allele were found to be associated with PNR (Table 2) and were incorporated into a weighted genetic risk score. Six of these alleles were associated with increased risk of PNR, while 2 were inversely associated. The alleles with the strongest effect sizes were rs6679677 (potential genes PTPN22, PHTF1; odds ratio [OR], 2.26, P = 0.041) and rs3851228 (potential genes TRAF3IP2-AS1; OR, 2.33; P = 0.027).
Table 2:
Single Nucleotide Polymorphisms Associated With Primary Nonresponse in Ulcerative Colitis
| Chromosome | SNP | Risk Allele | Odds Ratio | P | Potential Genes |
|---|---|---|---|---|---|
| 1 | rs6679677 | A | 2.26 | 0.041 | PTPN22, PHTF1 |
| 6 | rs3851228 | T | 2.33 | 0.027 | TRAF3IP2-AS1 |
| 9 | rs4743820 | C | 1.81 | 0.044 | NFIL3 |
| 11 | rs568617 | T | 0.39 | 0.042 | FIBP |
| 12 | rs653178 | C | 1.78 | 0.049 | SH2B3, ATXN2 |
| 13 | rs3742130 | A | 1.98 | 0.023 | UBAC2, GPR18 |
| 21 | rs2284553 | A | 1.80 | 0.037 | IFNGR2, IFNAR1, IL10RB |
| 9 | rs1330307 | C | 0.23 | 5.65E-06 |
Multivariable analysis including clinical and genetic features revealed the genetic risk score as the only significant predictor of primary nonresponse (P = 3.87 × 10-8) (Table 3). Age at ulcerative colitis diagnosis, disease duration, sex, disease extent, and active smoking were not significantly associated with primary nonresponse. Using area under the receiver operating characteristic curves, both a combined clinical-genetic model (AUROC, 0.87) and genetic risk score alone (AUROC, 0.86) more accurately predicted PNR compared with a clinical-only model (AUROC, 0.57; P < 0.0001) (Fig. 1A). The combined clinical and genetic model was well calibrated in our cohort (Hosmer and Lemeshow P = 0.153). The genetic risk score was not independently associated with pancolonic disease extent (OR, 1.08; 95% confidence interval [CI], 0.91–1.29). However, it did predict colectomy (OR, 1.56; 95% CI, 1.20–2.04; P = 0.001), when adjusted for sex, disease duration, age at diagnosis, smoking, and disease extent.
Table 3:
Multivariable Analysis of Predictors of Primary Nonresponse to Anti-TNF Therapy in Ulcerative Colitis
| Odds Ratio | 95% Confidence Interval | P | |
|---|---|---|---|
| Age at diagnosis | 0.980 | 0.940–1.019 | 0.319 |
| Disease duration | 0.959 | 0.887–1.026 | 0.249 |
| Sex | 1.055 | 0.383–2.888 | 0.916 |
| Disease extent (pancolitis vs not) | 0.680 | 0.236–1.935 | 0.467 |
| Active tobacco use | 0.135 | 0.002–2.316 | 0.284 |
| Genetic risk score (per 1-unit increase) | 3.419 | 2.294–5.562 | 3.87 × 10-8 |
FIGURE 1.

Receiver operating curves comparing a combined genetic and clinical model with genetic and clinical models alone for PNR (A) and DR (B).
Within the subset of patients (n = 104) who had laboratory markers of disease severity drawn within 1 month prior to starting anti-TNF therapy, 94 patients were responders and 10 patients were primary nonresponders. Adjusting for serum albumin, C-reactive protein, and erythrocyte sedimentation rate slightly improved the discrimination of the model in the primary cohort (AUROC, 0.90). However, genetic risk score remained the only independently significant predictor of primary nonresponse (P = 0.0019).
External Validation of the Primary Nonresponse Genetic Risk Score
One-hundred thirty-one patients were included in the validation cohort. Of these, 32 patients (24.4%) experienced primary nonresponse and 99 patients (75.6%) were responders. Increasing tertiles of genetic risk score were associated with a trend to increased risk of PNR (P = 0.052). Compared with patients in the lowest genetic risk tertile, patients in the highest-risk tertile had a trend to increased odds of primary nonresponse (OR, 2.30; P = 0.088).
Predictors of Durable Response
A total of 12 candidate loci were associated with DR and were included in a weighted genetic risk score: 11 IBD-associated alleles and 1 non-IBD associated allele (Table 4). Six alleles had a positive association with DR, whereas the other 6 were associated with a decreased risk of DR. Multivariable analysis including clinical and genetic features revealed the genetic risk score as the only significant predictor of durable response (P = 4.74 × 10-11) (Table 5). AUROC curves demonstrated that both a combined genetic-clinical model (AUROC, 0.80) and genetic risk score alone (AUROC, 0.79) more accurately predicted DR as compared with a clinical-only model (AUROC, 0.57; P < 0.0001) (Fig. 1B).
Table 4:
Single Nucleotide Polymorphisms Associated With Durable Response in Ulcerative Colitis
| Chromosome | SNP | Risk Allele | Odds Ratio | P | Potential Genes |
|---|---|---|---|---|---|
| 1 | rs670523 | A | 0.64 | 0.021 | UBQLN4, RIT1, MSTO1 |
| 2 | rs6716753 | C | 0.61 | 0.026 | SP140 |
| 4 | rs4692386 | T | 0.57 | 0.004 | RBP-J |
| 7 | rs1077773 | G | 0.68 | 0.035 | AHR |
| 8 | rs921720 | A | 0.67 | 0.042 | TRIB1 |
| 10 | rs2790216 | A | 1.56 | 0.048 | CISD1, IPMK |
| 11 | rs907611 | A | 1.54 | 0.039 | TNNI2, LSP1 |
| 16 | rs529866 | T | 2.18 | 0.001 | SOCS1, LITAF, RMI2 |
| 16 | rs5743289 | T | 1.79 | 0.033 | NOD2 |
| 17 | rs3091315 | G | 0.63 | 0.024 | CCL2, CCL7 |
| 18 | rs9319943 | C | 1.66 | 0.037 | |
| 16 | rs12051532 | C | 2.35 | 8.44E-06 |
Table 5:
Multivariable Analysis of Predictors of Durable Response to Anti-TNF Therapy in Ulcerative Colitis
| Odds Ratio | 95% Confidence Interval | P | |
|---|---|---|---|
| Age at diagnosis | 0.978 | 0.952–1.005 | 0.116 |
| Disease duration | 0.997 | 0.956–1.039 | 0.873 |
| Sex | 1.132 | 0.603–2.125 | 0.700 |
| Disease extent | 1.195 | 0.636–2.244 | 0.580 |
| Active tobacco use | 1.817 | 0.428–7.712 | 0.418 |
| Genetic risk score (per 1-unit increase) | 2.799 | 2.060–3.803 | 4.74 × 10-11 |
Within the subset of patients (n = 104) who had laboratory markers of disease severity drawn within 1 month before starting anti-TNF therapy, 55 patients were durable responders and 49 patients were nonresponders. The addition of serum albumin, C-reactive protein, and erythrocyte sedimentation rate to the combined clinical/genetic model did not substantially improve the discrimination of the model (AUROC, 0.82). Genetic risk score remained the only independent predictor of durable response (P = 7.28 × 10-6).
Internal Validation of the Durable Response Genetic Risk Score
For the genetic-clinical model, bootstrapping with 10 000 replications internally validated a mean c-statistic of 0.806 (95% CI, 0.806–0.807), with potential optimism of 0.020 (95% CI, 0.019–0.020). Genetic risk score quartiles for durable response demonstrated a strong dose-response relationship in predicting duration of treatment, with higher quartiles exhibiting the longest duration of therapy (Plog-rank < 0.0001) (Fig. 2).
FIGURE 2.

Time to cessation of anti-TNF biologic therapy, stratified by risk burden of 12 SNPs associated with durable response. Increasing quartile indicates increasing genetic burden for durable response.
Predictors of Primary Nonresponse and Durable Response Are Mutually Exclusive
In univariate analysis, genetic risk score for durable response did not predict and was not protective against primary nonresponse (OR, 1.01; 95% CI, 0.83–1.24). Similarly, genetic risk score for primary nonresponse did not predict and was not protective against durable response (OR, 0.92; 95% CI, 0.78–1.08), emphasizing likely distinct biologic mechanisms for the 2 different outcomes.
There Is No Association Between Genetic Risk Score and Infliximab Levels or Antibodies
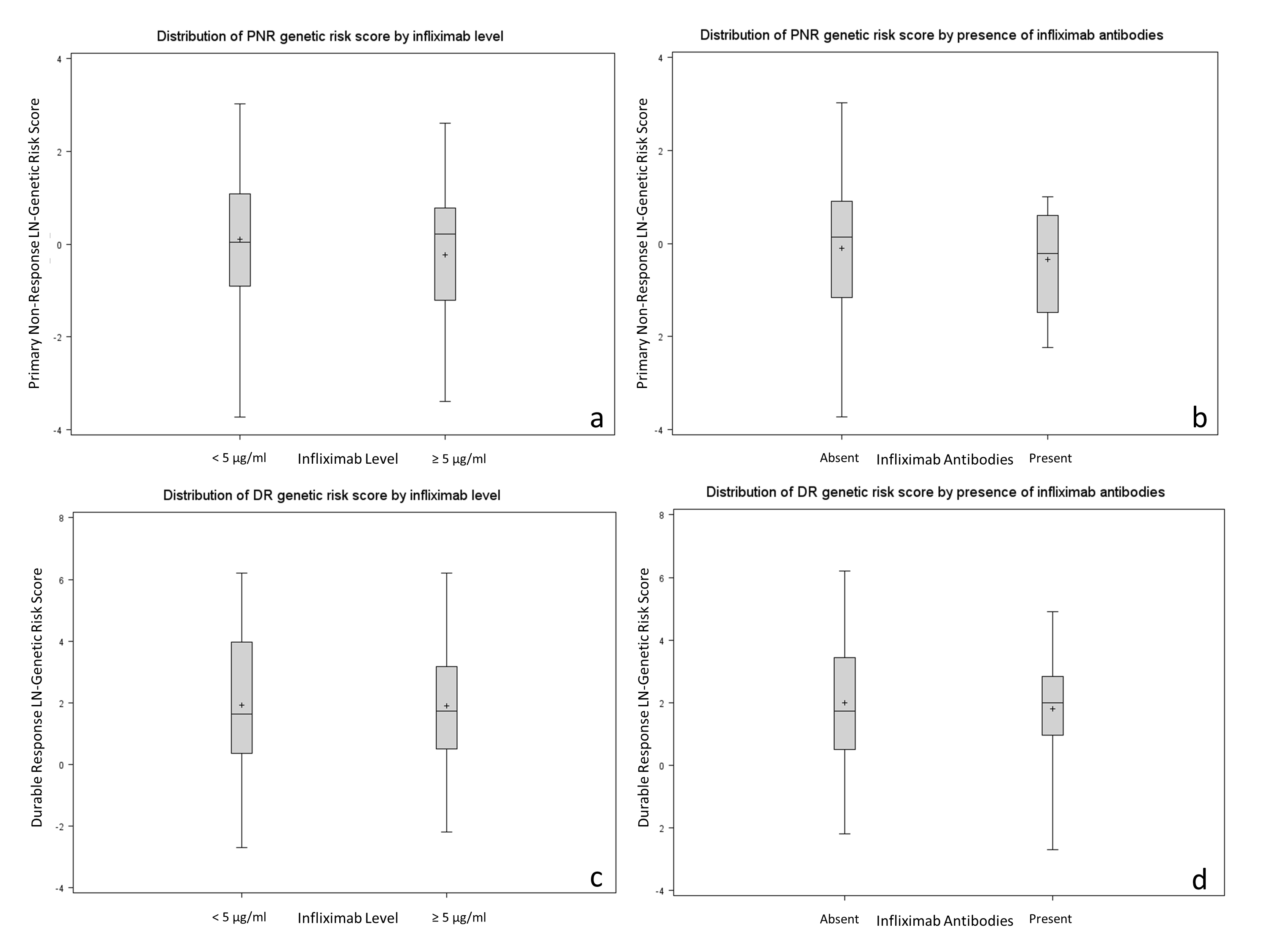
Multivariable-adjusted logistic regression demonstrated no association between quartiles of genetic risk score for primary nonresponse and therapeutic infliximab trough level ≥5 μg/mL (P = 0.41) (Supplementary Fig. 1). Similarly, there was no association between genetic risk score for durable response and infliximab level ≥5 μg/mL (P = 0.65).
On multivariable-adjusted logistic regression, there was a no association between genetic risk score for primary nonresponse and presence of infliximab antibodies (P = 0.97) (Supplementary Fig. 1). There was no association between genetic risk score for durable response and presence of antibodies (P = 0.47).
DISCUSSION
Personalizing therapy in ulcerative colitis and a priori prediction of response to a particular therapeutic class are attaining greater urgency with the availability of treatments with comparable efficacy targeting distinct therapeutic pathways. Thus far, clinical predictors and single gene candidate studies have yielded inconsistent results and inadequate predictive value. In this study of a large prospective registry of patients with ulcerative colitis, we identified SNPs associated with primary nonresponse and durable response and demonstrated that a combined clinical and genetic risk model utilizing these SNPs more accurately predicted outcomes than clinical factors alone.
In prior studies, extensive disease10 and markers of severe disease4, 7 have been associated with anti-TNF nonresponse and subsequent colectomy. However, the results remain poorly replicated. For example, although Morita et al.13 demonstrated an inverse association between week 2 CRP level and week 8 response to anti-TNF therapy, Lee et al.11 found that CRP >10 mg/dl predicted colectomy, but not primary nonresponse to anti-TNF therapy. Ferrante et al.8 found no association between age at diagnosis, duration of disease, elevated CRP, or active smoking and 10-week response to infliximab. We similarly did not find an association between clinical risk factors and anti-TNF response in our cohort.
Nearly all prior studies of polymorphisms associated with response to anti–tumor necrosis factor therapies have been performed in Crohn’s disease, or have assessed individual candidate genes with small sample sizes.15–18, 21–24 Several of the SNPs identified in our analysis are consistent with prior observations relating to the same pathway of effect. Jurgens et al.21 demonstrated that patients homozygous for IL-23 receptor SNPs that increased risk of inflammatory bowel disease were more likely to respond to anti-TNF therapy as compared with patients with IL-23 receptor SNPs that decreased risk. In our cohort, a polymorphism of CCL2, a chemokine part of the IL-23 pathway that decreases T-cell migration and impacts downstream TNF-α levels, decreased risk of durable response. It is possible that polymorphisms in this SNP decrease the effect of this protective chemokine.30–34Suppressor Of Cytokine Signaling 1 (SOCS1), a component of the IL-12 pathway, was associated with higher likelihood of durable response in our cohort.34 TNF-α mediates type 1 inflammatory response through the regulation of IL-12.35, 36 Polymorphisms in the IL-12 pathway may change the sensitivity of this pathway to TNF-α regulation, increasing the likelihood of durable response to anti-TNF therapy. PTPN22 (rs6679677), associated with a 2.26-fold increased odds of primary nonresponse to anti-TNF therapy in our cohort, is a gene associated with several immune signaling pathways.37 It has been associated with increased disease severity and extent in ulcerative colitis and need for escalation of therapy to agents including azathioprine and anti-TNF medications.38 However, it is associated with decreased serum TNF-α levels.33, 39, 40 Polymorphisms in this gene may impact TNF and other cytokine levels, placing patients at increased risk for more severe disease and nonresponse to anti-TNF therapy.
We did not identify an association between therapeutic infliximab trough levels or infliximab antibodies and primary nonresponse or durable response. While we acknowledge that this may be due to lower statistical power, it may also imply that the associations of these SNPs may be mediated by mechanisms other than drug pharmacokinetics or antibody formation. This should be further addressed in larger studies to more robustly define the mechanistic relationship of these SNPs with nonresponse to anti-TNF therapy.
Our study has many strengths. Utilizing a prospective registry of patients newly initiated on anti-TNF therapy, we were able to accurately characterize outcomes of primary nonresponse and durable response, in addition to other clinical risk factors for these outcomes. Whereas other studies of genetics and anti-TNF response in ulcerative colitis have focused on targeted SNPs, genotyping using the Illumina immunochip allowed for an unbiased assessment of SNPs associated with anti-TNF response in ulcerative colitis. We also tested our primary nonresponse genetic risk score in an external validation cohort of patients and demonstrated promising results, providing a basis for further validation in future studies.
Our findings should be interpreted within the limitations of our study design. Within our large prospective registry, a small proportion of patients initiated anti-TNF therapy while followed at our institution, limiting the power of our study. As genome-wide level of significance was not used in this hypothesis-generating study, it is possible that some SNP associations are by chance. However, several SNPs did show a significant association with plausible mechanisms of action, providing a further basis for mechanistic exploration. Response to treatment was characterized based on global physician impression through careful review of subjective symptoms and objective testing. Due to the retrospective study design, standardized disease activity scores, fecal calprotectin, and endoscopic severity indices were not routinely utilized, as these were not obtained at each visit, leading to subjectivity in defining response. The entire population in this study was Caucasian, possibly limiting the generalizability of the SNPs identified.
In summary, we present a large comprehensive study of both genetic and clinical risk factors for anti-TNF response in ulcerative colitis. Clinical factors alone do not predict response to anti-TNF therapy. However, we have demonstrated that a weighted genetic risk score significantly enhances prediction of response to anti-TNF therapy and have shown the applicability of this score in an external validation cohort of primary nonresponse. By identifying loci implicated in anti-TNF nonresponse, this study provides new markers to further basic research in the pathogenesis of this disease. Larger prospective studies are required to confirm the predictive utility of these SNPs. As more data on genetic predictors become available for different classes, it is possible that it may be useful to select therapy in the future.
SUPPLEMENTARY DATA
Supplementary data are available at Inflammatory Bowel Diseases online.
{kind=link}
ACKNOWLEDGMENTS
Guarantor of the article: Ashwin Ananthakrishnan. Specific author contributions: Kristin E. Burke – study concept and design, data acquisition, analysis and interpretation of data, drafting manuscript, critically revising manuscript. Hamed Khalili – data acquisition, critically revising manuscript. John J. Garber – data acquisition, critically revising manuscript. Talin Haritunians – data acquisition, analysis and interpretation of data, critically revising manuscript. Dermot P. B. McGovern – study concept and design, data acquisition, analysis and interpretation of data, critically revising manuscript. Ramnik J. Xavier – study concept and design, data acquisition, critically revising manuscript. Ashwin N. Ananthakrishnan – study concept and design, data acquisition, analysis and interpretation of data, critically revising manuscript. All authors have approved the final version of this manuscript, including the authorship list.
Conflicts of interest: Hamed Khalili receives consulting fees from Abbvie, Takeda, and Samsung Bioepis. Hamed Khalili also receives grant support from Takeda. Ashwin N. Ananthakrishnan serves on the scientific advisory board of Abbvie, Takeda, and Merck. Dermot McGovern consults for Janssen, Celgene, Pfizer, and Merck. The remaining authors have no conflicts to disclose.
Supported by: K. Burke is supported by T32 DK007191. H. Khalili is supported by K23 DK099681 and a career development award from the American Gastroenterological Association. A. Ananthakrishnan is supported by R03 DK112909. D.P.B. McGovern is supported by DK062413, AI067068, and The Leona M. and Harry B. Helmsley Charitable Trust. The Prospective Registry in IBD Study at Massachusetts General Hospital cohort is supported by P30DK043351 to the Center for the Study of Inflammatory Bowel Disease at Massachusetts General Hospital. The Material and Information Resources for Inflammatory And Digestive Diseases IBD Biobank at Cedars-Sinai is supported by the Widjaja Foundation Inflammatory Bowel and Immunobiology Research Institute, US Public Health Service grant P01DK046763, National Institute of Diabetes and Digestive and Kidney Diseases Grant DK062413, and The Leona M. and Harry B. Helmsley Charitable Trust.
REFERENCES
- 1. Bressler B, Marshall JK, Bernstein CN et al. ; Toronto Ulcerative Colitis Consensus Group Clinical practice guidelines for the medical management of nonhospitalized ulcerative colitis: the Toronto consensus. Gastroenterology. 2015;148:1035–58.e3. [DOI] [PubMed] [Google Scholar]
- 2. Dignass A, Lindsay JO, Sturm A et al. . Second European evidence-based consensus on the diagnosis and management of ulcerative colitis part 2: current management. J Crohns Colitis. 2012;6:991–1030. [DOI] [PubMed] [Google Scholar]
- 3. D’Haens GR, Panaccione R, Higgins PD et al. . The London Position Statement of the World Congress of Gastroenterology on Biological Therapy for IBD with the European Crohn’s and Colitis Organization: when to start, when to stop, which drug to choose, and how to predict response?Am J Gastroenterol. 2011;106:199–212; quiz 213. [DOI] [PubMed] [Google Scholar]
- 4. Oussalah A, Evesque L, Laharie D et al. . A multicenter experience with infliximab for ulcerative colitis: outcomes and predictors of response, optimization, colectomy, and hospitalization. Am J Gastroenterol. 2010;105:2617–25. [DOI] [PubMed] [Google Scholar]
- 5. Papamichael K, Rivals-Lerebours O, Billiet T et al. . Long-term outcome of patients with ulcerative colitis and primary non-response to infliximab. J Crohns Colitis. 2016;10:1015–23. [DOI] [PubMed] [Google Scholar]
- 6. O’Donnell S, Stempak JM, Steinhart AH et al. . Higher rates of dose optimisation for infliximab responders in ulcerative colitis than in Crohn’s disease. J Crohns Colitis. 2015;9:830–36. [DOI] [PubMed] [Google Scholar]
- 7. Sandborn WJ, Rutgeerts P, Feagan BG et al. . Colectomy rate comparison after treatment of ulcerative colitis with placebo or infliximab. Gastroenterology. 2009;137:1250–60; quiz 1520. [DOI] [PubMed] [Google Scholar]
- 8. Ferrante M, Vermeire S, Katsanos KH et al. . Predictors of early response to infliximab in patients with ulcerative colitis. Inflamm Bowel Dis. 2007;13:123–28. [DOI] [PubMed] [Google Scholar]
- 9. Jakobovits SL, Jewell DP, Travis SP. Infliximab for the treatment of ulcerative colitis: outcomes in Oxford from 2000 to 2006. Aliment Pharmacol Ther. 2007;25:1055–60. [DOI] [PubMed] [Google Scholar]
- 10. Gonzalez-Lama Y, Fernandez-Blanco I, Lopez-SanRoman A et al. ; Group for the Study of Inflammatory Bowel Diseases from Madrid Open-label infliximab therapy in ulcerative colitis: a multicenter survey of results and predictors of response. Hepatogastroenterology. 2008;55:1609–14. [PubMed] [Google Scholar]
- 11. Lee KM, Jeen YT, Cho JY et al. ; IBD study Group of Korean Association for the Study of Intestinal Diseases Efficacy, safety, and predictors of response to infliximab therapy for ulcerative colitis: a Korean multicenter retrospective study. J Gastroenterol Hepatol. 2013;28:1829–33. [DOI] [PubMed] [Google Scholar]
- 12. Ferrante M, Vermeire S, Fidder H et al. . Long-term outcome after infliximab for refractory ulcerative colitis. J Crohns Colitis. 2008;2:219–25. [DOI] [PubMed] [Google Scholar]
- 13. Morita Y, Bamba S, Takahashi K et al. . Prediction of clinical and endoscopic responses to anti-tumor necrosis factor-α antibodies in ulcerative colitis. Scand J Gastroenterol. 2016;51:934–41. [DOI] [PubMed] [Google Scholar]
- 14. Arijs I, Li K, Toedter G et al. . Mucosal gene signatures to predict response to infliximab in patients with ulcerative colitis. Gut. 2009;58:1612–19. [DOI] [PubMed] [Google Scholar]
- 15. Dubinsky MC, Mei L, Friedman M et al. . Genome wide association (GWA) predictors of anti-tnfalpha therapeutic responsiveness in pediatric inflammatory bowel disease. Inflamm Bowel Dis. 2010;16:1357–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hlavaty T, Pierik M, Henckaerts L et al. . Polymorphisms in apoptosis genes predict response to infliximab therapy in luminal and fistulizing Crohn’s disease. Aliment Pharmacol Ther. 2005;22:613–26. [DOI] [PubMed] [Google Scholar]
- 17. Louis E, El Ghoul Z, Vermeire S et al. . Association between polymorphism in IgG Fc receptor IIIa coding gene and biological response to infliximab in Crohn’s disease. Aliment Pharmacol Ther. 2004;19:511–19. [DOI] [PubMed] [Google Scholar]
- 18. Pierik M, Vermeire S, Steen KV et al. . Tumour necrosis factor-alpha receptor 1 and 2 polymorphisms in inflammatory bowel disease and their association with response to infliximab. Aliment Pharmacol Ther. 2004;20:303–10. [DOI] [PubMed] [Google Scholar]
- 19. Urcelay E, Mendoza JL, Martinez A et al. . IBD5 polymorphisms in inflammatory bowel disease: association with response to infliximab. World J Gastroenterol. 2005;11:1187–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Billiet T, Dreesen E, Cleynen I et al. . A genetic variation in the neonatal Fc-receptor affects anti-TNF drug concentrations in inflammatory bowel disease. Am J Gastroenterol. 2016;111:1438–45. [DOI] [PubMed] [Google Scholar]
- 21. Jürgens M, Laubender RP, Hartl F et al. . Disease activity, ANCA, and IL23R genotype status determine early response to infliximab in patients with ulcerative colitis. Am J Gastroenterol. 2010;105:1811–19. [DOI] [PubMed] [Google Scholar]
- 22. Koder S, Repnik K, Ferkolj I et al. . Genetic polymorphism in ATG16L1 gene influences the response to adalimumab in Crohn’s disease patients. Pharmacogenomics. 2015;16:191–204. [DOI] [PubMed] [Google Scholar]
- 23. Siegel CA, Melmed GY. Predicting response to anti-TNF agents for the treatment of Crohn’s disease. Therap Adv Gastroenterol. 2009;2:245–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Taylor KD, Plevy SE, Yang H et al. . ANCA pattern and LTA haplotype relationship to clinical responses to anti-TNF antibody treatment in Crohn’s disease. Gastroenterology. 2001;120:1347–55. [DOI] [PubMed] [Google Scholar]
- 25. Barber GE, Yajnik V, Khalili H et al. . Genetic markers predict primary non-response and durable response to anti-Tnf biologic therapies in Crohn’s disease. Am J Gastroenterol. 2016;111:1816–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ananthakrishnan AN, Huang H, Nguyen DD et al. . Differential effect of genetic burden on disease phenotypes in Crohn’s disease and ulcerative colitis: analysis of a North American cohort. Am J Gastroenterol. 2014;109:395–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Feuerstein JD, Nguyen GC, Kupfer SS et al. ; American Gastroenterological Association Institute Clinical Guidelines Committee American Gastroenterological Association institute guideline on therapeutic drug monitoring in inflammatory bowel disease. Gastroenterology. 2017;153:827–34. [DOI] [PubMed] [Google Scholar]
- 28. Purcell S, Neale B, Todd-Brown K et al. . PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yoon SM, Haritunians T, Chhina S et al. . Colonic phenotypes are associated with poorer response to anti-TNF therapies in patients with IBD. Inflamm Bowel Dis. 2017;23:1382–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Iwakura Y, Ishigame H. The IL-23/IL-17 axis in inflammation. J Clin Invest. 2006;116:1218–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cho JH, Feldman M. Heterogeneity of autoimmune diseases: pathophysiologic insights from genetics and implications for new therapies. Nat Med. 2015;21:730–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu JZ, van Sommeren S, Huang H et al. ; International Multiple Sclerosis Genetics Consortium; International IBD Genetics Consortium Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015;47:979–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jostins L, Ripke S, Weersma RK et al. ; International IBD Genetics Consortium (IIBDGC) Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. de Lange KM, Moutsianas L, Lee JC et al. . Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet. 2017;49:256–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Berger S, Chandra R, Balló H et al. . Immune complexes are potent inhibitors of interleukin-12 secretion by human monocytes. Eur J Immunol. 1997;27:2994–3000. [DOI] [PubMed] [Google Scholar]
- 36. Zakharova M, Ziegler HK. Paradoxical anti-inflammatory actions of Tnf-alpha: inhibition of Il-12 and Il-23 via Tnf receptor 1 in macrophages and dendritic cells. J Immunol. 2005;175:5024–33. [DOI] [PubMed] [Google Scholar]
- 37. Maine CJ, Marquardt K, Cheung J et al. . Ptpn22 controls the germinal center by influencing the numbers and activity of T follicular helper cells. J Immunol. 2014;192:1415–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Spalinger MR, Zeitz J, Biedermann L et al. ; Swiss IBD Cohort Study Group Genotype-phenotype associations of the Cd-associated single nucleotide polymorphism within the gene locus encoding protein tyrosine phosphatase non-receptor type 22 in patients of the swiss Ibd cohort. PLoS One. 2016;11:e0160215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bank S, Andersen PS, Burisch J et al. . Associations between functional polymorphisms in the NFκB signaling pathway and response to anti-TNF treatment in Danish patients with inflammatory bowel disease. Pharmacogenomics J. 2014;14:526–34. [DOI] [PubMed] [Google Scholar]
- 40. Kariuki SN, Crow MK, Niewold TB. The PTPN22 C1858T polymorphism is associated with skewing of cytokine profiles toward high interferon-alpha activity and low tumor necrosis factor alpha levels in patients with lupus. Arthritis Rheum. 2008;58:2818–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.