

Abstract
Hypertriglyceridemia (HTG) is a common lipid abnormality in humans. However, its etiology remains largely unknown. It was shown that severe HTG can be induced in mice by overexpression of wild-type (WT) apolipoprotein E (apoE) or specific apoA-I mutants. Certain mutations in apoE4 were found to affect plasma triglyceride (TG) levels in mice overexpressing the protein. HTG appeared to positively correlate with the ability of the apoE4 variants to bind to TG-rich particles, protein destabilization, and the exposure of protein hydrophobic surface in solution. Here, we propose that the apoA-I mutations that cause HTG may also lead to changes in the conformation and stability that promote binding of apoA-I to TG-rich lipoproteins. To test this hypothesis, we studied binding to TG-rich emulsion and biophysical properties of the apoA-I mutants that induce HTG, apoA-I[E110A/E111A] and apoA-I[Δ(61–78)], and compared them to those of WT apoA-I and another apoA-I mutant, apoA-I[Δ(89–99)], that does not induce HTG but causes hypercholesterolemia in mice. We found that the apoAI[E110A/E111A] and apoA-I[Δ(61–78)] mutations lead to enhanced binding of apoA-I to TG-rich particles, destabilization, and greater exposure of the hydrophobic surface of the protein. The apoA-I[Δ(89–99)] mutant did not show enhanced binding to the emulsion or a more exposed hydrophobic surface. Thus, like apoE4, the apoA-I variants that cause HTG in mice have the altered conformation and stability that facilitate their binding to TG-rich lipoproteins and thereby may lead to the reduced level of lipolysis of these lipoproteins. While many factors may be involved in induction of HTG, we suggest that an increased level of association of destabilized loosely folded apolipoproteins with TG-rich lipoproteins may contribute to some cases of HTG in humans.
Graphical Abstract
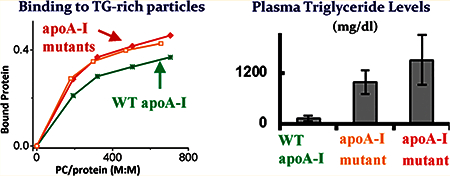
Hypertriglyceridemia (HTG) is a common lipid disorder that is characterized by increased plasma concentrations of triglycerides (TG) and TG-rich lipoproteins, mainly very low-density lipoproteins (VLDL). HTG is a hallmark of many disorders, including metabolic syndrome, diabetes, atherosclerosis, and obesity.1,3,2,4 The etiology of HTG in humans is largely unknown. Elevated plasma apolipoprotein E (apoE) concentrations in humans and animal models have been correlated positively with HTG.5 In addition, it has been shown that overexpression of wild-type (WT) apoE2, apoE3, or apoE4 in apoE-deficeint (apoE−/−) mice triggers HTG because of impaired VLDL lipolysis.5,6 Deletion of the C-terminus (residues 260–299) of apoE and certain amino acid substitutions in this region prevented the induction of HTG.6,7 ApoA-I is the major protein constituent of high-density lipoproteins (HDL), and variations in plasma concentrations of this apolipoprotein mainly affect plasma concentrations of total cholesterol and HDL cholesterol and generally do not correlate with plasma TG concentrations.8,9 Many natural and engineered mutations in apoA-I affect plasma levels of cholesterol and HDL cholesterol by altering the conformation of apoA-I and consequently the protein intravascular fate.10,11,12 However, two engineered mutations of apoA-I, apoA-I[E110A/E111A] and apoA-I[Δ(61–78)], have been reported to cause severe HTG when expressed in apoAI-deficient (apoA−/−) mice13,14 (Figure 1A). The molecular mechanisms of the effect of the apoA-I mutations on TG homeostasis remain poorly understood.
Figure 1.

Plasma triglyceride levels in mice expressing the apoA-I and apoE4 variants.7,13,14 (A) Plasma triglyceride concentrations in apoA-I−/− mice were measured 4 days after infection with 1 × 109 plaque-forming units (pfu) of recombinant adenovirus expressing WT apoA-I, apoA-I[Δ(61–78)], or apoA-I[E110A/E111A].13,14 (B) Plasma triglyceride concentrations in apoE4−/− mice were measured 4 days after infection with 2 × 109 pfu of recombinant adenovirus expressing WT apoE4, apoE4-mut2, or apoE4mut1.7 The values shown are means ± the standard deviation. Arrows point to the protein forms that induce HTG.
We have recently studied the biophysical characteristics of WT apoE4 and apoE4 mutants designated as apoE4-mut1, apoE4[L261A/W264A/F265A/L268A/V269A], and apoE4-mut2, apoE4[W276A/L279A/V280A/V283A], that induce severe HTG, induce mild HTG, and prevent induction of HTG, respectively, when overexpressed in apoE−/− mice15 (Figure 1B). We have found that the ability of the apoE4 forms to bind to TG-rich emulsion, protein destabilization, and the exposure of the hydrophobic surface in solution correlated positively with HTG, suggesting that enhanced binding of apoE4 to TG-rich lipoproteins resulting from the protein having a destabilized conformation with a more exposed hydrophobic surfacemaycontributetothedevelopmentofHTG.Here,wepropose that the apoA-I mutations that cause HTG in mice may also lead to enhancedbindingofapoA-ItoTG-richlipoproteinsandtherebyaffect the metabolism of the lipoproteins. To test this hypothesis, we studied in vitro the ability of the apoA-I[E110A/E111A] and apoA-I[Δ(6178)] mutantstobindtosyntheticTG-richparticlesandcompareditto that of WT apoA-I. We also included in the study an apoA-I variant, apoA-I[Δ(89–99)], that was reported to cause hypercholesterolemia with normal plasma TG concentrations in apoA-I−/− mice,14 as we wanted to examine whether this mutation alters binding of apoA-I to TG-rich particles in a manner similar to that of the mutations that do induce HTG. To understand the molecular basis for the different binding of the apoA-I variants to TG-rich particles, we correlated the binding ability to the conformation and stability of the proteins. We also compared in parallel the findings for the apoA-I variants with the previously reported results for the apoE4 variants15 and found similar changes in the properties of the apoA-I and apoE4 forms associated with HTG.
EXPERIMENTAL PROCEDURES
Protein Sample Preparation.
The recombinant WT apoA-I, apoA-I[Δ(61–78)], and apoA-I[Δ(89–99)] were expressed in the baculovirus expression system and purified as described previously.14,16 Recombinant apoA-I[E110A/E111A] and WT apoA-I were expressed in the adenovirus expression system and purified as described previously.13,16 All the proteins were at least 95% pure as assessed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Before the experiments, the proteins were freshly refolded by subsequent dialysis against guanidine hydrochloride (GdnHCl) solutions of 4, 2.5, and 1.25 M in Tris buffer followed by extensive dialysis against Tris buffer. The protein samples used in the circular dichroism (CD) experiments were refolded and then extensively dialyzed against 10 mM sodium phosphate and 0.02% NaN3 (pH 7.4).
Preparation of TG-Rich Emulsion Particles and Binding Assays.
TG-rich emulsion particles were made from triolein and egg yolk phosphatidylcholine (PC) by sonication and isolated by ultracentrifugation as previously described.15 After isolation, the particles were assayed for their TG, PC, and protein content and observed by electron microscopy to examine morphology and determine particle size. The emulsions were used for binding assays within 2 days of isolation; no changes in particle size or morphology were detected during this period. The binding of apoA-I to the emulsion was studied as described for the binding assays for apoE415 with minor modifications made because of a weaker binding ability of apoA-I compared to that of apoE4. Briefly, 120 μg of WT apoA-I or one of the apoA-I mutants was incubated with increasing amounts of emulsion in 1.8 mL of Tris buffer to give a PC:protein molar ratio ranging from 150 to 730. After incubation for 1 h in a water bath at 27 °C with gentle shaking, emulsion-bound and unbound proteins were separated by ultracentrifugation. Each mixture was overlaid with a NaCl solution at a density of 1.006 g/cm3 and spun in an SW60 rotor for 80 min at 40000 rpm and 25 °C. After the samples had been spun, four fractions (1 mL each) were recovered from each centrifuge tube; the top fraction was obtained by tube slicing, and the other fractions were obtained by pipetting from the surface of the solution. All the fractions were assayed for their protein, phospholipid, and TG content and, in most cases, analyzed by electron microscopy. Control samples containing only protein with no emulsion or only emulsion without protein were spun and analyzed to determine the amount of background free apoAI and background emulsion in each fraction. Binding assays were repeated with emulsions from four different preparations; three to five samples with various PC: protein ratios were studied for each protein with each emulsion.
Circular Dichroism (CD) Spectroscopy.
Far-UV spectra of lipid-free proteins were recorded at 25 °C on AVIV 62DS or AVIV 215 spectropolarimeters (AVIV Associates, Inc.) at protein concentrations of 25–70 μg/mL as described previously.16 The α-helical content was determined from the mean residue ellipticity at 222 nm.17 Thermally induced and GdnHCl-induced unfolding of lipid-free apoA-I was monitored by ellipticity at 222 nm; the melting temperature (Tm) and van’t Hoff enthalpy (ΔHv) were determined fromvan’tHoffanalysisof themeltingcurves,andtheconformational stability (ΔGD°) was determined from the GdnHCl-induced denaturation curves as described previously.16,18
ANS Fluorescence Measurements.
Fluorescence spectra of 8-anilino-2-naphthalenesulfonate (ANS) at a concentration of 0.25 mM were recorded in the presence of 0.05 mg/mL WT apoA-I or apoA-I[Δ(89–99)] using a FluoroMax-2 fluorescence spectrometer (Instruments S.A., Inc.) as described previously.16 The fluorescence intensity at the wavelength of maximal fluorescence (I) was measured and expressed inrelative units, taking the ANS fluorescence intensity in the presence of WT apoA-I to be 1.
Electron Microscopy.
Isolated emulsions and fractions recovered after spinning the protein/emulsion mixtures were visualized by negative staining electron microscopy to analyze morphology and to estimate the size of the particles. The emulsions and the top fractions recovered after ultracentrifugation were appropriately diluted before processing; other fractions recovered after ultracentrifugation were processed without dilution. The analysis was performed using a CM12 electron microscope (Philips Electron Optics, Eindhoven, The Netherlands) as described previously.18 The major diameter of the particles was determined from the electron micrographs as an average of 100200 particles.
Analytical Procedures.
Protein concentrations were determined by the modified Lowry assay.19 TG concentrations were determined with the InfinityTM TG Kit (Thermo Electron). PC concentrations were determined by the Bartlett phosphorus assay.20
RESULTS
Binding of the ApoA-I Variants to Emulsion Particles.
The emulsion particles were prepared from triolein and egg york PC to be used in binding assays as a model for TG-rich lipoproteins. The isolated emulsions had a triolein:PC weight ratio of 4.1 ± 0.3 [mean ± standard deviation (SD) of four isolated emulsions]. The average size of the emulsion particles measured from electron micrographs was 54 ± 17 nm (mean ± SD; n = 207), which is similar to the size distribution of plasma VLDL and justifies the use of these particles as a model for plasma TG-rich lipoproteins.
To compare binding of the apoA-I mutants and WT apoA-I to TG-rich particles, we incubated each protein with the emulsion at various PC:protein ratios and then separated emulsion-bound and unbound protein by ultracentrifugation. Protein, phospholipid, and TG assays and electron microscopy analysis of the fractions recovered after centrifugation of the protein/emulsion mixtures showed that emulsion particles floated to the top and were recovered in the top fraction. The bottom fractions contained lipid-free protein. The TG and PC concentrations and the particle size distribution in the fractions collected after centrifugation of the protein/emulsion mixtures were very close to those in the corresponding fractions collected after centrifugation of the control samples containing emulsion only. These results are consistent with binding of the protein to emulsion particles and suggest that no detectable solubilization of emulsion lipids by the proteins occurs during incubation. The concentration of emulsion-bound apoA-I was calculated by subtracting the background free apoA-I concentration in the top fraction from the apoA-I concentration assayed in the top fraction for each incubation mixture as described previously.21 Figure 2 shows the portion of the emulsion-bound apoA-I from the total amount of added protein at various emulsion PC:protein molar ratios in the incubation mixtures. The data for WT apoA-I are shown for the protein expressed in the adenovirus expression system. The binding parameters for WT apoA-I expressed in the baculovirus expression system were essentially the same as those for WT apoA-I expressed in the adenovirus expression system. The plots illustrate representative results of binding assays performed with one of four isolated emulsions. Because binding assays performed with emulsions from different preparations might be conducted at slightly different PC:protein ratios,15 the standard deviations for the portion of bound protein in Figure 2 are shown only for the PC:protein ratios at which at least three binding assays were performed. The portions of bound protein (mean ± SD) at these PC:protein ratios are listed in Table 1. Regardless of the initial proportion between apoA-I and emulsion during the incubation, the portion of the bound protein is higher for apoA-I[E110A/ E111A] and apoA-I[Δ(61–78)] than for WT apoA-I, indicating that the apoA-I mutations that induce HTG in mice lead to enhanced binding of apoA-I to TG-rich particles. The inset in Figure 2 shows similar data for the apoE4 forms that induce HTG15 (discussed below).
Figure 2.

Binding of the apoA-I variants to emulsion particles. Emulsion/ protein mixtures were incubated for 1 h, and then bound and unbound proteins were separated. A portion of bound protein was determined for various PC:protein molar ratios in the incubation mixtures: WT apoA-I (*), apoA-I[Δ(61–78)] (□), and apoA-I[E110A/E111A] (⊠). The inset shows the binding of the apoE4 variants to emulsion particles:15 WT apoE4 (*), apoE4-mut2 (Δ), apoE4-mut1 (○). Black symbols are used for the data for the proteinsthat induce HTG; graysymbols are usedfor the data for the proteins that do not induce HTG. Arrows point to the curves for the proteins that induce HTG.
Table 1.
Binding of the ApoA-I Variants to TG-Rich Emulsion Particlesa
| 188 ± 9 PC:protein molar ratio |
310 ± 13 PC:protein molar ratio |
688 ± 20 PC:protein molar ratio |
||||
|---|---|---|---|---|---|---|
| portion of the bound proteinb | protein on the emulsion particlesc (no. of amino acids/mol of PC) | portion of the bound proteinb | protein on the emulsion particlesc (no. of amino acids/mol of PC) | portion of the bound proteinb | protein on the emulsion particlesc (no. of amino acids/mol of PC) | |
| WT apoA-I | 0.21 ± 0.02 | 0.30 ± 0.01 | 0.29 ± 0.01 | 0.24 ± 0.01 | 0.37 ± 0.01 | 0.18 ± 0.01 |
| apoA-I [E110A/E111A] | 0.28 ± 0.03d | 0.42 ± 0.02f | 0.37 ± 0.0e | 0.34 ± 0.0f | 0.46 ± 0.02f | 0.26 ± 0.02d |
| apoA-l[Δ(61–78)] | 0.28 ± 0.03d | 0.40 ± 0.03f | 0.35 ± 0.02d | 0.33 ± 0.03f | 0.43 ± 0.02d | 0.24 ± 0.02d |
| apoA-I[Δ(89–99)] | – | – | 0.26 ± 0.03 | 0.22 ± 0.03 | 0.34 ± 0.03 | 0.16 ± 0.01 |
Values are means ± SD from at least three experiments.
Portion of the bound protein of the total amount of protein added to emulsion.
Parameter derived from composition of apoA-I–emulsion complexes recovered by ultracentrifugation following the incubation of apoA-I with emulsion.
p < 0.05, compared to the value for WT apoA-I.
p < 0.01, compared to the value for WT apoA-I.
p < 0.005, compared to the value for WT apoA-I.
To characterize the relative content of apoA-I on the resultant apoA-I-emulsion particles formed during the incubation, we determined the weight ratio of the bound protein to PC in the top fractions recovered after ultracentrifugation of the protein/ emulsion mixtures. The values for this ratio in terms of a number of amino acids of bound protein per molecule of PC are listed for each protein in Table 1. At each indicated ratio of emulsion PC to protein in incubation mixtures, the number of amino acids per PC molecule in the resultant protein-emulsion particle is significantly higher for apoA-I[E110A/E111A] and apoA-I[Δ(61–78)] than for WT apoA-I. We also calculated the number of protein molecules per protein-emulsion particle on the basis of the molar ratio of the bound apoA-I to PC in the top fractions recovered after ultracentrifugation, using the average emulsion particle diameter and the known values for the emulsion surface area per amino acid residue in a helix (0.15 nm2)22 and for the emulsion surface area covered by one PC molecule (0.7 nm2).23 The numbers of bound protein molecules per emulsion particle at various ratios of PC to protein in incubation mixtures are shown in Figure 3. At each emulsion PC to protein ratio, the particles containing the apoAI[E110A/E111A] or apoA-I[Δ(61–78)] variant have more protein molecules per particle than particles containing WT apoA-I. A similar analysis was previously performed for the apoE4 forms15 (inset of Figure 3; see Discussion).
Figure 3.

Number of bound apoA-I molecules per emulsion particle. Numbers of the bound protein molecules per emulsion particle were determined at various PC to protein molar ratios in the incubation mixtures: WT apoA-I (*), apoA-I[Δ(61–78)] (□), and apoA-I- [E110A/E111A] (⊠). The inset shows the number of bound apoE-4 molecules per emulsion particle:15 WT apoE4 (*) and apoE4-mut1 (○). Black symbols are used for the data for the proteins that induce HTG; gray symbols are used for the data for the proteins that do not induce HTG. Arrows point to the curves for the proteins that induce HTG.
To examine how a mutation that does not cause HTG but leads to hypercholesterolemia affects binding of apoA-I to TGrich particles, we performed the binding studies with the apoAI[Δ(89–99)] mutant. Because of a limited amount of this mutant available for the binding experiments, the binding assays with this protein were performed only at PC to protein molar ratios of 310 ± 13 and 688 ± 20. The values for the portion of bound protein and for the number of amino acids per molecule of PC on the resultant protein-emulsion particles for apoA-I[Δ(89–99)] are close to those for WT apoA-I (Table 1). No significant differences between apoA-I[Δ(89–99)] and WT apoA-I were found when the binding characteristics were expressed in terms of the number of bound protein molecules per emulsion particle (data not shown). The data indicate that in contrast to the apoA-I mutations that induce HTG, the apoA-I[Δ(89–99)] mutation does not affect significantly the binding of apoA-I to TG-rich particles.
Conformation and Stability of the ApoA-I Forms.
To improve our understanding of how the mutations introduced into apoA-I alter the affinity of the protein for TG-rich particles, we analyzed the biophysical properties of the apoA-I forms. The CD and fluorescence studies for WT apoA-I and apoA-I[Δ(8999)] were performed in this work, and similar measurements for apoA-I[Δ(61–78)], apoA-I[E110A/E111A], and also WT apoA-I were performed previously.16 The parameters for each apoA-I form are summarized in Table 2. As some of the mutants were expressed in the baculovirus expression system and the others in the adenovirus expression system, the data for WT apoA-I from both expression systems are included, and the observations are based on the comparison of the apoA-I mutants to WT apoA-I expressed in the same expression system, as in previous studies.16 The data indicate that the mutations in apoAI[Δ(61–78)] and apoA-I[E110A/E111A] lead to a decreased R-helical content of apoA-I and increased ANS fluorescence intensity. As fluorescence of the hydrophobic dye ANS is insignificant in solution and is increased when the dye binds to hydrophobic surfaces, the data indicate that the apoA-I[Δ(61–78)] and apoAI[E110A/E111A] mutants have more exposed hydrophobic surface area than WT apoA-I. The significantly reduced values for the midpoint of thermal denaturation (Tm) and the free energy of denaturation (ΔGD°) for apoA-I[Δ(61–78)] and apoA-I[E110A/ E111A] indicate the lower stability of these mutants compared to WT apoA-I. The reduced effective enthalpy (ΔHv) is consistent with low cooperativity of unfolding of these mutants and suggests, together with the increased exposure of hydrophobic surface and destabilization, that the apoA-I[Δ(61–78)] and apoA-I[E110A/ E111A] mutants have a more loosely folded conformation than WT apoA-I. The mutation in apoA-I[Δ(89–99)] leads to a decreased Rhelical content and significantly reduced stability, as well. However, in contrast to those in apoA-I[Δ(61–78)] and apoA-I[E110A/ E111A], the mutation in apoA-I[Δ(89–99)] leads to slightly reduced ANS fluorescence, suggesting slightly less exposed hydrophobic surfaces in this mutant compared to WT apoA-I. Thus, the two apoA-I forms that induce HTG exhibit enhanced ANS binding compared to the two apoA-I forms that do not induce HTG.
Table 2.
α-Helical Content, Thermodynamic Parameters, and ANS Binding for the ApoA-I Variants in Solution
| α-helixc (%) | Tmd (°C) | ΔHvd (kcal/mol) | ΔGD°e (kcal/mol) | Pf (relative units) | |
|---|---|---|---|---|---|
| WT apoA-Ia | 58 ± 2 | 60 ± 0.5 | 41 ± 1 | 2.5 ± 0.1 | 1.0 |
| apoA-I[Δ(89–99)]a | 52 ± 2g | 48 ± 1i | 9 ± 2i | 1.7 ± 0.1h | 0.8 |
| apoA-I[Δ (61–78)]a | 50 ± 3h | 47 ± 1i | 16 ± 1i | 1.0 ± 0.2i | 1.6 |
| WT apoA-Ib | 59 ± 2 | 59 ± 1 | 48 ± 1 | 3.0 ± 0.2 | 1.0 |
| apoA-I[E110A/E111A]b | 52 ± 2h | 54 ± 0.5h | 30 ± 1h | 2.0 ± 0.2g | 1.5 |
Proteins were expressed in the baculovirus expression system.
Proteins were expressed in the adenovirus expression system.
The α-helical content was calculated from the mean residue ellipticity at 222 nm determined from the normalized far-UV CD spectra.
The melting temperature (Tm) and van’t Hoff enthalpy (ΔHv) were determined from van’t Hoff analysis of thermal unfolding curves monitored by CD.
The conformational stability (ΔGD°) was determined from the CD-monitored GdnHCl-induced unfolding.
The ANS fluorescence intensity (I) is shown relative to the ANS intensity in the presence of WT apoA-I expressed in the baculovirus expression system.
p < 0.05, compared to the values for WT apoA-I expressed in the same expression system.
p < 0.01, compared to the values for WT apoA-I expressed in the same expression system.
p < 0.005, compared to the values for WT apoA-I expressed in the same expression system. The experimental data for WT apoA-I, apoA-I[Δ(61–78)], and apoA-I[E110A/E111A] can be found in ref 16.
DISCUSSION
The hypothesis tested in this study was that the apoA-I forms that cause HTG in apoA−/− mice have increased affinity for TGrich particles. We investigated two apoA-I mutants, apoA-I[Δ(61–78)] and apoA-I[E110A/E111A], that have been shown to trigger severe HTG when expressed in apoA−/− mice and compared them to WT apoA-I that does not induce HTG at similar levels of expression13,14 (Figure 1A). Our studies indicate that both apoA-I mutants that cause HTG exhibit enhaned binding to TG-rich emulsion particles compared to WT apoA-I (Figure 2). On the other hand, the apoA-I[Δ((89–99)] mutant, which does not induce HTG (but causes hypercholesterolemia), does not exhibit enhanced binding to TG-rich particles. This suggests a correlation between enhanced binding of apoA-I to TG-rich lipoproteins and induction of HTG. In agreement with the results of the binding experiments, in plasma of apoA-I−/− mice expressing apoA-I[E110A/E111A] or apoA-I[Δ(61–78)], a portion of these proteins was detected in TGrich lipoprotein fractions, VLDL and intermediate-density lipoprotein (IDL).13,14 In contrast, no detectable apoA-I was found in the TG-rich lipoprotein fractions of apoA−/− mice expressing WT apoA-I or apoA-I[Δ(89–99)].14
Potential Implications of the Enhanced Binding of ApoA-I to TG-Rich Lipoproteins.
Larger numbers of amino acids per molecule of PC (Table 1) and larger numbers of protein molecules per emulsion particle (Figure 3) for apoA-I[Δ(61–78)] and apoA-I[E110A/E111A] indicate a higher ratio of protein to phospholipid on the surface of the particles containing these mutants. In mice expressing the apoA-I[Δ(61–78)] or apoA-I[E110A/ E111A] mutant, the higher affinity of these apoA-I variants for TGrich lipoproteins may result in a higher ratio of total protein to phospholipid in the surface monolayer of VLDL. The increased ratio of protein to phospholipid may lead to reduced fluidity of the surface monolayer of thelipoproteins.24,25 These surface changes mayinhibit insertion of lipoprotein lipase (LPL) into VLDL particles and ensuing hydrolysis of TG. Reduced fluidity of the surface monolayer resulting from the increased protein content may be one of the factors accounting for the reported reduced level of hydrolysis of VLDL isolated from plasma of mice expressing apoA-I[Δ(61–78)] and apoA-I[E110A/E111A] compared to VLDL isolated from plasma of mice expressing WT apoAI, when lipolysis was promoted by purified recombinant LPL.13,14
Another consequence of the enhanced association of apoA-I with TG-rich lipoproteins may be a diminished area on the lipoprotein surface available for apoE molecules. Under these conditions, apoE molecules may adopt a more compact conformation or embed themselves more deeply into the lipid monolayer of the particles that may result in masking or burying of the receptor binding region of apoE. Alternatively, under the conditions of the more “dense” protein packing on the particle surface, the N-terminal domain of apoE may be displaced from the lipid surface and adopt an inactive four-helix bundle conformation.21 In both cases, the conformational changes in lipoprotein-bound apoE may impair the interaction of the receptorbinding region of apoE with the LDL receptor26 and clearance of remnant lipoproteins from the circulation. The enhanced binding of apoA-I to TG-rich lipoproteins may also lead to displacement from these lipoproteins of apoC-II that is required for activation of LPL.5 Displacement of apoC-II from TG-rich lipoproteins may account for the lower apoC-II content in VLDL and IDL fractions of mice expressingapoA-I[E110A/E111A] or apoA-I[Δ(61–78)] compared to the corresponding fractions of mice expressing WT apoA-I.13,14
Molecular Basis for the Enhanced Binding of the ApoA-I Mutants to TG-Rich Particles.
To understand the molecular basis for the enhanced ability of the apoA-I mutants to bind to TG-rich particles, we examined the conformational and stability characteristics of the proteins (Table 2). The mutations in apoAI[E110A/E111A] and apoA-I[Δ(61–78)] destabilize apoA-I and enhance ANS binding by the protein, indicating increased exposure of the hydrophobic surface in the protein tertiary structure. These structural changes are associated with enhanced binding of apoA-I to TG-rich particles and induction of HTG. In contrast, the mutation in apoA-I[Δ(89–99)] also destabilizes apoA-I but does not increase exposure of the protein hydrophobic surface. This mutation does not lead to enhanced binding of apoA-I to TG-rich particles and is not associated with HTG. These observations imply that for the apoA-I forms studied here, destabilization is not always associated with the increased exposure of the hydrophobic surface of the protein, and the apoA-I mutants that have the destabilized conformation with more exposed hydrophobic surface exhibit enhanced binding to TG-rich emulsion particles. Although destabilized apoA-I forms often have more exposed hydrophobic surface,12,16,27,28 it was shown that for some apoA-I variants, the exposure of hydrophobic surface did not correlate with protein destabilization.16,27–31 It was found that an apoA-I[Δ(88–98)] mutant, which differs from the apoA-I[Δ(89–99)] mutant only by two amino acids, is destabilized and, like the latter mutant, has less exposed hydrophobic surface than WT apoA-I.29 The authors suggested that the apoA-I[Δ(88–98)] mutant has highly uncooperative and unstable structure and attributed these characteristics to a rather unfolded structure with low conformational plasticity.29 While we realize that it is difficult to predict the structure of the apoAI[Δ(89–99)] mutant on the basis of the available characteristics (Table 2),thestudies of this mutantimply thatapoA-Idestabilization not associated with increased exposure of hydrophobic surface does not enhance binding of apoA-I to TG-rich particles. Protein conformations with more exposed hydrophobic surface are expected to enhance protein-lipid interactions, as they lead to diminished exposure of the hydrophobic surface to solution. Being an indicator of exposed hydrophobic surface, significant ANS binding by a protein has been shown to be a characteristic property of the molten globulelike or partially folded state of the protein.32,33 Molten globule-like conformations can provide proteins flexibility and adaptability for substantial conformational changes that accompany their binding to the lipid surface.34 Thus, the ANS fluorescence data suggest that compared to WT apoA-I and the apoA-I[Δ(89–99)] mutant, the apoA-I[E110A/E111A] and apoA-I[Δ(61–78)] mutants have more molten globule-like folding that may facilitate their binding to TG-rich particles. ApoE studies also show that more molten globule-like folding of the protein (and enhanced ANS binding by the proteins) is associated with enhanced binding of the protein to TG-rich particles. Of the tree common apoE isoforms (apoE2, apoE3, and apoE4), apoE4 was shown to have the strongest propensity to form a molten globule35 and the greatest ability to bindtoTG-richparticles.36 Our studiesof theapoE4forms15 showed that ANS binding to the proteins correlated positively with their binding to TG-rich particles. In a recent study of apoE3, apoE4, and their variants by Nguyen et al.,37 the protein forms that exhibited enhanced binding of ANS also had increased affinity for TG-rich particles (both emulsion and isolated plasma VLDL). Importantly, both apoA-I mutants that show enhanced binding to TG-rich particles, apoA-I[E110A/E111A] and apoA-I[Δ(61–78)], are destabilized. Apparently, lower conformational stability promotes binding of apoA-I to lipoprotein particles by facilitating a conformational change on binding to the particle surface. Various studies indicate that destabilization promotes lipid binding by apoA-I and other exchangeable apolipoproteins.28,30,31,35,38,39 For apoA-I, it was shown specifically that destabilization of the N-terminal helix bundle significantly increased the lipid affinity of the protein.28 It is likely that the mutations in apoA-I[E110A/E111A] and apoA-I[Δ(61–78)] destabilize the structure of the N-terminal domain16 and thereby promote binding of these mutants to the phospholipid-covered surface of TG-rich emulsion particles.
The biophysical characteristics of the apoA-I[E110A/E111A] and apoA-I[Δ(61–78)] mutants can aid in our understanding of why in plasma of apoA−/− mice expressing these proteins, these apoA-I variants partition between the HDL and VLDL/IDL fractions, while WT apoA-I is predominantly located in the HDL fraction.13,14 In contrast to the surface of TG-rich lipoprotein particles that is largely covered by phospholipids, up to 80% of the HDL particle surface is covered by protein.37 A surface plasmon resonance analysis of binding of apoA-I to HDL suggests that the initial binding of apoA-I to HDL can occur via either the interaction of theN-terminalhelix bundle with a protein site on the HDL particle surface or the interaction of the C-terminal domain with a lipid site on the HDL surface.40 As the mutations in apoA-I[E110A/E111A] and apoA-I[Δ(61–78)] are likely to disturb the structure of the N-terminal domain,16 the protein-protein interactions between the N-terminal domain and a protein site on HDL are probably weakened for these mutants, and their binding to HDL particles is mainly mediated by the protein-lipid interactions between the C-terminal domain and a lipid site on HDL. At the same time, the destabilized and partially folded conformation of these mutants with more exposed hydrophobic surface (probably resulting from destabilization of the N-terminal region) can promote protein-lipid interactions with the largely lipid-covered surface of VLDL.28,30 The intact N-terminal helix bundle of WT apoA-I mediates strong protein-protein interactions with a protein site of HDL, and the relatively small degree of exposure of the hydrophobic surface in WT apoA-I makes interactions of this protein with the largely lipidcovered surface of VLDL unfavorable.
Similar Changes in the Properties of the ApoA-I and ApoE4 Forms Associated with HTG.
After we have investigated the properties of the apoA-I forms associated with HTG, it is interesting to compare them with the properties of the apoE4 forms associated with HTG. We have previously studied the apoE4 forms that trigger mild and severe HTG when overexpressed in apoE−/− mice, apoE4-mut2 and WT apoE4, and the apoE4-mut1 that does not cause HTG at similar levels of expression15 (Figure 1B). The ability of the apoE4 forms to bind to TG-rich emulsion particles was examined in binding assays in a manner similar to that for the apoA-I forms. The results of this analysis (inset of Figure 2) show that like apoA-I, the apoE4 forms that induce HTG had an enhanced ability to bind to TGrich emulsion compared to the form that does not induce HTG. The trend in binding for the apoE4 forms (apoE4-mut1 < apoE4mut2 < WT apoE4) correlated positively with the trend in plasma TG concentrations in mice expressing the corresponding apoE4 form (Figure 1B). Similarly, the trend in emulsion binding for the apoA-I forms {WT apoA-I < apoA-I[Δ(61–78)] ~ apoA-I[E110A/E111A] (Figure 2)} correlates positively with the trend in plasma TG concentrations in mice expressing the corresponding apoA-I form (Figures 1A). These observations support the notion that that enhanced binding of apoA-I, as well as apoE, to TG-rich lipoproteins may be associated with induction of HTG. In the apoE4 binding studies, it was also found that the number of protein molecules bound to one emulsion particle is higher for the form that induces HTG (WT apoE4) compared to the form that does not induce HTG (apoE4-mut1) (inset of Figure 3). In Vivo, the increased content of apoE4 on VLDL and IDL, similar to the increased content of apoA-I, may be associated with structural changes in the lipoprotein particles that may reduce the level of lipolysis of the lipoproteins and impair clearance of lipoprotein remnants.15 Moreover, compared to apoE4-mut1, WT apoE4 and apoE4-mut2 have a destabilized conformation with more exposed hydrophobic surface in solution that may facilitate binding of the protein to TG-rich lipoproteins.15 Thus, the apoA-I forms and apoE4 forms that induce HTG share similar changes in the biophysical properties as compared to their counterparts that do not induce HTG. Taken together, these observations suggest that in mice, an increased level of association of apoA-I or apoE4 with TG-rich lipoproteins resulting from the proteins having a destabilized conformation with increased exposure of hydrophobic surface may contribute to the development of HTG. While various metabolic events may be involved into the development of HTG in humans, it is possible that structural mutations that increase the affinity of apoA-I, apoE, and perhaps some other apolipoproteins for TG-rich lipoproteins may contribute to some cases of this disorder. Analysis of newly discovered mutations in apolipoproteins associated with the development of HTG in animal models or humans will help to further test this hypothesis.
ACKNOWLEDGMENT
We are grateful to Dr. Vassilis I. Zannis and his lab for generation of the recombinant apoA-I forms and helpful comments on the manuscript. We thank Donald L. Gantz for help with electron microscopy analysis and Cheryl England and Michael Gigliotti for help with biochemical assays.
Funding Sources
This work was supported by National Institutes of Heath Grants POHL 26335 and RO1 HL 48739.
ABBREVIATIONS
- ANS
8-anilino-2-naphthalenesulfonate
- apo
apolipoprotein
- apoA-I−/−
apoA-I-deficient
- apoE−/−
apoE-deficient
- CD
circular dichroism
- GdnHCl
guanidine hydrochloride
- HDL
highdensity lipoprotein(s)
- HTG
hypertriglyceridemia
- IDL
intermediate-density lipoprotein(s)
- LPL
lipoprotein lipase
- PC
phosphatidylcholine
- TG
triglyceride
- VLDL
very low-density lipoprotein(s)
- WT
wild type
REFERENCES
- (1).Hokanson JE, and Austin MA (1996) Plasma triglyceride level is a risk factor for cardiovascular disease independent of highdensity lipoprotein cholesterol level: A meta-analysis of populationbased prospective studies. J. Cardiovasc. Risk 3 (2), 213–219. [PubMed] [Google Scholar]
- (2).Therond P (2009) Catabolism of lipoproteins and metabolic syndrome. Curr. Opin. Clin. Nutr. Metab. Care 12, 366–371. [DOI] [PubMed] [Google Scholar]
- (3).Gupta A, and Gupta V (2010) Metabolic syndrome: What are the risks for humans? Biosci. Trends 4 (5), 204–212. [PubMed] [Google Scholar]
- (4).Havel RJ (2010) Triglyceride-rich lipoproteins and plasma lipid transport. Arterioscler., Thromb., Vasc. Biol 30, 9–19. [DOI] [PubMed] [Google Scholar]
- (5).Huang Y, Liu XQ, Rall SC, Taylor JM, von Eckardstein A, Assmann G, and Mahley RW (1998) Overexpression and accumulation of apolipoprotein E as a cause of hypertriglyceridemia. J. Biol. Chem 273, 26388–26393. [DOI] [PubMed] [Google Scholar]
- (6).Kypreos KE, van Dijk KW, van Der Zee A, Havekes LM, and Zannis VI (2001) Domains of apolipoprotein E contributing to triglyceride and cholesterol homeostasis In Vivo. Carboxyl-terminal region 203–299 promotes hepatic very low density lipoprotein-triglyceride secretion. J. Biol. Chem 276, 19778–19786. [DOI] [PubMed] [Google Scholar]
- (7).Kypreos KE, van Dijk KW, Havekes LM, and Zannis VI (2005) Generation of a recombinant apolipoprotein E variant with improved biological functions: Hydrophobic residues (Leu-261, Trp264, Phe-265, Leu-268, Val-269) of apoE can account for the apoEinduced hypertriglyceridemia. J. Biol. Chem 280, 6276–6284. [DOI] [PubMed] [Google Scholar]
- (8).Pownall HJ, and Ehnholm C (2006) The unique role of apolipoprotein A-I in HDL remodeling and metabolism. Curr. Opin. Lipidol 17, 209–213. [DOI] [PubMed] [Google Scholar]
- (9).Navab M, Anantharamaiah GM, Reddy ST, Van Lenten BJ, Ansell BJ, and Fogelman AM (2006) Mechanisms of disease: Proatherogenic HDL—an evolving field. Nat. Clin. Pract. Endocrinol. Metab 2 (9), 504–511. [DOI] [PubMed] [Google Scholar]
- (10).Sorci-Thomas MG, and Thomas MJ (2002) The effects of altered apolipoprotein A-I structure on plasma HDL concentration. Trends Cardiovasc. Med 12, 121–128. [DOI] [PubMed] [Google Scholar]
- 11).Sviridov D, Hoang A, Huang W, and Sasaki J (2002) Structure-function studies of apoA-I variants: Site-directed mutagenesis and natural mutations. J. Lipid Res 43 (8), 1283–1292. [PubMed] [Google Scholar]
- (12).Alexander ET, Tanaka M, Kono M, Saito H, Rader DJ, and Phillips MC (2009) Structural and functional consequences of the Milano mutation (R173C) in human apolipoprotein A-I. J. Lipid Res 50 (7), 1409–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Chroni A, Kan H-Y, Kypreos KE, Gorshkova IN, Shkodrani A, and Zannis VI (2004) Substitutions of Glu110 and Glu111 in the middle helix 4 of human apoA-I by alanine affect the structure and in vitro functions of apoA-I and induce severe hypertriglyceridemia in apoA-I-deficient mice. Biochemistry 43, 10442–10457. [DOI] [PubMed] [Google Scholar]
- (14).Chroni A, Kan H-Y, Shkodrani A, Liu T, and Zannis VI (2005) Deletions of helices 2 and 3 of human apoA-I are associated with severe dyslipidemia following adenovirus-mediated gene transfer in apoA-I-deficient mice. Biochemistry 44, 4108–4117. [DOI] [PubMed] [Google Scholar]
- (15).Gorshkova IN, Kypreos KE, Gantz DL, Zannis VI, and Atkinson D (2008) Biophysical properties of apolipoprotein E4 variants: Implications in molecular mechanisms of correction of HTG. Biochemistry 47, 12644–12654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Gorshkova IN, Liu T, Kan HY, Chroni A, Zannis VI, and Atkinson D (2006) Structure and stability of apolipoprotein A-I in solution and in discoidal high-density lipoprotein probed by double charge ablation and deletion mutation. Biochemistry 45, 1242–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Chen YH, Yang JT, and Martinez HM (1972) Determination of the secondary structures of proteins by circular dichroism and optical rotatory dispersion. Biochemistry 11 (22), 4120–4131. [DOI] [PubMed] [Google Scholar]
- (18).Gorshkova IN, Liu T, Zannis VI, and Atkinson D (2002) Lipid-free structure and stability of apolipoprotein A-I: Probing the central region by mutation. Biochemistry 41, 10529–10539. [DOI] [PubMed] [Google Scholar]
- (19).Lowry OH, Rosebrough NJ, Farr AL, and Randall RJ (1951) Protein measurement with the Folin phenol reagent. J. Biol. Chem 193, 265–275. [PubMed] [Google Scholar]
- (20).Bartlett GR (1959) Phosphorus assay in column chromatography. J. Biol. Chem 234, 466–468. [PubMed] [Google Scholar]
- (21).Saito H, Dhanasekaran P, Baldwin F, Weisgraber KH, Lund-Katz S, and Phillips MC (2001) Lipid binding-induced conformational change in human apolipoprotein E. Evidence for two lipid-bound states on spherical particles. J. Biol. Chem 276, 40949–40954. [DOI] [PubMed] [Google Scholar]
- (22).Wang L, Atkinson D, and Small DM (2003) Interfacial properties of an amphipathic α-helix consensus peptide of exchangeable apolipoproteins at air/water and oil/water interfaces. J. Biol. Chem 278, 37480–37491. [DOI] [PubMed] [Google Scholar]
- (23).Nagle JF, and Tristram-Nagle S (2000) Lipid bilayer structure. Curr. Opin. Struct. Biol 10, 474–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Gorshkova IN, Menschikowski M, and Jaross W (1996) Alterations in the physicochemical characteristics of low and high density lipoproteins after lipolysis with phospholipase A2. A spin-label study. Biochim. Biophys. Acta 1300, 103–113. [DOI] [PubMed] [Google Scholar]
- (25).Tetali SD, Budamagunta MS, Simion C, den Hartigh LJ, Kalai T, Hideg K, Hatters DM, Weisgraber KH, Voss JC, and Rutledge JC (2010) VLDL lipolysis products increase VLDL fluidity and convert apolipoprotein E4 into a more expanded conformation. J. Lipid Res 51 (6), 1273–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Narayanaswami V, and Ryan RO (2000) Molecular basis of exchangeable apolipoprotein function. Biochim. Biophys. Acta 1483, 15–36. [DOI] [PubMed] [Google Scholar]
- (27).Tanaka M, Dhanasekaran P, Nguyen D, Ohta S, LundKatz S, Phillips MC, and Saito H (2006) Contributions of the Nand C-terminal helical segments to the lipid-free structure and lipid interaction of apolipoprotein A-I. Biochemistry 45, 10351–10358. [DOI] [PubMed] [Google Scholar]
- (28).Tanaka M, Dhanasekaran P, Nguyen D, Nickel M, Takechi Y, Lund-Katz S, Phillips MC, and Saito H (2011) Influence of N-terminal helix bundle stability on the lipid-binding properties of human apolipoprotein A-I. Biochim. Biophys. Acta 1811, 25–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Rogers D, Roberts LM, Lebowitz J, Datta G, Anantharamaiah GM, Engler J, and Brouillette CG (1998) The lipid-free structure of apolipoprotein A-I: Effect of amino-terminal deletions. Biochemistry 37, 11714–11725. [DOI] [PubMed] [Google Scholar]
- (30).Tanaka M, Koyama M, Dhanasekaran P, Nguyen D, Nickel M, Lund-Katz S, Saito H, and Phillips MC (2008) Influence of tertiary structure domain properties on the functionality of apolipoprotein A-I. Biochemistry 47 (7), 2172–2180. [DOI] [PubMed] [Google Scholar]
- (31).Koyama M, Tanaka M, Dhanasekaran P, Lund-Katz S, Phillips MC, and Saito H (2009) Interaction between the N- and C-terminal domains modulates the stability and lipid binding of apolipoprotein A-I. Biochemistry 48, 2529–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Semisotnov GV, Rodionova NA, Razgulyaev OI, Uversky VN,Gripas AF,andGilmanshin RI(1991)Studyofthe”moltenglobule” intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers 31, 119–128. [DOI] [PubMed] [Google Scholar]
- (33).Uversky VN, and Ptitsyn OB (1996) All-or-none solventinduced transitions between native, molten globule and unfolded states in globular proteins. Folding Des. 1 (2), 117–122. [DOI] [PubMed] [Google Scholar]
- (34).Ptitsyn OB (1995) Molten globule and protein folding. Adv. Protein Chem 47, 83–229. [DOI] [PubMed] [Google Scholar]
- (35).Morrow JA, Hatters DM, Lu B, Hochtl P, Oberg KA, Rupp B, and Weisgraber KH (2002) Apolipoprotein E4 forms a molten globule. A potential basis for its association with disease. J. Biol. Chem 277 (52), 50380–50385. [DOI] [PubMed] [Google Scholar]
- (36).Saito H, Dhanasekaran P, Baldwin F, Weisgraber KH, Phillips MC, and Lund-Katz S (2003) Effects of polymorphism on the lipid interaction of human apolipoprotein E. J. Biol. Chem 278, 40723–40729. [DOI] [PubMed] [Google Scholar]
- (37).Nguyen D, Dhanasekaran P, Nickel M, Nakatani R, Saito H, Phillips MC, and Lund-Katz S (2010) Molecular basis for the differences in lipid and lipoprotein binding properties of human apolipoproteins E3 and E4. Biochemistry 49, 10881–10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Hatters DM, Peters-Libeu CA, and Weisgraber KH (2005) Engineering conformational destabilization into mouse apolipoprotein E. A model for a unique property of human apolipoprotein E4. J. Biol. Chem 280, 26477–26482. [DOI] [PubMed] [Google Scholar]
- (39).Weers PM, Abdullahi WE, Cabrera JM, and Hsu TC (2005) Role of buried polar residues in helix bundle stability and lipid binding of apolipophorin III: Destabilization by threonine 31. Biochemistry 44, 8810–8816. [DOI] [PubMed] [Google Scholar]
- (40).Lund-Katz S, Nguyen D, Dhanasekaran P, Kono M, Nickel M, Saito H, and Phillips MC (2010) Surface plasmon resonance analysis of the mechanism of binding of apoA-I to high density lipoprotein particles. J. Lipid Res 51, 606–617.</References> [DOI] [PMC free article] [PubMed] [Google Scholar]